
Alveolar Macrophages Are Essential for Protection from Respiratory Failure and Associated Morbidity following Influenza Virus Infection
Acute respiratory viral infections can cause severe morbidity and pneumonia in infected individuals. Alveolar macrophages and various subsets of dendritic cells have been implicated in innate immunity and induction of anti-viral T cell responses that contribute to host defense against influenza virus infection. However, their relative importance in protection from pathology and disease severity has never been compared side by side. In this report, we demonstrate that mice lacking alveolar macrophages succumb to infection with low dose influenza virus and vaccinia virus infection due to respiratory failure. In contrast, mice lacking lymphoid CD8α+ and lung CD103+ DCs survived and showed little if any differences in disease severity compared to infected wild-type mice. These results indicate that therapies supporting AM and lung function may be beneficial during severe respiratory viral infection.
Published in the journal:
. PLoS Pathog 10(4): e32767. doi:10.1371/journal.ppat.1004053
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004053
Summary
Acute respiratory viral infections can cause severe morbidity and pneumonia in infected individuals. Alveolar macrophages and various subsets of dendritic cells have been implicated in innate immunity and induction of anti-viral T cell responses that contribute to host defense against influenza virus infection. However, their relative importance in protection from pathology and disease severity has never been compared side by side. In this report, we demonstrate that mice lacking alveolar macrophages succumb to infection with low dose influenza virus and vaccinia virus infection due to respiratory failure. In contrast, mice lacking lymphoid CD8α+ and lung CD103+ DCs survived and showed little if any differences in disease severity compared to infected wild-type mice. These results indicate that therapies supporting AM and lung function may be beneficial during severe respiratory viral infection.
Introduction
Alveolar macrophages (AM) are lung-resident macrophages important for the maintenance of surfactant homeostasis in the alveolar space [1]. Their importance for lung physiology becomes evident in a rare human syndrome termed “pulmonary alveolar proteinosis” (PAP), which is characterized by the accumulation of surfactant material and a varying degree of respiratory insufficiency [2]. PAP patients have a higher risk for pulmonary infections with opportunistic pathogens [3]. PAP typically occurs in patients that spontaneously develop GM-CSF autoantibodies [4] or carrying mutations in the GM-CSF receptor α chain [5] associated with impaired function and/or reduced numbers of AM. Similarly, mice lacking GM-CSF (Csf2−/−) or the receptor β chain (Csf2rb−/−) develop PAP [6], [7], [8], [9] and display increased susceptibility to a range of bacterial and fungal infections, which is associated with impaired innate functions of AM [10], [11], [12], [13], [14]. High phagocytic activity and expression of pattern recognition receptors provides them with the capacity to respond to bacterial and fungal pathogens. However, AM were also described to have anti-inflammatory properties based on direct inhibition of the antigen-presenting function of lung DCs [15] and production of immunosuppressive mediators such as IL-10 and nitric oxide [16]. In addition, AM were proposed to sequester pulmonary antigen and thereby interfere with efficient priming of immune responses by DCs [17]. In contrast to the well-described functions of AM in bacterial and fungal infections, their precise role during viral infections is poorly understood. AM have the capacity to endocytose adenovirus particles, which was impaired in cells isolated from Csf2-deficient mice [10]. Furthermore, AM are potent producers of type I IFNs upon pulmonary virus infection [18], [19]. A beneficial role of AM in influenza infection has been proposed based on AM depletion experiments [20], [21], [22] and treatment of mice with GM-CSF that increased numbers of AM [23], [24]. However, besides the described role of GM-CSF for AM function under steady-state conditions, GM-CSF also influences DCs as demonstrated by its positive effects on the homeostasis of CD103+ DCs [25] or the upregulation of CD103 on CD11b− DCs [26]. Indeed, a recent report proposed that GM-CSF protects from lethal influenza virus infection by enhancing CD103+ DC mediated anti-viral T cell responses [27].
In this study, we revisited these controversies by comparing DC subpopulations and anti-viral CD8+ T cell responses in the presence or absence of AM with the outcome of respiratory viral infection in Csf2−/−, Csfrb2−/−, Batf3−/−, CD11c-Cre/Ppargfl/fl (with a defect in AM) and wild-type mice. Csf2-deficient mice developed severe morbidity and hypersuceptibility to influenza virus and vaccinia virus infection due to the complete absence of AM, although CD103+ DC mediated anti-viral T cell responses were unaffected. In contrast, virus-induced morbidity was moderately affected in Batf3-deficient mice, which mounted impaired anti-viral T cell responses due to the absence of CD8α+ DCs and lung CD103+ DCs. Selective depletion of AM using clodronate liposomes prior influenza virus infection resulted in morbidity comparable to Csf2-deficient mice. Moreover, mice with functionally impaired AM due to conditional deletion of Pparg showed increased disease severity and respiratory failure despite normal T cell responses. In contrast, selective postnatal AM reconstitution in Csfrb2−/−mice prevented pulmonary pathology and morbidity following influenza virus infection. Overall, our study provides evidence for a vital function of AM for the maintenance of lung function upon pulmonary viral infections.
Results
Alveolar macrophages fail to develop in the absence of GM-CSF production by non-hematopoietic cells
Mice lacking GM-CSF (Csf2−/−) or the β subunit of its receptor (Csf2rb−/−) have been reported to develop alveolar proteinosis due to a defect in terminal maturation and surfactant catabolism of AM [6], [8]. However, AM characterized as CD45+CD11c+Siglec-F+ cells with high autofluorescence were completely absent in lung and BAL of adult Csf2−/− mice (Figure 1A, B and Figure S1). Staining with a viability dye showed a striking enrichment of dead non-hematopoietic (CD45−) cells, presumably consisting mainly of lung epithelial cells (Figure 1C), indicating that the removal of dead cells (efferocytosis) in the respiratory tract is impaired in the absence of AM. Indeed, following tracheal instillation of labeled apoptotic thymocytes, we found that AM are by far the most potent cell type compared to DCs and neutrophils in removal of apoptotic cells from the bronchoalveolar space (Figure 1D and Figure S1). The few remaining cells that showed up in the CD11c+Siglec-F+ population of Csf2−/− mice were contaminating dead cells with high autofluorescence as demonstrated by comparing the analysis with and without prior exclusion of dead cells (Figure S1). Absence of AM resulted in the accumulation of lipoproteinaceous and eosinophilic material in the BAL and lung of Csf2−/− mice (Figure 1E–G). Moreover, while BAL of WT mice consisted almost entirely (>90%) of AM, BAL of Csf2−/− mice contained a substantial proportion of neutrophils (Figure 1H). This indicates low-grade inflammation possibly induced by the accumulation of dead cells (Figure 1C). Nonetheless, arterial oxygen saturation in Csf2−/− and WT mice was comparable indicating unimpaired gas exchange in the lung in the absence of AM under steady state (Figure 1I).
