
Targeting Human Transmission Biology for Malaria Elimination
Malaria remains one of the leading causes of death worldwide, despite decades of public health efforts. The recent commitment by many endemic countries to eliminate malaria marks a shift away from programs aimed at controlling disease burden towards one that emphasizes reducing transmission of the most virulent human malaria parasite, Plasmodium falciparum. Gametocytes, the only developmental stage of malaria parasites able to infect mosquitoes, have remained understudied, as they occur in low numbers, do not cause disease, and are difficult to detect in vivo by conventional methods. Here, we review the transmission biology of P. falciparum gametocytes, featuring important recent discoveries of genes affecting parasite commitment to gametocyte formation, microvesicles enabling parasites to communicate with each other, and the anatomical site where immature gametocytes develop. We propose potential parasite targets for future intervention and highlight remaining knowledge gaps.
Published in the journal:
. PLoS Pathog 11(6): e32767. doi:10.1371/journal.ppat.1004871
Category:
Review
doi:
https://doi.org/10.1371/journal.ppat.1004871
Summary
Malaria remains one of the leading causes of death worldwide, despite decades of public health efforts. The recent commitment by many endemic countries to eliminate malaria marks a shift away from programs aimed at controlling disease burden towards one that emphasizes reducing transmission of the most virulent human malaria parasite, Plasmodium falciparum. Gametocytes, the only developmental stage of malaria parasites able to infect mosquitoes, have remained understudied, as they occur in low numbers, do not cause disease, and are difficult to detect in vivo by conventional methods. Here, we review the transmission biology of P. falciparum gametocytes, featuring important recent discoveries of genes affecting parasite commitment to gametocyte formation, microvesicles enabling parasites to communicate with each other, and the anatomical site where immature gametocytes develop. We propose potential parasite targets for future intervention and highlight remaining knowledge gaps.
Introduction
Over 200 million people contract malaria each year, with nearly half of the global population at risk of infection [1]. Of the five malaria parasite species that infect humans, Plasmodium falciparum is responsible for the majority of the 600,000 malaria-related deaths occurring every year [1]. Despite repeated calls for global malaria eradication in the past century, the local and regional elimination programs that underlie this larger goal have proven to be challenging. In particular, the lack of an effective vaccine and the emergence of drug-resistant parasites suggests that interventions targeting the symptom-causing (asexual) parasite forms are insufficient to interrupt transmission. Indeed, many P. falciparum infections are not severe, and in highly endemic regions, most cause no symptoms at all but can nevertheless contribute to malaria transmission. The importance of these asymptomatic infections, in particular submicroscopic ones, for onward transmission remains controversial [2], although evidence suggests they contribute significantly [2–5]. With a view to defining the human infectious reservoir, research into the basic biology of parasite transmission, a process that remains poorly understood, has been recently reinvigorated.
The life cycle of P. falciparum is highly complex, involving several developmental stages in both the human host and the mosquito vector (Fig 1). The mature asexual blood stages within the human host are responsible for all the clinical symptoms of malaria, though they are unable to infect the mosquito and complete the parasite’s life cycle. Only the nonreplicating sexual blood stages of the parasite (male and female gametocytes), which circulate at much lower densities and peak at different times during infection than asexual parasites, are capable of developing in the mosquito vector and causing onward infection. Not only are the developmental processes and dynamics of gametocytes in the human poorly understood, making it difficult to correlate asexual parasitemia with gametocyte density [6], but also the association between gametocyte density in the blood and infectivity to mosquitoes appears to be nonlinear [7]. Unlike directly transmitted pathogens, therefore, parasite load per se is a poor indicator of infectiousness, and we currently lack efficient methods to measure the human infectious reservoir that underlies malaria transmission.

In order to target interventions during elimination programs and to design new transmission-blocking vaccines or drugs, we need a deeper understanding of the complex processes that underlie human infectiousness. Here, we will discuss recent advances in P. falciparum gametocyte biology, describe key knowledge gaps (Box 1) that must be addressed in order to direct future interventions, and identify potential parasite targets that may be explored for development of drugs and vaccines aiming to interrupt malaria transmission. We follow the parasite from commitment along the sexual pathway to hidden development in the human host and finally to transmission to the mosquito vector.
Box 1. Knowledge Gaps
Commitment
What factors activate the Apicomplexan Apetala 2 (ApiAP2)-mediated sexual conversion pathway (Fig 2)?
How are stress and the microvesicle pathway linked to epigenetic switch rates (Section 2)?
Which components within microvesicles modulate sexual conversion (Box 3)?
What drives reproductive restraint in P. falciparum, other Plasmodia, and Hemosporidia (Section 2)?
What determines the sex ratio, and how is it modulated (Section 2)?
Sequestration
How do gametocytes come to be in the bone marrow (Section 3, Fig 4)?
Do sexually committed merozoites preferentially invade young RBCs, whether reticulocytes in the vasculature or RBC progenitor cells in the extravascular compartment of the bone marrow (Fig 4)?
Does the presence of parasites and/or gametocytes in the bone marrow induce pathology of the hematopoietic system, such as dyserythropoiesis (Section 3)?
Is the bone marrow associated with immune tolerance, preventing immune recognition of parasites (Section 3)?
How does the mature (stage V) gametocyte exit the bone marrow and return to circulation? Can the parasite modulate the timing of exit (Section 3, Fig 4)?
Transmission
What is the best way to detect and measure infectious gametocytes (Section 4)?
What is the contribution of subdermal localization and aggregation of mature gametocytes towards infection (Section 4)?
How can gametocyte load be used to predict infectiousness to feeding mosquitoes (Section 4)?
How do the outcomes of feeding assays relate to human infectious reservoir (Section 4)?
What immune responses exist towards different stages of gametocytes in the human (Section 4)?
Developmental Decisions: To Proliferate or to Transmit?
The first step in the transmission of malaria parasites from the human host to the mosquito vector is the formation of male and female gametocytes. At this juncture, the parasite exhibits adaptive strategies with respect to two aspects of gametocyte formation: (i) the proportion of asexual parasites that develop into gametocytes, referred to as the conversion rate [8] and (ii) the ratio of male to female gametocytes formed [9]. The precise point within an individual parasite’s life cycle at which it commits to the sexual pathway and determines the ensuing gametocyte sex ratio remains unclear but is thought to occur prior to merozoite formation [10–13]. The molecular mechanism responsible for the switch from asexual to sexual development has remained elusive until the recent identification of a master gene, ap2-g, responsible for triggering a transcriptional cascade that initiates gametocytogenesis in both P. falciparum [14] and P. berghei [15], a malaria parasite of rodents (Box 2). This conserved member of the AP2-family of transcription factors appears to be generally epigenetically silenced in asexual parasites by P. falciparum histone deacetylase 2 (PfHda2) and P. falciparum heterochromatin protein 1 (PfHP1) but could be prone to stochastic activation, leading to the low level of “background” gametocyte formation commonly observed in vitro (Fig 2) [16,17]. Potential upstream mediators of the AP2 master switch include the schizont expressed proteins P. falciparum gametocyte development 1 (Pfgdv1) and Nima-related kinase (Pfnek4), of which the former has proven to be crucial for gametocyte formation [18,19]. At a population level, the timing of gametocyte appearance in asymptomatic and symptomatic infections is poorly understood.

