
Reovirus FAST Proteins Drive Pore Formation and Syncytiogenesis Using a Novel Helix-Loop-Helix Fusion-Inducing Lipid Packing Sensor
The fusogenic ortho - and aquareoviruses are the only known nonenveloped viruses that induce syncytium formation. Cell-cell fusion is a virulence determinant of fusogenic reoviruses, and is mediated by a singular family of fusion-associated small transmembrane (FAST) proteins, the smallest known viral fusogens. Unlike their enveloped virus counterparts, reovirus FAST proteins have exceptionally small ectodomains and considerable larger cytoplasmic endodomains, suggesting FAST protein interactions with the cytoplasmic leaflet of the plasma membrane likely play a prominent role in the fusion process. We determined that the baboon reovirus p15 FAST protein endodomain contains a novel type of helix-loop-helix lipid packing sensor that partitions into hydrophobic defects present in highly curved membranes. This fusion-inducing lipid packing sensor (FLiPS) is required for pore formation, and can be functionally replaced by heterologous lipid packing sensors. By masking hydrophobic defects appearing in the highly curved rim of nascent fusion pores, the FliPS would make the forward reaction to pore formation a more energetically favored means of resolving an unstable hemifusion intermediate. These results define a new role for curvature sensing motifs, and reveal how viral fusion proteins can drive pore formation without having to rely on membrane stresses induced by complex refolding of large ectodomains.
Published in the journal:
. PLoS Pathog 11(6): e32767. doi:10.1371/journal.ppat.1004962
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004962
Summary
The fusogenic ortho - and aquareoviruses are the only known nonenveloped viruses that induce syncytium formation. Cell-cell fusion is a virulence determinant of fusogenic reoviruses, and is mediated by a singular family of fusion-associated small transmembrane (FAST) proteins, the smallest known viral fusogens. Unlike their enveloped virus counterparts, reovirus FAST proteins have exceptionally small ectodomains and considerable larger cytoplasmic endodomains, suggesting FAST protein interactions with the cytoplasmic leaflet of the plasma membrane likely play a prominent role in the fusion process. We determined that the baboon reovirus p15 FAST protein endodomain contains a novel type of helix-loop-helix lipid packing sensor that partitions into hydrophobic defects present in highly curved membranes. This fusion-inducing lipid packing sensor (FLiPS) is required for pore formation, and can be functionally replaced by heterologous lipid packing sensors. By masking hydrophobic defects appearing in the highly curved rim of nascent fusion pores, the FliPS would make the forward reaction to pore formation a more energetically favored means of resolving an unstable hemifusion intermediate. These results define a new role for curvature sensing motifs, and reveal how viral fusion proteins can drive pore formation without having to rely on membrane stresses induced by complex refolding of large ectodomains.
Introduction
The fusogenic ortho - and aquareoviruses are the only known examples of nonenveloped viruses that induce syncytium formation. Reovirus-induced syncytiogenesis is mediated by a novel family of viral fusogens, the fusion-associated small transmembrane (FAST) proteins [1, 2]. Structurally and functionally, FAST proteins differ dramatically from the three well-characterized classes of enveloped virus fusion proteins [3]. Ranging from ~100–200 residues in size, FAST proteins are considerably smaller than enveloped virus fusion proteins, and they position the majority of their mass within their single transmembrane domain and cytoplasmic tail rather than in their ectodomains [4–9]. FAST protein-mediated membrane fusion must therefore proceed in the absence of the complex ectodomain refolding used by enveloped virus fusogens to drive the fusion process [10]. Furthermore, unlike enveloped virus fusogens, FAST proteins are nonessential for reovirus replication, and as nonstructural viral proteins are not involved in virus entry. Instead, FAST proteins localize to the plasma membrane of virus-infected cells where they mediate cell-cell, not virus-cell, membrane fusion. The fusogenic reoviruses use syncytium formation to promote direct cell-cell transmission of the infection and rapid release of intracellular progeny virions following disruption of syncytia, both of which may contribute to the natural pathogenicity of the fusogenic reoviruses [11–13]. The unique structural and functional attributes of the FAST proteins and their link to virulence underscore the need for a better understanding of how these diminutive viral fusogens induce syncytium formation.