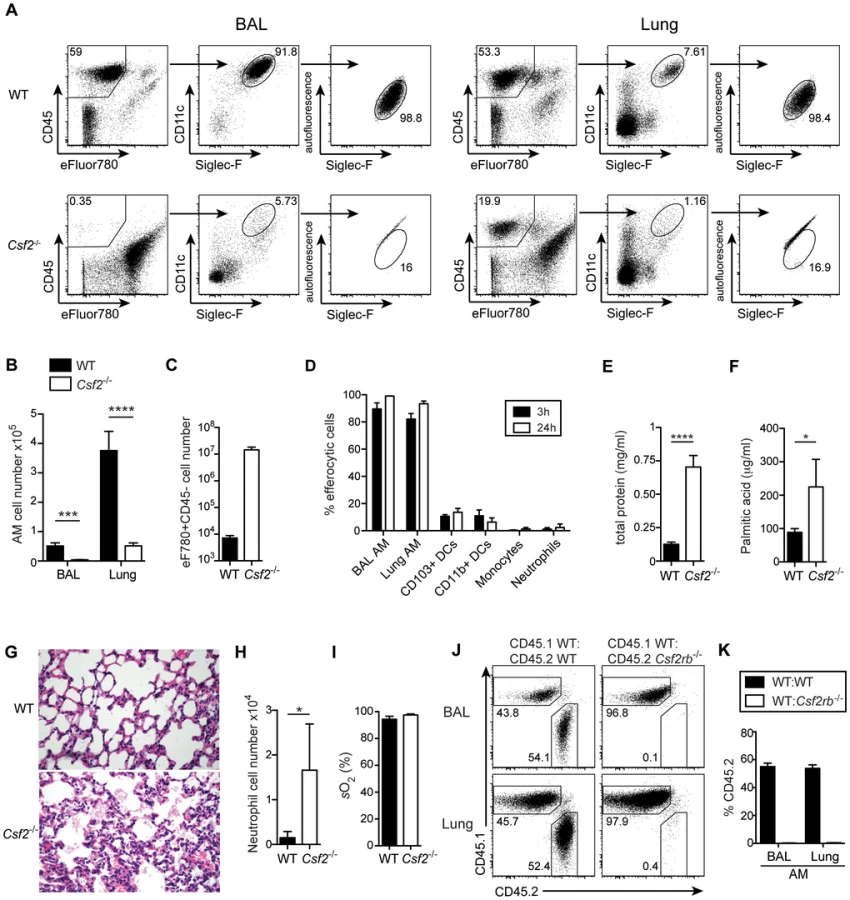
To determine whether AM require cell intrinsic GM-CSFR signaling for their development, we generated mixed bone marrow (BM) chimeras by adoptive transfer of a 1∶1 mixture of BM cells from Csf2rb−/− (CD45.2+) and WT (CD45.1+) mice (as well as CD45.2+WT and CD45.1+WT BM as controls). The reconstituted AM in the BAL and lung of Csf2rb−/−:WT chimeras were exclusively of WT donor origin and prevented development of alveolar proteinosis. (Figure 1 J, K and data not shown). In agreement with a recent report, these data show a requirement of GM-CSF signaling for the reconstitution of AM post-irradiation [28].
GM-CSF can be expressed by a variety of different cell types including T cells, macrophages, mast cells and non-hematopoietic cells [29]. To assess the source of GM-CSF for the development of alveolar macrophages, we generated 4-way Csf2−/− and WT crisscross BM chimeras. Irradiated WT recipients that received BM from either Csf2−/− or WT mice did not develop any pulmonary alveolar proteinosis during 16 weeks post-reconstitution. In contrast, Csf2−/− recipients reconstituted with WT or Csf2−/− BM showed a massive and comparable proteinosis (Figure S2). These results identify radio-resistant non-hematopoietic cells such as epithelial cells as the primary source of GM-CSF in the lung, which is absolutely essential for development of alveolar macrophages.
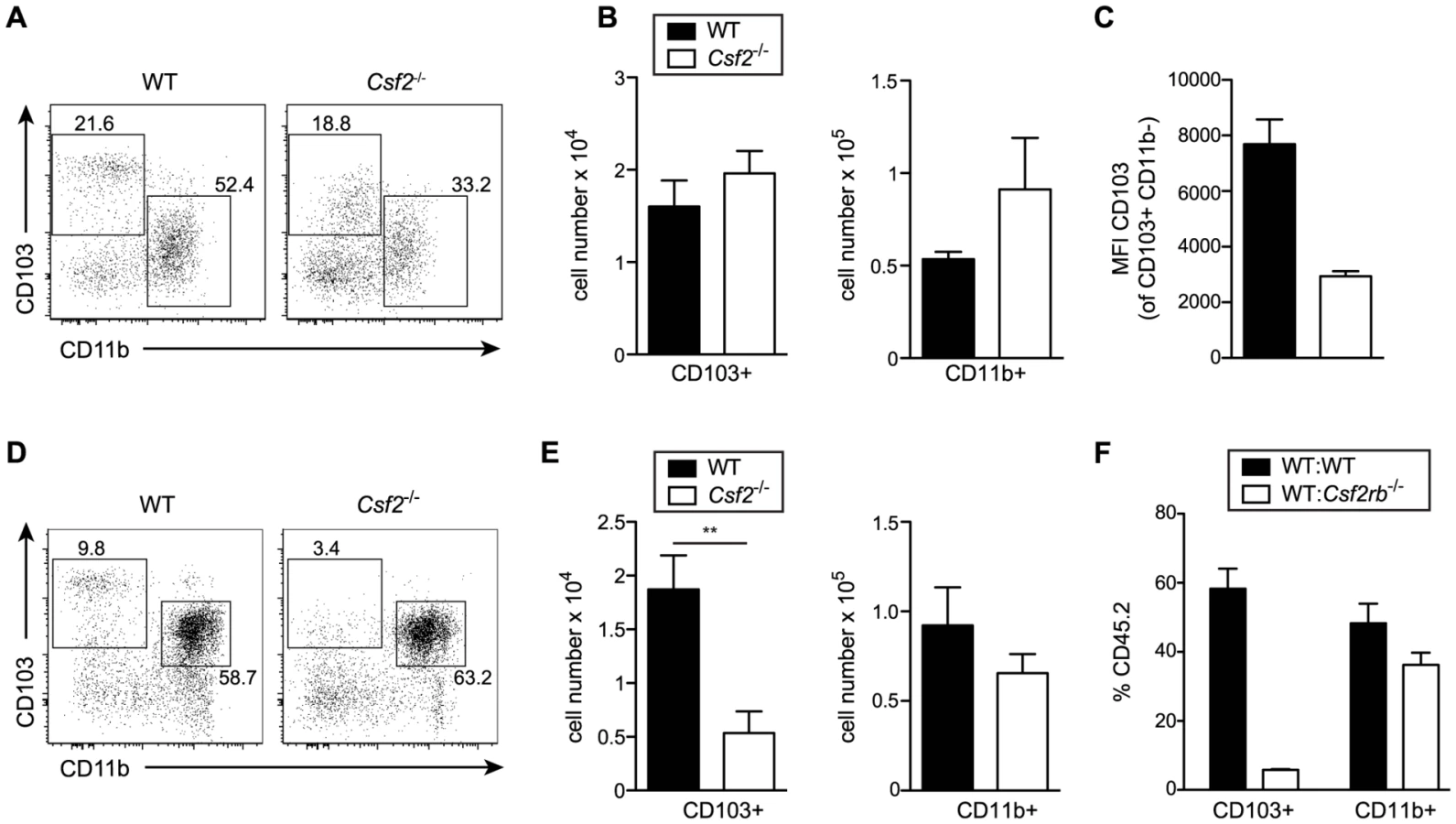
Development of lung CD103+ DCs has been reported to depend on GM-CSF [25]. However, comparing adult WT and Csf2−/− mice, we found similar numbers of pulmonary CD103+ DCs, although CD103 surface expression on CD11b−CD11c+ DCs was strongly impaired in Csf2−/− mice (Figure 2A–C) consistent with a previous study using Csf2rb−/− mice [26]. Interestingly, CD103+ DCs were strongly reduced in 6-day-old Csf2−/− pups (Figure 2D, E). In addition, injection of BM from Csf2rb−/− donors to irradiated WT recipients failed to reconstitute pulmonary CD103+ DCs (Figure 2F) [25], while other myeloid cell populations in the lung and peripheral blood were comparably well reconstituted from Csf2rb−/− and WT bone marrow cells (Figure S2). These data suggest that GM-CSF plays an important role in development of CD103+ DCs, although a population of CD103lowCD11b− DCs can substitute typical CD103+ DCs in the absence of GM-CSF possibly under inflammatory conditions.