Box 2. Epigenetic Control of Gametocyte Formation
Prior to the discovery of the Apicomplexan AP2 (ApiAP2) family of DNA-binding proteins, it was thought that Plasmodium parasites had a minimal set of transcription factors [101]. ApiAP2 proteins have been found to regulate a variety of developmental processes along the parasite cycle, including ookinete formation [102], sporozoite maturation [103], and development within hepatocytes [104]. AP2-G, a conserved member of the ApiAP2 transcription factor family, was recently identified as an essential regulator controlling the switch from asexual to sexual development in both P. falciparum [14] and P. berghei [15]. In P. falciparum, the Pfap2-g locus appears to be in an epigenetically silenced state in asexual parasites but could be prone to stochastic activation explaining the commonly observed low level of gametocyte formation under standard, noninducing culture conditions (Fig 2) [14]. Interestingly, recent evidence suggests that the expression of Pfap2-g could be mutually exclusive with that of var genes, the gene family encoding the major asexual surface ligand PfEMP1 responsible for asexual sequestration [14]. Indeed, PfHda2, an important histone-modifying enzyme involved in P. falciparum gene silencing, modulates not only expression of multigene families such as var, stevor, and rifs, but also sexual commitment by promoting transcriptional silencing of Pfap2-g [17]. Similarly, PfHP1, a regulator of heritable gene silencing, controls the mutually exclusive expression of antigenically varying genes and also regulates sexual conversion by maintaining Pfap2-g in an epigenetically silent state [16]. Taken together, these novel findings indicate that the decision between continuing to proliferate asexually and differentiating into nondividing but transmissible gametocytes is under transcriptional and epigenetic control and suggest a link between transmission and virulence.
Beyond this baseline rate of gametocyte formation, environmental modulation or “stress” (a poorly defined term used to indicate a variety of external factors detrimental to the parasite) has been reported to cause additional asexual parasites to commit to the sexual pathway [20,21]. A commonly used method to produce gametocytes has been developed based on this hypothesis [22] but has not been formally tested for an effect on conversion rate. It was thought that some unidentified parasite-derived factor found in this conditioned medium provides a stress signal that causes a subset of parasites to withdraw from the recurrent asexual replication and instead form gametocytes [23]. Microvesicles, derived from the membrane of parasitized red blood cells (pRBCs), have recently been shown to be the critical factor in conditioned media capable of inducing increased conversion to gametocytes [24,25]. Other putative triggers such as elevated levels of immature RBCs, serum, and immune cells from naturally infected donors have yet to be confirmed (Box 3, Fig 2) [6,26–29]. While the precise pathway of the microvesicle-mediated initiation of gametocytogenesis remains unresolved, it may occur via AP2-G or another member of this family of transcription factors. Microvesicles seem to be a method by which parasites communicate, initiating a response to high parasitemia or other unfavorable conditions and enhancing the prospect of rapid transmission to a new host [24,25]. A sensing mechanism of this type is likely to be of particular importance for pathogens that cause long, chronic infections, as they need to adapt to fluctuations in the host environment. Indeed, another protozoan parasite causing chronic infections, Trypanosoma brucei, has a density sensing mechanism associated with a life cycle switch [30–32].
Box 3. Communication between Parasites
Despite the ubiquitous in vitro usage of parasite-conditioned medium to induce gametocyte formation, it was unknown until recently that microscopic vesicles in conditioned media represent a critical factor responsible for the observed increase in gametocyte formation [24,25]. Microvesicles are released by both uninfected and parasitized RBCs, although the latter release about ten times more per cell and are the only ones capable of uptake of microvesicles [24]. Interestingly, recent work demonstrated that pRBC-derived microvesicles promote sexual differentiation, arguing that parasites are able to sense their environment and respond accordingly [24,25]. The identity of the factor(s) in microvesicles responsible for stimulating the increased sexual commitment remains elusive, but it must either directly or indirectly affect the transcriptional program of the parasite, potentially through the Pfgdv1/PfHda2/PfHP1/ApiAP2 pathway (Box 2 and Fig 2). Indeed, internalized microvesicles were observed in close association with the parasite nucleus [24], and transfer of plasmid DNA into the parasite nucleus has been shown [25]. The ability of the parasites to communicate is likely of great importance for the parasite to optimize the balance between transmission success and persistence.
P. falciparum is unique among Plasmodium species in its low epigenetic conversion rate in vitro [8,33] and extended gametocyte maturation time [34,35]. While conversion rates cannot be directly quantified during natural infections, the gametocyte density in experimental P. falciparum infections is often very low [6,36,37], consistent with conversion rates of 1% to 5% of pRBCs in quantitative models [38,39] and low levels during in vitro experiments under noninducing culture conditions [8,40]. Several plausible hypotheses have been proposed for “reproductive restraint,” the observed low gametocyte density in vivo [41], such as minimizing the immune response directed specifically towards gametocytes or reducing the parasite burden in mosquitoes [41,42]. These hypotheses are consistent with the observed low gametocyte density in vivo, although neither are specific to low conversion rates, since low gametocyte densities can result from both very low and very high conversion rates [41]. In the latter case with high conversion rates, the asexual parasite population is not able to grow to high density, limiting the absolute level of gametocytes possible. Alternatively, competition between parasite genotypes within the human host may generate a low conversion rate in P. falciparum, if survival through asexual growth is prioritized over gametocyte production [41]. Although studies with the rodent malaria parasite P. chabaudi, with its shorter gametocyte maturation period, show evidence of a reduced gametocyte conversion rate in the presence of multiple genotypes [43–45], there is no evidence of reduced conversion between genotypes of human Plasmodium species. Mixed infections, however, between P. falciparum and P. malariae show evidence of increased gametocyte production [46], consistent with enhanced conversion to gametocytes during stress.
The gametocyte sex ratio is another important developmental decision that varies among P. falciparum parasites [47,48] and could provide a target for transmission-reducing interventions. Parasites ensure that fertilization will succeed within the mosquito through production of a sufficient number of male microgametes to fertilize all the female macrogametes. The ratio of their precursors, male and female gametocytes, has been well studied theoretically for Apicomplexan parasites [45,49,50], and in particular for Plasmodium [45,51,52]. Evolutionary theory suggests that sexual differentiation will be biased towards production of female gametocytes to ensure an equal ratio of micro - and macrogametes (since male gametocytes form up to eight microgametes upon activation in the mosquito midgut, Fig 1) [53]. Intriguingly, evolutionary theory suggests that the parasites would benefit from modulation of their sex ratio, increasing the proportion of male gametocytes when there is greater opportunity for outbreeding during mixed infections in the mosquito midgut [54]. Indeed, elevated fractions of male P. falciparum gametocytes have been observed in infections comprised of multiple genotypes as compared to single genotype infections [55]. Although it has been suggested that merozoites from a single schizont are destined to form either all male or all female gametocytes [12,13], the same studies actually observed mixed progeny emerging from the same schizont [10–12]. The developmental mechanisms underlying the shift in population-level gametocyte sex ratios are only beginning to be elucidated, with recent work demonstrating the requirement of Pfmdv1 [56] and PfPuf2 [57] for male gametocyte development. With improvements in detection methods such as single-cell analyses, it will be possible to revisit this and other important developmental questions.