Csf2−/− mice have a reduced resistance to influenza infection despite intact anti-viral T and B cell responses
To determine whether absence of AM and/or impaired CD103+ DC compartment affect resistance of Csf2−/− mice to respiratory viral infection, we infected WT and Csf2−/− mice with a sublethal dose of influenza virus PR8. Compared to WT mice, Csf2−/− animals showed an earlier onset of disease and a much more pronounced loss of body weight and body temperature (Figure 3A, B). Strikingly, whereas all WT mice recovered from infection, moribund Csf2−/− mice had to be euthanized between day 10 and 12 post-infection (Figure 3C). Moreover, Csf2−/− mice showed an increased lung virus titer at the peak of viral load (i.e. day 6) and a slight delay in viral clearance (Figure 3D), which were nevertheless to small to explain the moribund state of the Csf2−/− mice. Importantly, the fatal outcome of infection resulted from the absence of GM-CSF production by non-hematopoietic cells as demonstrated by a higher susceptibility of WT→Csf2−/− compared to Csf2−/−→WT BM chimeras (Figure S3).

Lung CD103+ DCs have been reported to induce CD8+ T cell responses by cross-presentation of influenza virus [19], [30]. Notably, Csf2−/− mice mounted intact anti-viral CD8+ T cell effector responses in BAL, lung, and draining LN (Figure 3E, F) despite a 2-fold reduction in lung CD103+ DC frequencies and decreased CD103 surface levels during influenza virus infection (Figure 3G). Nonetheless, total numbers of CD103+ DCs were similar in Csf2−/− compared to wild-type mice (Figure 3H). In contrast, influenza virus infected Batf3-deficient mice were almost entirely devoid of lung CD103+ DCs (Figure 3G, H) and showed a strong reduction in virus-specific CD8+ T cells and anti-viral effector cytokine (i.e. TNFα and IFNγ) production [19]. Regardless, morbidity and disease severity remained almost completely unaffected in Batf3-deficient compared to wild-type mice, while Csf2−/− mice succumbed to infection (Figure 3I). Notably, Batf3−/− mice were able to efficiently clear the virus despite impaired anti-viral CD8+ T cell responses (Figure S4).
To further characterize the adaptive response and rule out a possible defect in anti-viral antibody responses in the absence of GM-CSF, we assessed anti-viral B cell responses by measurement of influenza HA-specific antibodies. Csf2−/− mice showed increased levels of virus-specific IgA and IgG2a antibodies in BAL and serum compared to wild-type mice (Figure S4). Furthermore, we found similar NK cell recruitment and increased activation in Csf2−/− compared to wild-type mice (Figure S4).
Together these results demonstrate that CD103+ DC mediated anti-viral CD8+ T cell responses or B cell responses are intact and therefore not the underlying reason for the disease severity in Csf2−/− mice.
AM are crucial for the maintenance of respiratory function during influenza virus infection
As shown above, absence of AM in mice lacking GM-CSF results in accumulation of surfactant material and dead cells under homeostatic conditions. Interestingly, influenza virus infection strikingly aggravated pulmonary alveolar proteinosis in Csf2−/− mice indicated by an obstruction of alveoli with aggregates of eosinophilic material (Figure 4A), increased total protein concentration (Figure 4B) and accumulation of surfactant material in the BAL fluid at days 6 and 10 p.i. (Figure 4C). Moreover, the BAL of Csf2−/− mice was highly enriched in dead cells and cellular debris indicating impaired clearance of apoptotic cells (Figure 4D–F). Accordingly, respiratory function as measured by arterial oxygen levels was critically impaired at the time when Csf2−/− mice succumb to infection (Figure 4 G, H). These results suggest that AM prevent influenza induced morbidity by maintenance of lung function through removal of dead cells and surfactant material.

Selective restoration of AM development in Csf2rb−/− mice prevents severe morbidity and mortality following influenza virus infection
AM were described to maintain independently from blood monocytes through self-renewal [31]. We and others recently showed that AM originate from a fetal monocyte precursor in the lung around E17–19, which subsequently differentiates into mature AM during the following 5–7 days ([32]; Schneider et al., unpublished data). Single intranasal transfer of CD45+ lung cells from E18.5 fetuses into Csf2rb−/− newborns completely restored development of mature AM (Figure 5A, B) but did not contribute to the pool of BM-derived short-lived DCs including CD103+ DCs 6 weeks after transfer (Figure 5C). Thus, we generated mice lacking Csf2rb in every cell but AM. Presence of wild-type AM in Csf2rb−/− mice protected from pulmonary proteinosis, severe morbidity and mortality following influenza infection (Figure 5 D–F). Moreover, viral load in Csf2rb−/− mice with restored AM compartment was reduced to levels observed in wild type controls (Figure 5G). These results unequivocally demonstrate that the absence of AM is responsible for disease severity and rule out a putative contribution of DCs or other immune cells in mice lacking GM-CSF or its receptor.

Selective AM depletion results in respiratory failure and increased morbidity following influenza virus infection
To further rule out that a defect in CD103+ DCs contributed to the susceptibility of Csf2−/− mice to influenza virus infection, we selectively depleted AM in wild-type mice prior to infection using clodronate liposomes. Although DC subsets remained unaffected in clodronate-treated mice (Figure S5), AM-depletion resulted in increased morbidity (i.e. temperature and weight loss) (Figure 6A, B) and mortality (Figure 6C), which was associated with hypoxia (Figure 6D, E) basically resembling the phenotype observed in Csf2−/− mice. These results further support the conclusion that absence of AM and respiratory failure rather than impaired CD103+ DC-mediated CD8+ T cell responses are responsible for the high morbidity of Csf2−/− mice to influenza virus infection.
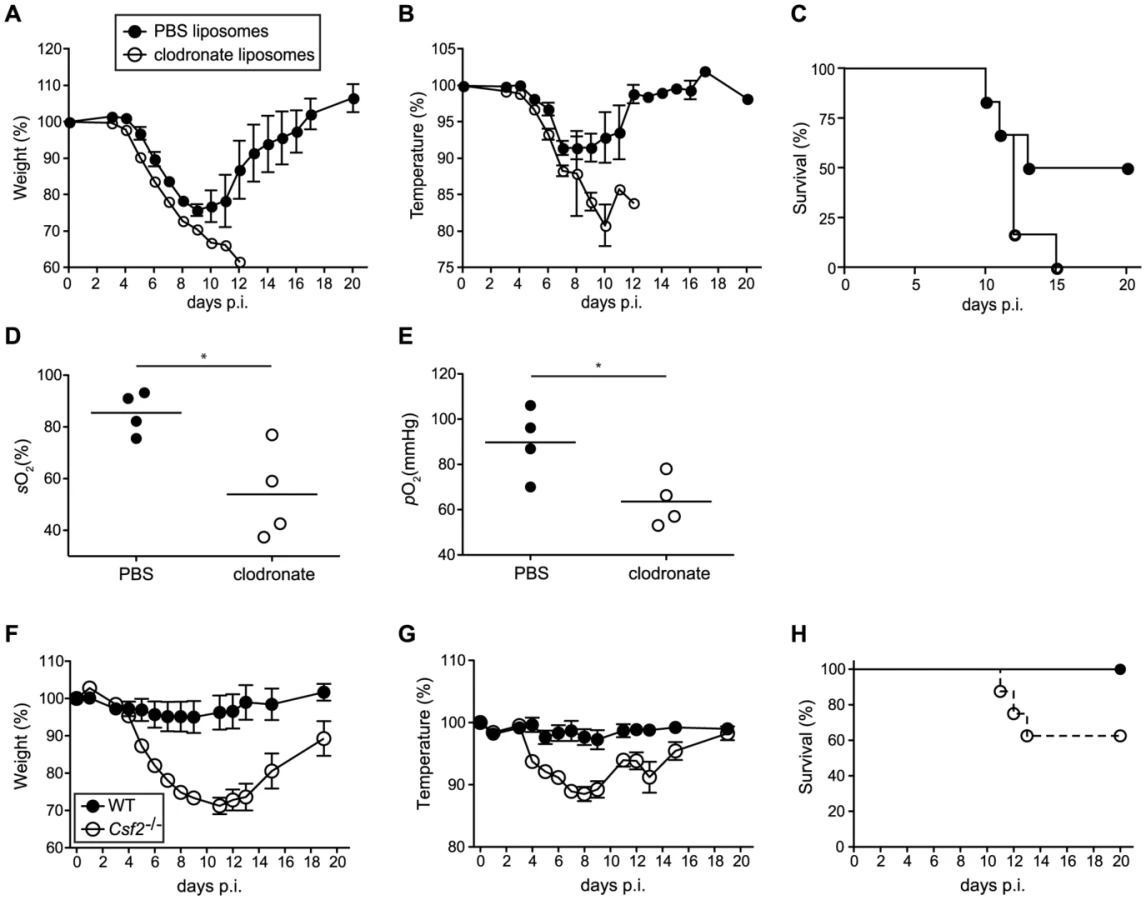
To assess whether the AM play an important role in resistance to other pulmonary viral infections, we assessed the outcome of infection with vaccinia virus (WR) in Csf2−/− and control WT mice. Control of vaccinia virus mainly depends on innate and T helper cell mediated B cell antibody responses, whereas the cytotoxic T cell response plays only a marginal role [33], [34]. When infected with a virus dose that did not induce any morbidity in WT mice, Csf2−/− mice showed a pronounced loss of body weight and temperature (Figure 6F, G) starting already during the innate immune response at day 4 p.i. and displayed increased lethality (Figure 6H), reminiscent to the outcome of influenza virus infection. Altogether, these results establish a direct role of AM in prevention of fatal respiratory viral infections.
Mice with immature and dysfunctional alveolar macrophages due to the absence of PPARγ show increased mortality and reduced lung function following influenza infection
Mice lacking Pparg specifically in macrophages (LysM-Cre/Ppargfl/fl) develop pulmonary alveolar proteinosis [35]. Moreover, using CD11c-Cre/Ppargfl/fl mice that lack Pparg in AM and DCs, we recently found that development of AM is arrested in an immature state with a defect in surfactant catabolism and lipid metabolism while development of DC subsets remained unaffected (data not shown). To determine whether absence of functional AM affects resistance of CD11c-Cre/Ppargfl/fl mice to respiratory viral infection, we infected WT and CD11c-Cre/Ppargfl/fl mice with influenza virus PR8. Compared to WT, CD11c-Cre/Ppargfl/fl mice showed an earlier onset and more pronounced loss of body weight and body temperature (Figure 7A, B) associated with increased lethality (Figure 7C), although lung virus titers were insignificantly elevated (Figure 7D). Besides a slightly increased number of neutrophils, recruitment of inflammatory cells to the lung was in general normal in CD11c-Cre/Ppargfl/fl mice (Figure S6). Moreover, we found similar frequencies and total numbers of anti-viral CD8+ T cells as well as IFNγ-producing CD4+ and CD8+ T cells (Figure 7E and Figure S6) indicating that DC-mediated T cell priming and effector responses were unaffected in CD11c-Cre/Ppargfl/fl mice. Likewise, levels of anti-influenza HA-specific antibody levels (i.e. IgG2c, IgA and IgG1) were comparable in BAL and serum of CD11c-Cre/Ppargfl/fl and WT mice (Figure 7F and Figure S6). Together these results indicate that innate and adaptive anti-viral immune responses were largely intact in CD11c-Cre/Ppargfl/fl mice.

Lungs of infected CD11c-Cre/Ppargfl/fl mice contained increased amounts of total protein in BAL fluid with a high proportion of lipid-engorged foam cells and alveoli filled with cellular infiltrate and debris (Figure 7G–I and Figure S6). Moreover, CD11c-Cre/Ppargfl/fl mice displayed significantly lower lung function as measured by arterial oxygen levels (Figure 7J, K). Taken together, CD11c-Cre/Ppargfl/fl mice with immature and dysfunctional AM show increased morbidity and respiratory failure due to an impaired removal of dead cells and debris following influenza virus infection similar to, but not as pronounced as Csf2−/− mice that are completely devoid of AM.
Influenza virus infection potently upregulates expression of interferon-induced transmembrane protein 3 in alveolar macrophages
GM-CSF-deficient mice lacking alveolar macrophages showed 5–10-fold increased viral titers and a delayed viral clearance. Therefore, we next asked whether alveolar macrophages might contribute directly to viral clearance besides their role in prevention of respiratory failure following viral infection. Infection of C57BL/6 mice with PR8 or NS1-GFP influenza virus showed a considerable proportion of AM containing viral material as determined by staining of viral NP or expression of NS1-GFP (Figure 8A, B). In addition, comparison of gene expression profiles of sorted AM from naïve and influenza virus infected mice by microarray analysis showed that the latter displayed a typical interferon signature with a 4 - to 65-fold upregulation of several interferon-induced genes including the interferon-induced transmembrane protein 3 (Ifitm3) and Ifitm6, which were upregulated 42 - and 9-fold, respectively (Figure 8C). Ifitm3 has recently been described as a key component in restricting viral spread and the morbidity and mortality following influenza virus infection [36]. Thus it is tempting to speculate that AM act like a virus sink and prevent morbidity at least partially through Ifitm3.

Discussion
In this study, we revisited the role of GM-CSF in AM homeostasis and function of this cell population in respiratory viral infection. According to the current understanding, Csf2 - and Csf2rb-deficient mice develop pulmonary alveolar proteinosis (PAP) due to a defect in terminal maturation of AM involving impaired lipid catabolism. However, using 8-parameter flow cytometry in combination with dead cell exclusion, we found that Csf2−/− mice were completely devoid of AM and presented a massive accumulation of dead CD45-negative cells, which are presumably epithelial cells, in the BAL and lung consistent with an important role of AM in removal of dead cells (efferocytosis). Indeed, usage of a viability dye in combination with CD45 staining was inevitable for the exclusion of highly auto-fluorescent dead cells and avoidance of misidentification as AM. We also observed increased numbers of neutrophils in the BAL of naive Csf2−/− mice supporting an association of impaired efferocytosis and chronic inflammatory lung disease [37]. Using mixed bone marrow chimeras we demonstrated that AM exclusively differentiated from WT and not from Csf2rb−/− BM. The absence of Csf2rb−/− AM in mixed BM chimeras also excludes the possibility that these cells have developed and died subsequently due to a functional defect in surfactant metabolism and accumulation of cellular debris, as the mixed chimeras did not develop PAP pathology. These results demonstrate a cell intrinsic requirement of GM-CSFR signaling for licensing development of AM from a precursor cell. Our results are in line with a recent report describing highly reduced numbers of AM in Csf2−/− mice and a requirement of GM-CSF signaling for the reconstitution of AM post-irradiation [28]. The license is provided by GM-CSF secretion of radio-resistant lung cells, as shown by our 4-way BM chimeras. Whether during homeostasis GM-CSF also influences the functionality of AM needs to be determined using inducible knock out strategies or antibody-mediated neutralization.