Recent breakthroughs in our understanding of the molecular mechanisms responsible for commitment to the sexual pathway offer new insights into ways to obstruct or prevent infectiousness to mosquitoes [14,15,18,24,25]. The identification of a transcriptionally regulated mechanism responsible for sexual commitment provides an opportunity to reduce or even abrogate the stochastic activation of the sexual pathway by blocking these key molecular players (Fig 2). Moreover, the discovery of how parasites communicate (Box 3), conceivably about conditions that are unfavorable for asexual growth, has opened up opportunities to specifically target sexual commitment by preventing parasite communication. For example, it may be possible to target parasite-derived microvesicles for vaccines, thereby blocking their ability to enhance sexual conversion rates.
Gametocyte Sequestration: Where Do They Hide?
Gametocytes are difficult to quantify in vivo since they sequester in tissues during the majority of their maturation [58–60]. Some of the most detailed in vivo dynamical data come from the early 20th century when malaria was used as a therapy for tertiary neurosyphilis [36–39]. Parasite and gametocyte densities were recorded daily for each patient with some infections lasting for hundreds of days [36]. In these infections, the peak density of mature gametocytes occurred around day 40 of patent parasitemia, approximately 13 days after the peak in asexual parasitemia (Fig 3), consistent with in vitro experiments in which gametocytes mature in eight to ten days and with previous theoretical work [61]. In some infections, gametocytes appeared as early as 13 days post–sporozoite inoculation, suggesting that gametocytes can form immediately upon release of merozoites from the liver into the blood stream (Fig 3).
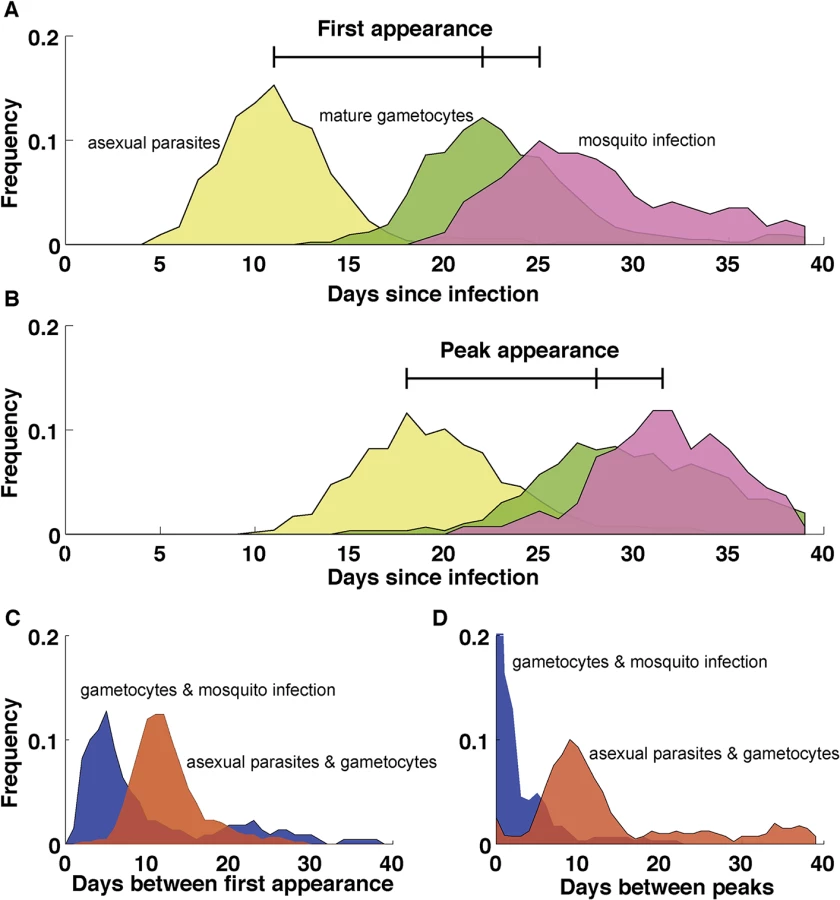
Several studies have observed immature gametocytes in the human bone marrow [59,60,62], but not in highly vascularized organs such as the heart, brain, and placenta [63–66], a strikingly different pattern from the sequestration of asexual stages. A recent quantitative analysis further confirmed the enrichment of immature gametocytes in bone marrow [67], although a secondary reservoir in the spleen could not be excluded. In addition to the distinct anatomical sequestration sites of asexual and gametocyte parasites, asexual pRBCs adhere to vascular endothelial cells but remain in the vasculature while some of the immature gametocytes develop in the extravascular space of the human bone marrow, a site where hematopoietic stem cells mature into RBCs [67,68]. Within this space, most gametocytes were found in close association with erythroblastic islands, structures that facilitate the development of cells of the erythroid lineage [69]. Interestingly, conditions such as anemia and dyserythropoiesis are associated with elevated gametocyte levels, demonstrating a potential interaction between gametocytes and the production of healthy RBCs within the hematopoietic system [70]. Despite the observed enrichment of gametocytes in the human bone marrow, successful in vitro culture of gametocytes demonstrates that their maturation does not require conditions unique to the bone marrow and is possible in both mature and immature RBCs.
There are potential benefits for extravascular sequestration of gametocytes: presence of a nutrient-rich environment with an abundance of young RBCs and potential reprieve from human immune responses. As gametocytes appear to thrive in blood rich with young RBCs [26,71–74], maturation within this main site for RBC production provides the parasites with an ample source of young host cells to invade. Although increased levels of young RBCs are associated with enhanced gametocyte densities, it remains unclear whether sexually committed merozoites preferentially invade RBC progenitor cells [28,67]. During development in the bone marrow, P. falciparum gametocytes may procure protection from the immune system. Recent in vivo data indicate that gametocytes are significantly less susceptible than asexual parasites to phagocytosis in the bone marrow [67,68], confirming previous in vitro data [75]. Similarly, a recent report indicated that latent Mycobacterium tuberculosis resides in the human bone marrow mesenchymal cells and hypothesizes that the bone marrow functions as a protected niche for the nonreplicating form of the bacteria [76].
It is currently unclear how immature gametocytes reach the bone marrow: whether gametocyte commitment and invasion occurs in the extravascular environment (causing gametocyte progeny to be born there) or young gametocytes home to the bone marrow and subsequently enter the extravascular space (Fig 4). As it has been demonstrated that gametocytes cannot cytoadhere efficiently to endothelial cells [77,78], a switch in cellular rigidity during both the initial and final stages of gametocyte maturation provides a potential explanation for how immature gametocytes are able to enter and mature ones are able to exit the bone marrow extravascular space [79–81]. Indeed rigidity-mediated gametocyte retention in the extravascular space could serve as a potential mechanism for the extended gametocyte maturation of P. falciparum.