The role of GM-CSF for the development of DCs has been extensively studied. Most lymphoid tissue DCs develop normally in the absence of GM-CSF in vivo [38]. In contrast, GM-CSF has been shown to play a pivotal role for the development of non-lymphoid tissue DCs in the lamina propria [39] and the skin [40]. A recent report suggested that GM-CSF is critical for the homeostasis of tissue-resident CD103+ DCs in particular in the lung and skin [25], while others found that GM-CSF merely regulates CD103 expression levels on DCs and not development of this DC subset [26]. In keeping with the latter study by Edelson et al., we found that total CD11b−CD103+ DC cell numbers were not affected in the lung of adult Csf2-deficient mice, while CD103 surface expression on CD11b− DCs was clearly reduced. Interestingly, in lungs of neonatal Csf2−/− mice the CD103+ DC subpopulation was strongly reduced in cell numbers. Similarly, the vast majority of CD103+ DCs present in lungs of mixed bone marrow chimeras originated from WT but not Csf2rb−/− bone marrow and the few Csf2rb−/− BM-derived CD11b− lung DCs were CD103low. Whether impaired CD103 expression affects DC function is not clear. CD103+ DCs possess a unique potential in phagocytosis of virus-infected epithelial cells and capacity in cross-presentation of viral antigens to CD8+ T cells [41]. Several reports suggested that CD103+ DCs are the main inducers of CD8+ T cell responses to influenza virus infection with supportive evidence mainly based on depletion experiment using langerin-DTR mice [19], [30], [42]. It should be noted, however, that langerin is also expressed on CD8α+ DCs from spleen and LNs [43], [44], which are accordingly also depleted in langerin-DTR mice [45]. Indeed, LN-resident CD8α+ DCs as well as CD11b+ lung tissue DCs have also been implicated in influenza antigen transport, presentation and CD8+ T cell priming [30], [46], [47]. Batf3-deficient mice that are devoid of both CD8α+ DCs and CD103+ DCs showed a substantial reduction in anti-viral CD8+ T cell responses indicating that both CD8α+ DCs in spleen and lymph node together with lung migrating CD103+ DCs contribute to the induction and maintenance of T cell responses to influenza virus. In contrast, despite an almost 2-fold reduction in frequencies, total lung CD103+ DC numbers in influenza virus-infected Csf2−/− mice were comparable to wild-type mice and accordingly, antiviral CD8+ T cell numbers and cytokine responses in the BAL, lung, and the draining LN remained unaffected. However, the influenza-induced pathology and morbidity was far worse in Csf2-deficient mice lacking AM but having intact B and T cell responses as compared to Batf3-deficient mice with constrained CD8+ T cell responses due to the absence of both CD103+ DCs and CD8α+ DCs but intact AM indicating that only the latter are critical for survival of respiratory viral infection.
PAP was strikingly aggravated in influenza virus infected compared to naive Csf2−/− mice. The accumulation of cellular debris, dead epithelial cells and surfactant material in the respiratory tract in the absence of AM resulted in impaired gas exchange and eventually in a fatal hypoxia. Similar results were obtained by clodronate-mediated depletion of macrophages without affecting DCs in wild-type mice before viral infection. In keeping with our data, transgenic mice with lung-restricted GM-CSF overexpression (SPC-GM) and increased AM numbers [48] were better protected from influenza virus-induced morbidity [24]. However, using the same transgenic mice, another study linked the GM-CSF-induced protection to enhanced CD103+ DC-mediated antiviral T cell responses, although numbers of DCs and virus-specific CD4+ and CD8+ T cells in SPC-GM mice were comparable to non-transgenic WT mice and the viral load was significantly reduced as early as day 3 post-infection prior CD8+ T cell expansion and effector function [27]. Moreover, this study concluded that increased susceptibility of Csf2−/− mice is a consequence of defective CD103+ DC-mediated CD8+ T cell responses [27]. In contrast, protection from virus-induced pathology and mortality by reconstitution of AM development in Csf2rb−/− mice (Figure 5) unequivocally demonstrates that the absence of AM is the underlying reason for hypersusceptibility to influenza virus in mice lacking GM-CSF or its receptor. Notably, Csf2−/− mice showed also increased morbidity and pronounced lethality in response to pulmonary vaccinia virus infection, which does not depend on CD8+ T cells [33], [34]. These data suggest a vital role of AM in resistance to respiratory viral infection in general, although it should be noted that depletion of AM in mice infected with RSV did not alter subsequent disease development[49].
Induction of host cell death is a hallmark of viral infection including influenza virus infection. Indeed, increased cell death has been associated with the pathogenicity of highly virulent influenza virus [50]. Moreover, we found a correlation between viral burden, PAP, and severity of hypoxia in wild-type mice. Our findings suggest that the pivotal function of AM is the removal of dead cells and cellular debris to prevent clogging of the airways and maintain gas exchange during respiratory viral infection. In addition, AM may contribute to viral clearance or interfere with virus-induced morbidity by yet unknown mechanisms. Consistent with previous reports [51] [19], we found that influenza virus can directly infect AM (Figure 8A,B). Interestingly, transcriptome analysis of sorted AM from infected and uninfected mice revealed a striking upregulation of interferon response signature genes including high levels of Ifitm3. Ifitm3 has recently been described as a key component in restricting viral spread and the morbidity and mortality following influenza virus infection [36]. Moreover, high Ifitm3 expression in influenza-specific lung-resident CD8+ memory T cells confers resistance to infection and enhances survival of these cells upon recall infection with the virus [52]. Thus, induction of Ifitm3 in AM could serve as a mechanism to promote AM survival and thereby limit the loss of this vital cell type during influenza infection. Furthermore and in addition to their crucial role in maintaining respiratory function, AM could have a direct antiviral role serving as a sink for influenza virus consistent with slightly elevated virus titers in mice lacking AM.
Taken together, we identified a key function of alveolar macrophages in phagocytosis of dead cells and maintenance lung function in respiratory viral infections. Mice lacking Csf2 or Csf2rb are highly vulnerable to influenza virus infection due to the absence of AM but not potentially impaired DC/T cell immunity. These results have implications for therapies targeting Csf2 (GM-CSF).
Materials and Methods
Mice
Csf2−/− mice (originally kindly provided by A. Dunn, Ludwig Institute for Cancer Research, Royal Melbourne Hospital, Victoria, Australia) [7] were backcrossed to BALB/c for 7 generations [53]. Csf2−/− (C57BL/6) and Csf2rb−/− (C57BL/6) mice were kindly provided by B. Becher (University Zurich). C57BL/6 and BALB/c mice were purchased from Charles River (Germany). It should be noted that most of the results shown were done with Csf2−/− (C57BL/6) and validated with Csf2−/− (BALB/c). B6.129S(C)-Batf3tm1Kmm/J (Batf3−/−) mice [54] were purchased from the Jackson Laboratory. Ppargfl/fl mice [55] originally kindly provided by P. Chambon (Université Louis Pasteur, Illkirch Cedex, France) and backcrossed for 6 generations to C57BL/6 before crossing to CD11c-Cre [56] mice in our facility to generate CD11c-Cre/Ppargfl/fl mice. All animals were housed in individually ventilated cages under specific pathogen free conditions at BioSupport AG (Zurich, Switzerland) and used for experiments at between 8 and 12 weeks of age.
Ethics statement
All animal experiments were approved by the local animal ethics committee (Kantonales Veterinärsamt Zürich, licenses 167/2011 and 113/2012), and performed according to local guidelines (TschV, Zurich) and the Swiss animal protection law (TschG).
Cells suspension preparations
Mice were sacrificed by an overdose of pentobarbital sodium i.p. BAL was isolated by canalization of the trachea with a catheter. The lungs were flushed with 3×400 µl PBS and BAL cells were harvested by centrifugation. Lungs were digested with 2 mg/ml of type IV collagenase (Worthington) and 0.02 mg/ml DNaseI (Sigma) at 37°C for 45 minutes and subsequently passed through a 70 µm cell strainer.
Flow cytometry
Multiparameter analysis was performed on a FACSCanto II or LSR Fortessa (BD) and analyzed with FlowJo software (Tree Star). Monoclonal antibodies specific to mouse CD11c (N418), CD11b (M1/70), Ly-6C (HK1.4), Siglec-F (E50-2440, BD Biosciences), CD103 (2E7), CD115 (AFS98, eBioscience), CD45 (30-F11), CD45.1 (A20), CD45.2 (104), CD4 (GK1.5), CD8α (53-6.7), MHC class II (M5/114.15.2, eBioscience), Gr-1 (RB6-8C5, eBioscience), CD49b (DX5, eBioscience), CD69 (H1.2F3), TNF-α (MP6-XT22), IFN-γ (XMG1.1) were purchased from Biolegend unless otherwise stated. Dead cells were stained using eFluor780 (eBioscience). PE-conjugated peptide-MHC class I tetramers (H-2Db/NP34) with the peptide NP34 (NP366-374; ASNENMETM) from the nucleoprotein of influenza virus A/PR/8/34 were generated as described [57]. For detection of intracellular NP expression, cells were incubated with a monoclonal mouse anti-influenza NP antibody (HB-65, homemade) followed by staining with AF647-conjugated anti-mouse IgG (Life Technologies Co.). Prior to all stainings, FcγIII/II receptors were blocked by incubation with homemade anti-CD16/32 (2.4G2).
Phagocytosis of apoptotic cells
Thymocytes were isolated from C57BL/6 mice and apoptosis was induced by exposure to 60 mJ UV radiation (Spectrolinker XL-1500; Spectronics Corporation). After 2 h incubation at 37°C in IMDM+10% FCS, cells were labeled with 5 µM eFluor670 (eBioscience) according to the manufacturer's instructions, washed extensively with IMDM+10% FCS and delivered i.t. in PBS. 3 and 24 h after administration, efferocytosis by cells in the BAL and lung was assessed by flow cytometry.
Bone marrow chimeras
For mixed bone marrow chimeras, CD45.1+CD45.2+ mice were lethally irradiated (9.5 Gy, using a caesium source) and reconstituted with 5–10×106 BM cells of a 1∶1 mixture of CD45.1+WT∶CD45.2+WT or CD45.1+WT∶CD45.2+Csf2rb−/−. Mice were analysed 10 weeks post-reconstitution. For 4-way Csf2−/−/WT bone marrow chimeras, WT and Csf2−/− mice were lethally irradiated and reconstituted with 5–10×106 Csf2−/− or WT BM cells. Mice were analysed 16 weeks post-reconstitution.
BAL protein and fatty acid analysis
Total BAL protein concentration was measured by BCA Protein Assay (Thermo Scientific) according to the manufacturer's instructions. For fatty acid analysis, BAL lipids were extracted as described by Moser et. al [58] using methanol-methylene chloride (3∶1, v/v). Thereafter, two internal standards, deuterated C17 : 0 (Cambridge Isotope Laboratories, Inc., Andover, MA, USA) and deuterated C22 : 0 (Dr. Ehrenstorfer GmbH, Augsburg, Germany) were added to the samples. Derivatization was done by adding acetyl chloride followed by an incubation at 100°C for 1 h. The reaction was stopped with 7% K2CO3 and the extraction of fatty acid methyl esters was performed with hexane. After centrifugation for 20 min at 2500 rpm, the hexane layer was dried under a continuous nitrogen stream and resuspended in heptane. For the fatty acid analysis, gas chromatography-mass spectrometry was applied using a Finnigan PolarisQ ion trap gas chromatography-mass spectrometry system (Thermo Quest, Austin, TX, USA). For quantification of fatty acid methyl esters, the specific masses were extracted. Analytes were identified with authentic standards by comparison of their retention time and their mass spectrum.
Viruses and infection
Influenza virus strain PR8 (A/Puerto Rico/34, H1N1) was originally provided by J. Pavlovic, University Zurich. The influenza strain PR8 NS1-GFP carrying a GFP reporter in the NS segment was kindly provided by A. García-Sastre [51]. Vaccinia virus WR was kindly provided by J. Mercer, ETH Zurich. For infections, the mice were anaesthetized and intratracheally inoculated with indicated doses of virus in 50 µl endotoxin-free PBS. To determine influenza virus titers in the lungs, samples were collected on various days after infection, homogenized and serially diluted with MDCK cells as described [59]. Infected cells were detected using a monoclonal mouse anti-influenza NP antibody (HB-65, homemade). For the depletion of AM, mice were treated with 100 µl clodronate liposomes i.t. 2 days prior to infection. Clodronate was a gift from Roche Diagnostics GmbH and liposomes were prepared as previously described [60]. Control mice were treated with PBS liposomes.
In vitro stimulation
For restimulation, 1.5×105 bone marrow-derived dendritic cells (BMDC) were incubated overnight with 1×106 pfu UV-inactivated PR8 virus in 96-well plates. BMDC were pulsed with 1 µg/mL NP147 (Balb/c) or NP34 (C57BL/6) peptide for 2 hours before BAL, lung or LN cells from individual mice were added and restimulation was performed for 4–5 h in the presence of 2 µM monensin (Sigma-Aldrich). After surface staining and formalin-fixation, intracellular cytokine staining was done in the presence of 0.5% saponin using anti-mouse TNF-α FITC and IFN-γ APC and analysed by flow cytometry.
Detection of virus-specific antibodies
Serum or BAL fluid from indicated time points post-infection was measured for influenza HA-specific antibody levels. Ninety-six well plates (Maxisorp; Nunc) were coated with 5 µg/ml recombinant PR8 influenza virus HA (a kind gift of M. Bachmann, Cytos) in PBS overnight at 4°C. After blocking, serum and BAL fluid from individual mice were serially diluted and incubated at RT for 2 hours. Plates were washed and incubated with alkaline-phosphatase-labelled goat anti-mouse isotype-specific antibodies (Southern Biotech Technologies, Inc.) and developed using substrate p-nitrophenyl phosphate (Sigma-Aldrich). Optical densities were measured on an enzyme-linked immunosorbent assay reader (Bucher Biotec) at 405 nm.
Measurement of arterial oxygen saturation
The femoral artery was catheterized in anaesthetized (2% isoflurane in oxygen) mice and the wound was locally anaesthetized by the application of 2% lidocaine before the cut was closed and the catheter was sewn to the thigh to be held in place. The application of isoflurane was stopped and mice regained consciousness and were kept restrained in a dark card tube while normally breathing room air for 10 min to equilibrate blood gas. Subsequently, 100 µL arterial blood was taken from the catheter and blood gas composition was measured on an ABL800Flex blood gas analyzer (Radiometer, Denmark) before mice were sacrificed.
Lung histology
The lungs were removed, fixed in formalin and processed for Hematoxylin and Eosin (H&E) staining. Histological sections were evaluated according to general inflammation.