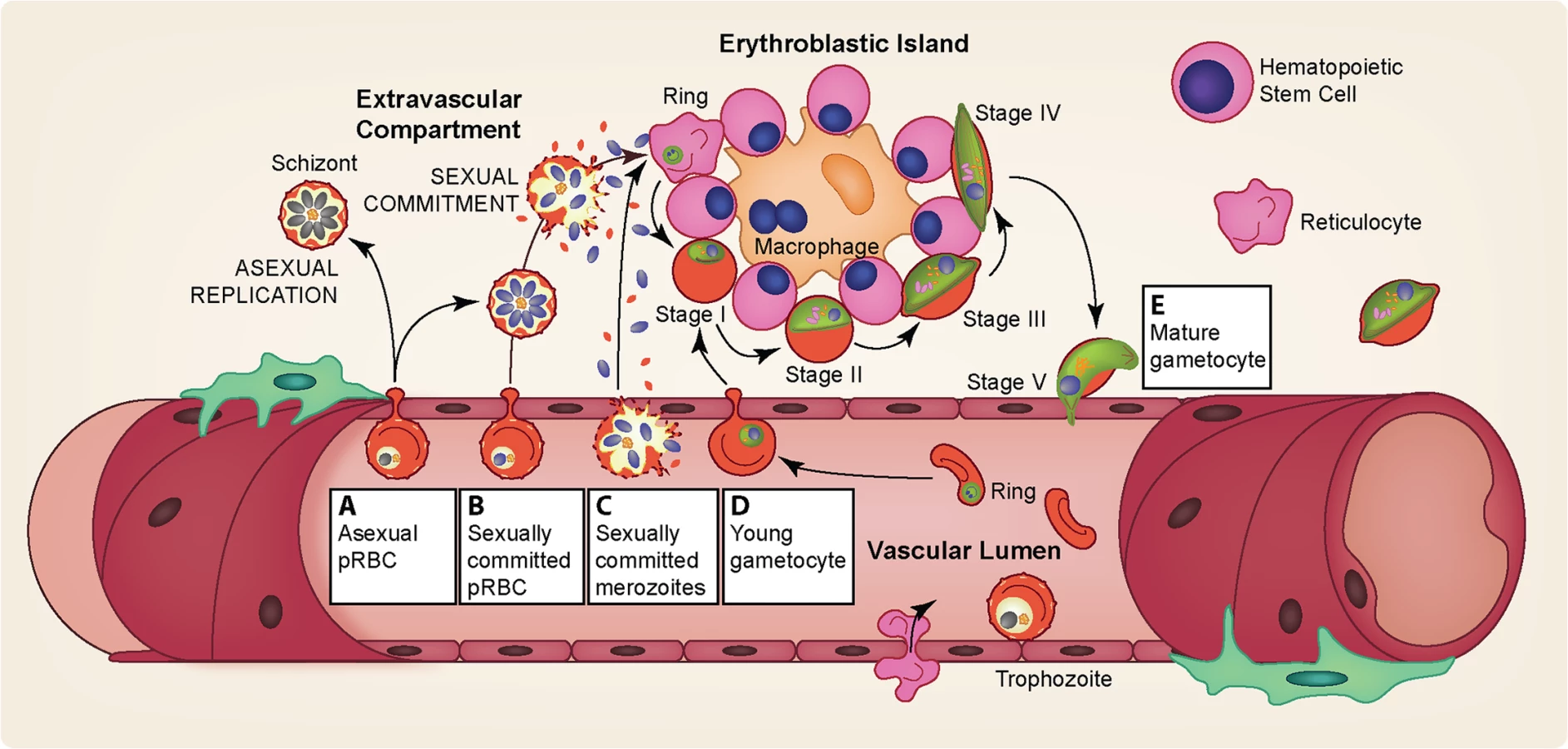
The sequestration of immature gametocytes from peripheral circulation during their prolonged development [58–60] provides potential targets for interventions at multiple points: the entrance into the extravascular space, the maturation within the bone marrow, and the re-entry into the peripheral circulation. For example, targeting surface antigens on pRBCs homing to the bone marrow could prevent parasites from even reaching this site. During development within the bone marrow, the close association of immature gametocytes with erythroblast islands (Fig 4) may play an important role in their maturation [67], and blocking this interaction by targeting potential gametocyte surface antigens could hinder proper gametocyte development. Drugs that prevent the rigidity switch during the end of maturation could potentially trap the gametocytes. Elucidation of the mechanistic basis and molecular factors involved in bone marrow homing, sequestration, and release will likely lead to ways of preventing successful gametocyte maturation and thereby reduce the reservoir of transmissible stages.
Determinants of Infectiousness to the Mosquito Vector
The nonlinear association between different parasite life cycle stages [6]—from asexual parasites in the human blood to sporozoites in the salivary glands of the mosquito—makes it difficult to use molecular data to determine the human infectious reservoir. Upon release from the extravascular compartment of the bone marrow, mature gametocytes appear to remain in circulation for around six days [38,82] and may require several days in circulation before becoming infectious to the mosquito (Fig 3) [36,83]. While in circulation, mature gametocytes must pass through microvasculature in the dermis in order to be accessible to feeding mosquitoes [84], but it remains to be seen whether mature gametocytes aggregate or preferentially localize in subdermal tissues to increase the likelihood of transmission [85]. Once ingested in a blood meal, both male and female gametocytes must be present for fertilization to occur. While the probability of transmission to the mosquito vector generally increases with higher gametocyte densities [7,86,87], mosquito infection has been observed at submicroscopic gametocyte levels [2–4,88]. These observations suggest that transmission is highly efficient even at very low gametocyte densities and that quantifying mature gametocytes capable of infecting mosquitoes remains challenging despite recent advancements in gametocyte detection [89].
Infectiousness of a P. falciparum–infected individual to a mosquito vector is difficult to reliably quantify by molecular measurements because of the low number of gametocytes detectable in peripheral blood [6,34,88] and the nonlinear relationship between mature gametocyte densities and oocyst presence in the mosquito [7]. Several different types of feeding assays are currently employed to measure infectiousness, but only direct skin feeding accurately recapitulates the environment during a normal blood feed [5]. For ethical reasons, this method is restricted to older children and adults, excluding the group with the highest prevalence of gametocytes [6,90,91] and thus limiting the scope of direct feeding as a tool for quantifying the infectious reservoir on a population level. The direct membrane-feeding assay (DMFA), in which mosquitoes feed on blood from a naturally infected gametocyte carrier through a membrane, is most commonly used, despite the unnatural transmission method [5]. The standard membrane-feeding assay (SMFA), in which mosquitoes are fed on gametocyte-rich cultured blood through a membrane and dissected to quantify oocyst burden following a gestation period, is the standard to test transmission-blocking activity in the lab [5,88,92]. SMFA, however, is an unnatural transmission method and often underestimates the infectivity of gametocytes [5]. Furthermore, all methods assume that the presence and level of oocysts in the mosquito midgut is indicative of mosquito infectivity. This relationship was recently demonstrated to be consistent for low-density infections despite a portion of the oocysts failing to release viable sporozoites [93,94]. Even in the cases in which oocysts burst and sporozoites successfully migrate to the salivary glands, not all bites from an infected mosquito are equally infectious. In general the heterogeneity in parasite maturation in the mosquito and the lack of standardized protocols for feeding assays makes it difficult to predict transmission efficiency from gametocyte levels [5,7].