Cell sorting and transfer
Fetal CD45+ cells were sorted from the lungs of CD45.1+ wild-type E18.5 fetuses using a FACSAria IIIu (BD). Neonatal Csf2rb−/− recipient mice were anesthetized using Isoflurane and 1×105 fetal cells were administered i.n. in 10 µl PBS. Reconstitution of AM in the BAL and lung was assessed by flow cytometry 6 weeks post-transfer and mice were used for infection experiments at 8 weeks of age.
Microarray analysis
Lungs of naive or influenza-infected animals at d5 post-infection were processed as described and stained with eF780, anti-mouse CD45, CD11c, CD11b and Siglec-F. AM were sorted as eF780−CD45+CD11chighautofluorescencehighSiglec-F+ (BD FACSAria IIIu) and RNA was prepared using PureLink RNA Mini Kit (ambion, Life Technologies Co.), amplified and hybridized on the Affymetrix Mouse Gene 1.1 ST.
Statistical analysis
Mean values, SD, SEM, and Student's t test (unpaired) were calculated with Prism (GraphPad Software, Inc). p<0.05 (*), p<0.01 (**), p<0.001 (***), p<0.0001 (****).
Supporting Information
Zdroje
1. TrapnellBC, WhitsettJA (2002) Gm-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Annual review of physiology 64 : 775–802.
2. TrapnellBC, WhitsettJA, NakataK (2003) Pulmonary alveolar proteinosis. The New England journal of medicine 349 : 2527–2539.
3. SeymourJF, PresneillJJ (2002) Pulmonary alveolar proteinosis: progress in the first 44 years. American Journal of Respiratory and Critical Care Medicine 166 : 215–235.
4. KitamuraT, TanakaN, WatanabeJ, Uchida, KanegasakiS, et al. (1999) Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. The Journal of experimental medicine 190 : 875–880.
5. SuzukiT, SakagamiT, RubinBK, NogeeLM, WoodRE, et al. (2008) Familial pulmonary alveolar proteinosis caused by mutations in CSF2RA. The Journal of experimental medicine 205 : 2703–2710.
6. DranoffG, CrawfordAD, SadelainM, ReamB, RashidA, et al. (1994) Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science 264 : 713–716.
7. StanleyE, LieschkeGJ, GrailD, MetcalfD, HodgsonG, et al. (1994) Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proceedings of the National Academy of Sciences of the United States of America 91 : 5592–5596.
8. NishinakamuraR, NakayamaN, HirabayashiY, InoueT, AudD, et al. (1995) Mice deficient for the IL-3/GM-CSF/IL-5 beta c receptor exhibit lung pathology and impaired immune response, while beta IL3 receptor-deficient mice are normal. Immunity 2 : 211–222.
9. RobbL, DrinkwaterCC, MetcalfD, LiR, KöntgenF, et al. (1995) Hematopoietic and lung abnormalities in mice with a null mutation of the common beta subunit of the receptors for granulocyte-macrophage colony-stimulating factor and interleukins 3 and 5. Proceedings of the National Academy of Sciences of the United States of America 92 : 9565–9569.
10. BerclazP-Y, ZsengellérZ, ShibataY, OtakeK, StrasbaughS, et al. (2002) Endocytic internalization of adenovirus, nonspecific phagocytosis, and cytoskeletal organization are coordinately regulated in alveolar macrophages by GM-CSF and PU.1. Journal of immunology 169 : 6332–6342.
11. LeVineAM, ReedJA, KurakKE, CiancioloE, WhitsettJA (1999) GM-CSF-deficient mice are susceptible to pulmonary group B streptococcal infection. The Journal of clinical investigation 103 : 563–569.
12. Gonzalez-JuarreroM, HattleJM, IzzoA, Junqueira-KipnisAP, ShimTS, et al. (2005) Disruption of granulocyte macrophage-colony stimulating factor production in the lungs severely affects the ability of mice to control Mycobacterium tuberculosis infection. Journal of Leukocyte Biology 77 : 914–922.
13. BallingerMN (2006) Role of Granulocyte Macrophage Colony-Stimulating Factor during Gram-Negative Lung Infection with Pseudomonas aeruginosa. American Journal of Respiratory Cell and Molecular Biology 34 : 766–774.
14. PaineR, PrestonAM, WilcoxenS, JinH, SiuBB, et al. (2000) Granulocyte-macrophage colony-stimulating factor in the innate immune response to Pneumocystis carinii pneumonia in mice. Journal of immunology (Baltimore, Md : 1950) 164 : 2602–2609.
15. HoltPG, OliverJ, BilykN, McMenaminC, McMenaminPG, et al. (1993) Downregulation of the antigen presenting cell function(s) of pulmonary dendritic cells in vivo by resident alveolar macrophages. The Journal of experimental medicine 177 : 397–407.
16. MartinezJA, KingTE, BrownK, JenningsCA, BorishL, et al. (1997) Increased expression of the interleukin-10 gene by alveolar macrophages in interstitial lung disease. The American journal of physiology 273: L676–683.
17. MacLeanJA, XiaW, PintoCE, ZhaoL, LiuHW, et al. (1996) Sequestration of inhaled particulate antigens by lung phagocytes. A mechanism for the effective inhibition of pulmonary cell-mediated immunity. The American journal of pathology 148 : 657–666.
18. KumagaiY, TakeuchiO, KatoH, KumarH, MatsuiK, et al. (2007) Alveolar Macrophages Are the Primary Interferon-α Producer in Pulmonary Infection with RNA Viruses. Immunity 27 : 240–252.
19. HelftJ, ManicassamyB, GuermonprezP, HashimotoD, SilvinA, et al. (2012) Cross-presenting CD103+ dendritic cells are protected from influenza virus infection. The Journal of clinical investigation 122 : 4037–4047.
20. TumpeyTM, García-SastreA, TaubenbergerJK, PaleseP, SwayneDE, et al. (2005) Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. Journal of virology 79 : 14933–14944.
21. KimHM, LeeY-W, LeeK-J, KimHS, ChoSW, et al. (2008) Alveolar macrophages are indispensable for controlling influenza viruses in lungs of pigs. Journal of virology 82 : 4265–4274.
22. TateMD, PickettDL, Van RooijenN, BrooksAG, ReadingPC (2010) Critical role of airway macrophages in modulating disease severity during influenza virus infection of mice. Journal of virology 84 : 7569–7580.
23. HuangH, LiH, ZhouP, JuD (2010) Protective effects of recombinant human granulocyte macrophage colony stimulating factor on H1N1 influenza virus-induced pneumonia in mice. Cytokine 51 : 151–157.
24. HuangF-F, BarnesPF, FengY, DonisR, ChroneosZC, et al. (2011) GM-CSF in the Lung Protects Against Lethal Influenza Infection. American Journal of Respiratory and Critical Care Medicine
25. GreterM, HelftJ, ChowA, HashimotoD, MorthaA, et al. (2012) GM-CSF Controls Nonlymphoid Tissue Dendritic Cell Homeostasis but Is Dispensable for the Differentiation of Inflammatory Dendritic Cells. Immunity 36 : 1031–1046.
26. EdelsonBT, BradstreetTR, WumeshKC, HildnerK, HerzogJW, et al. (2011) Batf3-Dependent CD11blow/ − Peripheral Dendritic Cells Are GM-CSF-Independent and Are Not Required for Th Cell Priming after Subcutaneous Immunization. PloS one 6: e25660.
27. UnkelB, HoegnerK, ClausenBE, Lewe-SchlosserP, BodnerJ, et al. (2012) Alveolar epithelial cells orchestrate DC function in murine viral pneumonia. The Journal of clinical investigation 122(10): 3652–64.