Quantifying infectivity is also hindered by both human and mosquito immune responses, as well as competition the parasite encounters within the mosquito vector. The immune system of the mosquito itself must play a role in elimination of the malaria parasite, although the molecular determinants of mosquito protection are poorly understood [95,96]. Transmission-blocking immunity, in which gamete-specific antibodies present in the circulating blood of the human host are taken up in the blood meal and alter the development of parasites within the mosquito, potentially contributes to the lack of correlation between gametocytes and transmissibility [5,97]. Competitive interactions between different pathogens within the mosquito could also be potential factors affecting parasite development. Initial experiments in which Anopheles mosquitoes infected with the maternally inherited symbiotic bacteria Wolbachia fed on gametocyte cultures have demonstrated that Wolbachia significantly inhibits parasite development, although by an unknown mechanism [98].
The portion of the parasite life cycle in the mosquito and the bottlenecks experienced during the transfer of parasites from the human host to the mosquito vector and back have been touted as the most effective intervention targets for over a century. In addition to conventional methods such as insecticides to kill mosquitoes and gametocytocidal or sporonticidal drugs to remove the parasites in the human and vector hosts, novel strategies of intervention are needed. A recent finding that treating mosquitoes with a naturally occurring mosquito hormone that prevents them from mating may be an alternative way to control mosquito populations as insecticide resistance spreads [99]. Within individual mosquitoes, compounds, such as kinase inhibitors or antimicrobial peptides that enhance the mosquito’s ability to fight the parasite, could reduce the prevalence of parasites within mosquitoes. Competition within the mosquito is also a promising intervention, following the recent discovery of Anopheles populations naturally infected with Wolbachia [100] and the reduction in malaria parasite development observed in experimentally infected Anopheles populations [98].
Conclusion
Reductions in malaria-related morbidity and mortality over the past few decades have shifted the focus of research from attenuation of disease towards strategies to reduce transmission as part of elimination and eradication campaigns. Without a highly effective vaccine, combinations of malaria prevention and treatment are required, and the sexual stage of the parasite’s life cycle represents a key bottleneck and an ideal target for novel strategies. Recent developments in the basic biology of P. falciparum transmission, including the identification of genes affecting parasite commitment to gametocyte production, microvesicles enabling the parasites to communicate among themselves, and the anatomical site where immature gametocytes develop, have led to the identification of novel intervention targets in addition to more established transmission-blocking strategies. We highlight key knowledge gaps that need to be addressed for the successful development of drugs and vaccines that interrupt malaria transmission. More fundamental research on gametocyte biology will enhance not only our understanding of the basic principles underlying parasite transmission but also our ability to identify infectious individuals.
Zdroje
1. WHO (2014) WHO Malaria Report 2014. http://www.who.int/malaria/publications/world_malaria_report_2014/en/
2. Lin JT, Saunders DL, Meshnick SR (2014) The role of submicroscopic parasitemia in malaria transmission: what is the evidence? Trends Parasitol 30 : 183–190. doi: 10.1016/j.pt.2014.02.004 24642035
3. Lindblade KA, Steinhardt L, Samuels A, Kachur SP, Slutsker L (2013) The silent threat: asymptomatic parasitemia and malaria transmission. Expert Rev Anti Infect Ther 11 : 623–639. doi: 10.1586/eri.13.45 23750733
4. Schneider P, Bousema T, Omar S, Gouagna L, Sawa P, et al. (2006) (Sub)microscopic Plasmodium falciparum gametocytaemia in Kenyan children after treatment with sulphadoxine-pyrimethamine monotherapy or in combination with artesunate. Int J Parasitol 36 : 403–408. 16500657
5. Bousema T, Dinglasan RR, Morlais I, Gouagna LC, van Warmerdam T, et al. (2012) Mosquito feeding assays to determine the infectiousness of naturally infected Plasmodium falciparum gametocyte carriers. PLoS One 7: e42821. doi: 10.1371/journal.pone.0042821 22936993
6. Bousema T, Drakeley C (2011) Epidemiology and infectivity of Plasmodium falciparum and Plasmodium vivax gametocytes in relation to malaria control and elimination. Clinical microbiology reviews 24 : 377–410. doi: 10.1128/CMR.00051-10 21482730
7. Churcher TS, Bousema T, Walker M, Drakeley C, Schneider P, et al. (2013) Predicting mosquito infection from Plasmodium falciparum gametocyte density and estimating the reservoir of infection. Elife 2: e00626. doi: 10.7554/eLife.00626 23705071
8. Graves PM, Carter R, McNeill KM (1984) Gametocyte production in cloned lines of Plasmodium falciparum. Am J Trop Med Hyg 33 : 1045–1050. 6391217
9. Schall JJ (1989) The sex ratio of Plasmodium gametocytes. Parasitology 98 Pt 3 : 343–350.
10. Bruce MC, Alano P, Duthie S, Carter R (1990) Commitment of the malaria parasite Plasmodium falciparum to sexual and asexual development. Parasitology 100 Pt 2 : 191–200.
11. Inselburg J (1983) Gametocyte formation by the progeny of single Plasmodium falciparum schizonts. J Parasitol 69 : 584–591. 6355424
12. Silvestrini F, Alano P, Williams JL (2000) Commitment to the production of male and female gametocytes in the human malaria parasite Plasmodium falciparum. Parasitology 121 Pt 5 : 465–471.
13. Smith TG, Lourenco P, Carter R, Walliker D, Ranford-Cartwright LC (2000) Commitment to sexual differentiation in the human malaria parasite, Plasmodium falciparum. Parasitology 121 (Pt 2): 127–133.
14. Kafsack BF, Rovira-Graells N, Clark TG, Bancells C, Crowley VM, et al. (2014) A transcriptional switch underlies commitment to sexual development in malaria parasites. Nature 507 : 248–252. doi: 10.1038/nature12920 24572369
15. Sinha A, Hughes KR, Modrzynska KK, Otto TD, Pfander C, et al. (2014) A cascade of DNA-binding proteins for sexual commitment and development in Plasmodium. Nature 507 : 253–257. doi: 10.1038/nature12970 24572359
16. Brancucci NM, Bertschi NL, Zhu L, Niederwieser I, Chin WH, et al. (2014) Heterochromatin protein 1 secures survival and transmission of malaria parasites. Cell Host Microbe 16 : 165–176. doi: 10.1016/j.chom.2014.07.004 25121746
17. Coleman BI, Skillman KM, Jiang RH, Childs LM, Altenhofen LM, et al. (2014) A Plasmodium falciparum histone deacetylase regulates antigenic variation and gametocyte conversion. Cell Host Microbe 16 : 177–186. doi: 10.1016/j.chom.2014.06.014 25121747
18. Eksi S, Morahan BJ, Haile Y, Furuya T, Jiang H, et al. (2012) Plasmodium falciparum gametocyte development 1 (Pfgdv1) and gametocytogenesis early gene identification and commitment to sexual development. PLoS Pathog 8: e1002964. doi: 10.1371/journal.ppat.1002964 23093935
19. Reininger L, Garcia M, Tomlins A, Muller S, Doerig C (2012) The Plasmodium falciparum, Nima-related kinase Pfnek-4: a marker for asexual parasites committed to sexual differentiation. Malar J 11 : 250. doi: 10.1186/1475-2875-11-250 22849771
20. Carter R, Miller LH (1979) Evidence for environmental modulation of gametocytogenesis in Plasmodium falciparum in continuous culture. Bull World Health Organ 57 Suppl 1 : 37–52. 397008
21. Kaushal DC, Carter R, Miller LH, Krishna G (1980) Gametocytogenesis by malaria parasites in continuous culture. Nature 286 : 490–492. 6250067
22. Fivelman QL, McRobert L, Sharp S, Taylor CJ, Saeed M, et al. (2007) Improved synchronous production of Plasmodium falciparum gametocytes in vitro. Mol Biochem Parasitol 154 : 119–123. 17521751
23. Williams JL (1999) Stimulation of Plasmodium falciparum gametocytogenesis by conditioned medium from parasite cultures. The American journal of tropical medicine and hygiene 60 : 7–13. 9988315
24. Mantel PY, Hoang AN, Goldowitz I, Potashnikova D, Hamza B, et al. (2013) Malaria-infected erythrocyte-derived microvesicles mediate cellular communication within the parasite population and with the host immune system. Cell Host Microbe 13 : 521–534. doi: 10.1016/j.chom.2013.04.009 23684304
25. Regev-Rudzki N, Wilson DW, Carvalho TG, Sisquella X, Coleman BM, et al. (2013) Cell-cell communication between malaria-infected red blood cells via exosome-like vesicles. Cell 153 : 1120–1133. doi: 10.1016/j.cell.2013.04.029 23683579
26. Trager W, Gill GS, Lawrence C, Nagel RL (1999) Plasmodium falciparum: enhanced gametocyte formation in vitro in reticulocyte-rich blood. Exp Parasitol 91 : 115–118. 9990338
27. Talman AM, Domarle O, McKenzie FE, Ariey F, Robert V (2004) Gametocytogenesis: the puberty of Plasmodium falciparum. Malar J 3 : 24. 15253774
28. Peatey CL, Watson JA, Trenholme KR, Brown CL, Nielson L, et al. (2013) Enhanced gametocyte formation in erythrocyte progenitor cells: a site-specific adaptation by Plasmodium falciparum. J Infect Dis 208 : 1170–1174. doi: 10.1093/infdis/jit309 23847056
29. Smalley ME, Brown J (1981) Plasmodium falciparum gametocytogenesis stimulated by lymphocytes and serum from infected Gambian children. Trans R Soc Trop Med Hyg 75 : 316–317. 7029805
30. Reuner B, Vassella E, Yutzy B, Boshart M (1997) Cell density triggers slender to stumpy differentiation of Trypanosoma brucei bloodstream forms in culture. Molecular and biochemical parasitology 90 : 269–280. 9497048
31. Vassella E, Reuner B, Yutzy B, Boshart M (1997) Differentiation of African trypanosomes is controlled by a density sensing mechanism which signals cell cycle arrest via the cAMP pathway. Journal of cell science 110 (Pt 21): 2661–2671. 9427384
32. Mony BM, MacGregor P, Ivens A, Rojas F, Cowton A, et al. (2014) Genome-wide dissection of the quorum sensing signalling pathway in Trypanosoma brucei. Nature 505 : 681–685. doi: 10.1038/nature12864 24336212
33. Bennett TN, Kosar AD, Roepe PD (2005) Plasmodium falciparum strain GC-03 exhibits hyper-gametocytogenesis in partially hemoglobin depleted red blood cells. Mol Biochem Parasitol 139 : 261–265. 15664660
34. Baker DA (2010) Malaria gametocytogenesis. Mol Biochem Parasitol 172 : 57–65. doi: 10.1016/j.molbiopara.2010.03.019 20381542
35. Sinden RE (2009) Malaria, sexual development and transmission: retrospect and prospect. Parasitology 136 : 1427–1434. doi: 10.1017/S0031182009990667 19660156
36. Collins WE, Jeffery GM (2003) A retrospective examination of mosquito infection on humans infected with Plasmodium falciparum. Am J Trop Med Hyg 68 : 366–371. 12685646
37. Simpson JA, Aarons L, Collins WE, Jeffery GM, White NJ (2002) Population dynamics of untreated Plasmodium falciparum malaria within the adult human host during the expansion phase of the infection. Parasitology 124 : 247–263. 11922427
38. Eichner M, Diebner HH, Molineaux L, Collins WE, Jeffery GM, et al. (2001) Genesis, sequestration and survival of Plasmodium falciparum gametocytes: parameter estimates from fitting a model to malariatherapy data. Trans R Soc Trop Med Hyg 95 : 497–501. 11706658
39. Diebner HH, Eichner M, Molineaux L, Collins WE, Jeffery GM, et al. (2000) Modelling the transition of asexual blood stages of Plasmodium falciparum to gametocytes. J Theor Biol 202 : 113–127. 10640432
40. Bhasin VK, Trager W (1984) Gametocyte-forming and non-gametocyte-forming clones of Plasmodium falciparum. Am J Trop Med Hyg 33 : 534–537. 6383092
41. Mideo N, Day T (2008) On the evolution of reproductive restraint in malaria. Proc Biol Sci 275 : 1217–1224. doi: 10.1098/rspb.2007.1545 18303001
42. Taylor LH, Read AF (1997) Why so few transmission stages? Reproductive restraint by malaria parasites. Parasitol Today 13 : 135–140. 15275099
43. Cameron A, Reece SE, Drew DR, Haydon DT, Yates AJ (2013) Plasticity in transmission strategies of the malaria parasite, Plasmodium chabaudi: environmental and genetic effects. Evol Appl 6 : 365–376. doi: 10.1111/eva.12005 23467678
44. Pollitt LC, Mideo N, Drew DR, Schneider P, Colegrave N, et al. (2011) Competition and the evolution of reproductive restraint in malaria parasites. Am Nat 177 : 358–367. doi: 10.1086/658175 21460544
45. Carter LM, Schneider P, Reece SE (2014) Information use and plasticity in the reproductive decisions of malaria parasites. Malar J 13 : 115. doi: 10.1186/1475-2875-13-115 24670151
46. Bousema JT, Drakeley CJ, Mens PF, Arens T, Houben R, et al. (2008) Increased Plasmodium falciparum gametocyte production in mixed infections with P. malariae. Am J Trop Med Hyg 78 : 442–448. 18337341
47. Robert V, Read AF, Essong J, Tchuinkam T, Mulder B, et al. (1996) Effect of gametocyte sex ratio on infectivity of Plasmodium falciparum to Anopheles gambiae. Trans R Soc Trop Med Hyg 90 : 621–624. 9015496
48. White NJ, Ashley EA, Recht J, Delves MJ, Ruecker A, et al. (2014) Assessment of therapeutic responses to gametocytocidal drugs in Plasmodium falciparum malaria. Malar J 13 : 483. doi: 10.1186/1475-2875-13-483 25486998
49. West SA, Smith TG, Read AF (2000) Sex allocation and population structure in apicomplexan (protozoa) parasites. Proc Biol Sci 267 : 257–263. 10714880
50. Ferguson DJ (2002) Toxoplasma gondii and sex: essential or optional extra? Trends Parasitol 18 : 355–359. 12380023
51. Reece SE, Ramiro RS, Nussey DH (2009) Plastic parasites: sophisticated strategies for survival and reproduction? Evol Appl 2 : 11–23. 20305703
52. West SA, Reece SE, Read AF (2001) Evolution of gametocyte sex ratios in malaria and related apicomplexan (protozoan) parasites. Trends Parasitol 17 : 525–531. 11872397
53. Read AF, Anwar M, Shutler D, Nee S (1995) Sex allocation and population structure in malaria and related parasitic protozoa. Proc Biol Sci 260 : 359–363. 7630901
54. Read AF, Narara A, Nee S, Keymer AE, Day KP (1992) Gametocyte sex ratios as indirect measures of outcrossing rates in malaria. Parasitology 104 (Pt 3): 387–395.