28. HashimotoD, ChowA, NoizatC, TeoP, BeasleyMB, et al. (2013) Tissue-Resident Macrophages Self-Maintain Locally throughout Adult Life with Minimal Contribution from Circulating Monocytes. Immunity 38 : 792–804.
29. GassonJC (1991) Molecular physiology of granulocyte-macrophage colony-stimulating factor. Blood 77 : 1131–1145.
30. GeurtsvanKesselCH, WillartMAM, van RijtLS, MuskensF, KoolM, et al. (2008) Clearance of influenza virus from the lung depends on migratory langerin+CD11b - but not plasmacytoid dendritic cells. The Journal of experimental medicine 205 : 1621–1634.
31. YonaS, KimK-W, WolfY, MildnerA, VarolD, et al. (2013) Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38 : 79–91.
32. GuilliamsM, De KleerI, HenriS, PostS, VanhoutteL, et al. (2013) Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. The Journal of experimental medicine 210 : 1977–1992.
33. SpriggsMK, KollerBH, SatoT, MorrisseyPJ, FanslowWC, et al. (1992) Beta 2-microglobulin-, CD8+ T-cell-deficient mice survive inoculation with high doses of vaccinia virus and exhibit altered IgG responses. Proceedings of the National Academy of Sciences of the United States of America 89 : 6070–6074.
34. XuR, JohnsonAJ, LiggittD, BevanMJ (2004) Cellular and humoral immunity against vaccinia virus infection of mice. Journal of immunology 172 : 6265–6271.
35. BonfieldTL, ThomassenMJ, FarverCF, AbrahamS, KolozeMT, et al. (2008) Peroxisome proliferator-activated receptor-gamma regulates the expression of alveolar macrophage macrophage colony-stimulating factor. Journal of immunology 181 : 235–242.
36. EverittAR, ClareS, PertelT, JohnSP, WashRS, et al. (2012) IFITM3 restricts the morbidity and mortality associated with influenza. Nature 484 : 519–523.
37. DonnellyLE, BarnesPJ (2012) Defective phagocytosis in airways disease. Chest 141 : 1055–1062.
38. VremecD, LieschkeGJ, DunnAR, RobbL, MetcalfD, et al. (1997) The influence of granulocyte/macrophage colony-stimulating factor on dendritic cell levels in mouse lymphoid organs. European journal of immunology 27 : 40–44.
39. BogunovicM, GinhouxF, HelftJ, ShangL, HashimotoD, et al. (2009) Origin of the lamina propria dendritic cell network. Immunity 31 : 513–525.
40. KingIL, KroenkeMA, SegalBM (2010) GM-CSF-dependent, CD103+ dermal dendritic cells play a critical role in Th effector cell differentiation after subcutaneous immunization. The Journal of experimental medicine 207 : 953–961.
41. DeschAN, RandolphGJ, MurphyK, GautierEL, KedlRM, et al. (2011) CD103+ pulmonary dendritic cells preferentially acquire and present apoptotic cell-associated antigen. The Journal of experimental medicine 208 : 1789–1797.
42. KimTS, BracialeTJ (2009) Respiratory dendritic cell subsets differ in their capacity to support the induction of virus-specific cytotoxic CD8+ T cell responses. PloS one 4: e4204.
43. McLellanAD, KappM, EggertA, LindenC, BommhardtU, et al. (2002) Anatomic location and T-cell stimulatory functions of mouse dendritic cell subsets defined by CD4 and CD8 expression. Blood 99 : 2084–2093.
44. ValladeauJ, Clair-MoninotV, Dezutter-DambuyantC, PinJJ, KissenpfennigA, et al. (2002) Identification of mouse langerin/CD207 in Langerhans cells and some dendritic cells of lymphoid tissues. Journal of immunology 168 : 782–792.
45. KissenpfennigA, HenriS, DuboisB, Laplace-BuilhéC, PerrinP, et al. (2005) Dynamics and function of Langerhans cells in vivo: dermal dendritic cells colonize lymph node areas distinct from slower migrating Langerhans cells. Immunity 22 : 643–654.
46. BelzGT, SmithCM, KleinertL, ReadingP, BrooksA, et al. (2004) Distinct migrating and nonmigrating dendritic cell populations are involved in MHC class I-restricted antigen presentation after lung infection with virus. Proceedings of the National Academy of Sciences of the United States of America 101 : 8670–8675.
47. Ballesteros-TatoA, LeónB, LundFE, RandallTD (2010) Temporal changes in dendritic cell subsets, cross-priming and costimulation via CD70 control CD8+ T cell responses to influenza. Nature Immunology 11 : 216–224.
48. HuffmanJA, HullWM, DranoffG, MulliganRC, WhitsettJA (1996) Pulmonary epithelial cell expression of GM-CSF corrects the alveolar proteinosis in GM-CSF-deficient mice. The Journal of clinical investigation 97 : 649–655.
49. PribulPK, HarkerJ, WangB, WangH, TregoningJS, et al. (2008) Alveolar macrophages are a major determinant of early responses to viral lung infection but do not influence subsequent disease development. Journal of virology 82 : 4441–4448.
50. KashJC, TumpeyTM, ProllSC, CarterV, PerwitasariO, et al. (2006) Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature 443 : 578–581.
51. ManicassamyB, ManicassamyS, Belicha-VillanuevaA, PisanelliG, PulendranB, et al. (2010) Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proceedings of the National Academy of Sciences of the United States of America 107 : 11531–11536.
52. WakimLM, GuptaN, MinternJD, VilladangosJA (2013) Enhanced survival of lung tissue-resident memory CD8+ T cells during infection with influenza virus due to selective expression of IFITM3. Nature Immunology 14 : 238–245.
53. SondereggerI, IezziG, MaierR, SchmitzN, KurrerM, et al. (2008) GM-CSF mediates autoimmunity by enhancing IL-6-dependent Th17 cell development and survival. The Journal of experimental medicine 205 : 2281–2294.
54. HildnerK, EdelsonBT, PurthaWE, DiamondM, MatsushitaH, et al. (2008) Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science (New York, NY) 322 : 1097–1100.
55. ImaiT, TakakuwaR, MarchandS, DentzE, BornertJ-M, et al. (2004) Peroxisome proliferator-activated receptor gamma is required in mature white and brown adipocytes for their survival in the mouse. Proceedings of the National Academy of Sciences of the United States of America 101 : 4543–4547.
56. CatonML, Smith-RaskaMR, ReizisB (2007) Notch-RBP-J signaling controls the homeostasis of CD8 - dendritic cells in the spleen. The Journal of experimental medicine 204 : 1653–1664.
57. AltmanJD, MossPA, GoulderPJ, BarouchDH, McHeyzer-WilliamsMG, et al. (1996) Phenotypic analysis of antigen-specific T lymphocytes. Science (New York, NY) 274 : 94–96.
58. MoserAB, JonesDS, RaymondGV, MoserHW (1999) Plasma and red blood cell fatty acids in peroxisomal disorders. Neurochemical research 24 : 187–197.
59. BachmannMF, EcabertB, KopfM (1999) Influenza virus: a novel method to assess viral and neutralizing antibody titers in vitro. Journal of immunological methods 225 : 105–111.
60. van RooijenN, SandersA (1994) Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. Journal of immunological methods 174 : 83–93.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 4
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- The 2010 Cholera Outbreak in Haiti: How Science Solved a Controversy
- , , , Genetic Variability: Cryptic Biological Species or Clonal Near-Clades?
- Efficient Parvovirus Replication Requires CRL4-Targeted Depletion of p21 to Prevent Its Inhibitory Interaction with PCNA
- An Overview of Respiratory Syncytial Virus