55. Sowunmi A, Gbotosho GO, Happi CT, Folarin OA, Balogun ST (2009) Population structure of Plasmodium falciparum gametocyte sex ratios in malarious children in an endemic area. Parasitol Int 58 : 438–443. doi: 10.1016/j.parint.2009.08.007 19723589
56. Furuya T, Mu J, Hayton K, Liu A, Duan J, et al. (2005) Disruption of a Plasmodium falciparum gene linked to male sexual development causes early arrest in gametocytogenesis. Proc Natl Acad Sci U S A 102 : 16813–16818. 16275909
57. Miao J, Li J, Fan Q, Li X, Li X, et al. (2010) The Puf-family RNA-binding protein PfPuf2 regulates sexual development and sex differentiation in the malaria parasite Plasmodium falciparum. J Cell Sci 123 : 1039–1049. doi: 10.1242/jcs.059824 20197405
58. Bachmann A, Esser C, Petter M, Predehl S, von Kalckreuth V, et al. (2009) Absence of erythrocyte sequestration and lack of multicopy gene family expression in Plasmodium falciparum from a splenectomized malaria patient. PLoS ONE 4: e7459. doi: 10.1371/journal.pone.0007459 19826486
59. Smalley ME, Abdalla S, Brown J (1981) The distribution of Plasmodium falciparum in the peripheral blood and bone marrow of Gambian children. Trans R Soc Trop Med Hyg 75 : 103–105. 7022784
60. Abdulsalam AH, Sabeeh N, Bain BJ (2010) Immature Plasmodium falciparum gametocytes in bone marrow. Am J Hematol 85 : 943. doi: 10.1002/ajh.21796 20687103
61. Johnston GL, Smith DL, Fidock DA (2013) Malaria's missing number: calculating the human component of R0 by a within-host mechanistic model of Plasmodium falciparum infection and transmission. PLoS Comput Biol 9: e1003025. doi: 10.1371/journal.pcbi.1003025 23637586
62. Aguilar R, Magallon-Tejada A, Achtman AH, Moraleda C, Joice R, et al. (2014) Molecular evidence for the localization of Plasmodium falciparum immature gametocytes in bone marrow. Blood 123 : 959–966. doi: 10.1182/blood-2013-08-520767 24335496
63. Seydel KB, Milner DA Jr., Kamiza SB, Molyneux ME, Taylor TE (2006) The distribution and intensity of parasite sequestration in comatose Malawian children. J Infect Dis 194 : 208–205. 16779727
64. Genrich GL, Guarner J, Paddock CD, Shieh WJ, Greer PW, et al. (2007) Fatal malaria infection in travelers: novel immunohistochemical assays for the detection of Plasmodium falciparum in tissues and implications for pathogenesis. Am J Trop Med Hyg 76 : 251–259. 17297032
65. Beeson JG, Amin N, Kanjala M, Rogerson SJ (2002) Selective accumulation of mature asexual stages of Plasmodium falciparum-infected erythrocytes in the placenta. Infect Immun 70 : 5412–5415. 12228265
66. Pongponratn E, Riganti M, Punpoowong B, Aikawa M (1991) Microvascular sequestration of parasitized erythrocytes in human falciparum malaria: a pathological study. Am J Trop Med Hyg 44 : 168–175. 2012260
67. Joice R, Nilsson SK, Montgomery J, Dankwa S, Egan E, et al. (2014) Plasmodium falciparum transmission stages accumulate in the human bone marrow. Sci Transl Med 6 : 244re245.
68. Farfour E, Charlotte F, Settegrana C, Miyara M, Buffet P (2012) The extravascular compartment of the bone marrow: a niche for Plasmodium falciparum gametocyte maturation? Malaria journal 11 : 285. doi: 10.1186/1475-2875-11-285 22905863
69. Lee SH, Crocker PR, Westaby S, Key N, Mason DY, et al. (1988) Isolation and immunocytochemical characterization of human bone marrow stromal macrophages in hemopoietic clusters. J Exp Med 168 : 1193–1198. 3049905
70. Aguilar R, Moraleda C, Achtman AH, Mayor A, Quinto L, et al. (2014) Severity of anaemia is associated with bone marrow haemozoin in children exposed to Plasmodium falciparum. Br J Haematol 164 : 877–887. doi: 10.1111/bjh.12716 24386973
71. Trager W, Gill GS (1992) Enhanced gametocyte formation in young erythrocytes by Plasmodium falciparum in vitro. J Protozool 39 : 429–432. 1640389
72. Roberts CH, Armstrong M, Zatyka E, Boadi S, Warren S, et al. (2013) Gametocyte carriage in Plasmodium falciparum-infected travellers. Malar J 12 : 31. doi: 10.1186/1475-2875-12-31 23347669
73. Meerman L, Ord R, Bousema JT, van Niekerk M, Osman E, et al. (2005) Carriage of chloroquine-resistant parasites and delay of effective treatment increase the risk of severe malaria in Gambian children. J Infect Dis 192 : 1651–1657. 16206082
74. Drakeley CJ, Secka I, Correa S, Greenwood BM, Targett GA (1999) Host haematological factors influencing the transmission of Plasmodium falciparum gametocytes to Anopheles gambiae s.s. mosquitoes. Trop Med Int Health 4 : 131–138. 10206267
75. Healer J, Graszynski A, Riley E (1999) Phagocytosis does not play a major role in naturally acquired transmission-blocking immunity to Plasmodium falciparum malaria. Infection and immunity 67 : 2334–2339. 10225892
76. Das B, Kashino SS, Pulu I, Kalita D, Swami V, et al. (2013) CD271(+) bone marrow mesenchymal stem cells may provide a niche for dormant Mycobacterium tuberculosis. Science translational medicine 5 : 170ra113.
77. Silvestrini F, Tiburcio M, Bertuccini L, Alano P (2012) Differential adhesive properties of sequestered asexual and sexual stages of Plasmodium falciparum on human endothelial cells are tissue independent. PloS one 7: e31567. doi: 10.1371/journal.pone.0031567 22363675
78. Tiburcio M, Silvestrini F, Bertuccini L, Sander AF, Turner L, et al. (2012) Early gametocytes of the malaria parasite Plasmodium falciparum specifically remodel the adhesive properties of infected erythrocyte surface. Cell Microbiol 15 : 647–659. doi: 10.1111/cmi.12062 23114006
79. Tiburcio M, Niang M, Deplaine G, Perrot S, Bischoff E, et al. (2012) A switch in infected erythrocyte deformability at the maturation and blood circulation of Plasmodium falciparum transmission stages. Blood 119: e172–e180. doi: 10.1182/blood-2012-03-414557 22517905
80. Aingaran M, Zhang R, Law SK, Peng Z, Undisz A, et al. (2012) Host cell deformability is linked to transmission in the human malaria parasite Plasmodium falciparum. Cellular microbiology 14 : 983–993. doi: 10.1111/j.1462-5822.2012.01786.x 22417683
81. Dearnley MK, Yeoman JA, Hanssen E, Kenny S, Turnbull L, et al. (2012) Origin, composition, organization and function of the inner membrane complex of Plasmodium falciparum gametocytes. Journal of cell science 125 : 2053–2063. doi: 10.1242/jcs.099002 22328505
82. Bousema T, Okell L, Shekalaghe S, Griffin JT, Omar S, et al. (2010) Revisiting the circulation time of Plasmodium falciparum gametocytes: molecular detection methods to estimate the duration of gametocyte carriage and the effect of gametocytocidal drugs. Malar J 9 : 136. doi: 10.1186/1475-2875-9-136 20497536
83. Lensen A, Bril A, van de Vegte M, van Gemert GJ, Eling W, et al. (1999) Plasmodium falciparum: infectivity of cultured, synchronized gametocytes to mosquitoes. Exp Parasitol 91 : 101–103. 9920049
84. van den BL, Chardome M (1951) An easier and more accurate diagnosis of malaria and filariasis through the use of the skin scarification smear. Am J Trop Med Hyg 31 : 411–413. 14857243
85. Pichon G, Awono-Ambene HP, Robert V (2000) High heterogeneity in the number of Plasmodium falciparum gametocytes in the bloodmeal of mosquitoes fed on the same host. Parasitology 121 (Pt 2): 115–120.
86. Jeffery GM, Eyles DE (1955) Infectivity to mosquitoes of Plasmodium falciparum as related to gametocyte density and duration of infection. Am J Trop Med Hyg 4 : 781–789. 13259002
87. Ross A, Smith T (2006) The effect of malaria transmission intensity on neonatal mortality in endemic areas. Am J Trop Med Hyg 75 : 74–81. 16931818
88. Bousema T, Okell L, Felger I, Drakeley C (2014) Asymptomatic malaria infections: detectability, transmissibility and public health relevance. Nat Rev Microbiol 12 : 833–840. doi: 10.1038/nrmicro3364 25329408
89. Joice R, Narasimhan V, Montgomery J, Sidhu AB, Oh K, et al. (2013) Inferring developmental stage composition from gene expression in human malaria. PLoS Comput Biol 9: e1003392. doi: 10.1371/journal.pcbi.1003392 24348235
90. Ouedraogo AL, Schneider P, de Kruijf M, Nebie I, Verhave JP, et al. (2007) Age-dependent distribution of Plasmodium falciparum gametocytes quantified by Pfs25 real-time QT-NASBA in a cross-sectional study in Burkina Faso. Am J Trop Med Hyg 76 : 626–630. 17426160
91. Drakeley CJ, Akim NI, Sauerwein RW, Greenwood BM, Targett GA (2000) Estimates of the infectious reservoir of Plasmodium falciparum malaria in The Gambia and in Tanzania. Trans R Soc Trop Med Hyg 94 : 472–476. 11132369
92. Miura K, Takashima E, Deng B, Tullo G, Diouf A, et al. (2013) Functional comparison of Plasmodium falciparum transmission-blocking vaccine candidates by the standard membrane-feeding assay. Infect Immun 81 : 4377–4382. doi: 10.1128/IAI.01056-13 24042109
93. Vaughan JA, Noden BH, Beier JC (1994) Sporogonic development of cultured Plasmodium falciparum in six species of laboratory-reared Anopheles mosquitoes. Am J Trop Med Hyg 51 : 233–243. 8074258
94. Stone WJ, Eldering M, van Gemert GJ, Lanke KH, Grignard L, et al. (2013) The relevance and applicability of oocyst prevalence as a read-out for mosquito feeding assays. Sci Rep 3 : 3418. doi: 10.1038/srep03418 24301557
95. Boissiere A, Tchioffo MT, Bachar D, Abate L, Marie A, et al. (2012) Midgut microbiota of the malaria mosquito vector Anopheles gambiae and interactions with Plasmodium falciparum infection. PLoS Pathog 8: e1002742. doi: 10.1371/journal.ppat.1002742 22693451
96. Blandin SA, Marois E, Levashina EA (2008) Antimalarial responses in Anopheles gambiae: from a complement-like protein to a complement-like pathway. Cell Host Microbe 3 : 364–374. doi: 10.1016/j.chom.2008.05.007 18541213
97. Bousema T, Sutherland CJ, Churcher TS, Mulder B, Gouagna LC, et al. (2011) Human immune responses that reduce the transmission of Plasmodium falciparum in African populations. International journal for parasitology 41 : 293–300. doi: 10.1016/j.ijpara.2010.09.008 20974145
98. Bian G, Joshi D, Dong Y, Lu P, Zhou G, et al. (2013) Wolbachia invades Anopheles stephensi populations and induces refractoriness to Plasmodium infection. Science 340 : 748–751. doi: 10.1126/science.1236192 23661760
99. Gabrieli P, Kakani EG, Mitchell SN, Mameli E, Want EJ, et al. (2014) Sexual transfer of the steroid hormone 20E induces the postmating switch in Anopheles gambiae. Proc Natl Acad Sci U S A 111 : 16353–16358. doi: 10.1073/pnas.1410488111 25368171
100. Baldini F, Segata N, Pompon J, Marcenac P, Robert Shaw W, et al. (2014) Evidence of natural Wolbachia infections in field populations of Anopheles gambiae. Nat Commun 5 : 3985. doi: 10.1038/ncomms4985 24905191
101. Painter HJ, Campbell TL, Llinas M (2011) The Apicomplexan AP2 family: integral factors regulating Plasmodium development. Molecular and biochemical parasitology 176 : 1–7. doi: 10.1016/j.molbiopara.2010.11.014 21126543
102. Yuda M, Iwanaga S, Shigenobu S, Mair GR, Janse CJ, et al. (2009) Identification of a transcription factor in the mosquito-invasive stage of malaria parasites. Mol Microbiol 71 : 1402–1414. doi: 10.1111/j.1365-2958.2009.06609.x 19220746
103. Yuda M, Iwanaga S, Shigenobu S, Kato T, Kaneko I (2010) Transcription factor AP2-Sp and its target genes in malarial sporozoites. Mol Microbiol 75 : 854–863. doi: 10.1111/j.1365-2958.2009.07005.x 20025671
104. Iwanaga S, Kaneko I, Kato T, Yuda M (2012) Identification of an AP2-family protein that is critical for malaria liver stage development. PLoS One 7: e47557. doi: 10.1371/journal.pone.0047557 23144823
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 6
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- HIV Latency Is Established Directly and Early in Both Resting and Activated Primary CD4 T Cells
- Battling Phages: How Bacteria Defend against Viral Attack
- A 21st Century Perspective of Poliovirus Replication
- Adenovirus Tales: From the Cell Surface to the Nuclear Pore Complex