Studies of archetypal enveloped virus membrane fusion proteins converge on a common pathway of protein-mediated membrane fusion [14–16]. In this fusion-through-hemifusion pathway, small hydrophobic or amphipathic fusion peptides or fusion loops are proposed to shallowly insert into the outer leaflet of the membrane bilayer, forcing apart lipid headgroups and resulting in formation of a protruding membrane dimple [17, 18]. Homotypic fusion of endoplasmic reticulum membranes, synaptic vesicle exocytosis, and formation of clathrin-coated pits all rely on membrane insertion of amphipathic helices (AHs) or hydrophobic loops to generate membrane curvature via a similar wedging mechanism [19–22]. In the case of membrane fusion, the resulting membrane stresses and lipid packing defects are partially relieved by lipid mixing between outer leaflets of the closely apposed bilayers to generate a hemifusion stalk intermediate [23, 24], with curvature stresses in the stalk being relieved by subsequent pore formation [18]. Theoretical and experimental studies suggest that the highly curved rim of nascent fusion pores forces apart lipid headgroups, exposing the membrane interior to water, and that pore formation is the rate-limiting step in the fusion process [25–28]. At present, there is no clear understanding of how protein fusogens breach this barrier.
Members of the FAST protein family have limited sequence similarity, but share a number of defining features. A single transmembrane domain separates exceedingly small N-terminal ectodomains of ~19–40 residues from equal sized or larger C-terminal cytoplasmic endodomains of ~40–140 residues [1]. Each FAST protein contains a cytoplasmic cluster of membrane-proximal basic residues, which in the case of the p14 FAST protein function as a Golgi export signal, [29], and all contain cites for acylation; either myristoylation of the N-terminal Gly residue or palmitoylation of endodomain Cys residues [4, 30, 31]. All three FAST protein domains (ecto-, endo - and transmembrane) are actively involved in the fusion process and are somewhat interchangeable between family members [32–34], indicating the FAST proteins evolved as modular fusogens. How each fusion module promotes membrane fusion is still unclear, but several essential structural motifs have been characterized. For example, ectodomains of the avian reovirus p10, reptilian reovirus p14 and baboon reovirus p15 FAST proteins contain a cystine loop, proline-hinged loop, or polyproline type II helix, respectively [35–38]. All three of these motifs are structurally distinct from, but functionally equivalent to, enveloped virus fusion peptides [3], using membrane insertion of hydrophobic residues to promote liposome fusion. While the fusion peptides of enveloped viruses and FAST proteins are clearly involved in inducing membrane curvature needed for hemifusion, how protein fusogens promote curvature changes required for pore formation has not been established.
A remarkable feature of FAST proteins is their asymmetric membrane topology, which positions the majority of their mass on the distal side of the contacting membrane bilayers [1]. These cytoplasmic tails play diverse roles in syncytiogenesis. Truncation and substitution studies revealed the membrane-distal region of the endodomain is required for pore formation and augments pore expansion [32]. The sequence-independence of the distal portion of the endodomain suggests this region may function as an intrinsically disordered motif, a common property of motifs with multiple interaction partners [39]. A recent study identified intracellular annexin A1 as one such interaction partner of the p14 FAST protein; annexin A1 functions in a calcium-specific manner to promote pore expansion [40]. Following proteolytic cleavage, the p14 FAST protein endodomain can also function in a sequence-specific fashion in trans to augment pore expansion following cell-cell fusion mediated by the full-length p14 protein [41]. As with annexin A1, the soluble p14 endodomain enhances pore expansion following membrane fusion induced by fusogens other than just FAST proteins [40, 41], suggesting syncytium formation may be a generalized cellular response to resolve the unfavorable architecture of intercellular pores.
We now report a new functional motif in the FAST protein cytoplasmic tails that is required for pore formation. In addition to their transmembrane domain, all FAST proteins contain a small cluster of predominantly hydrophobic or apolar residues, termed the hydrophobic patch (HP). While the HP is contained in the ectodomains of the p10 and p14 FAST proteins where it functions as a fusion peptide [35, 36, 42, 43], this motif is located in the endodomains of other FAST proteins and has no known function. Results indicate the membrane-proximal, endodomain HP of the p15 FAST protein (p15 HP) functions as a lipid packing sensor and preferentially partitions into highly curved membranes. By promoting or stabilizing such curvature in the inner leaflet of the plasma membrane, this curvature sensor provides a mechanism to lower the energy barrier to pore formation and promote cell-cell fusion.
Results
The cytosolic p15 HP is essential for syncytium formation
The majority of FAST proteins contain a membrane-proximal HP in their cytoplasmic endodomains [2]. The function of these motifs has not been investigated. To discern whether the p15 HP plays any role in syncytium formation, HP residues were replaced by Ala in groups of three consecutive amino acids. Syncytium formation in transfected QM5 cells expressing individual p15 HP mutant proteins was quantified using a standard syncytial assay based on the average number of syncytial nuclei/microscopic field [4]. Ala substitution of two regions in the p15 HP, GAG74-76 and LPL80-82, abrogated syncytium formation (Fig 1B). The non-syncytiogenic p15GAG74-76 and p15LPL80-82 constructs were also defective for pore formation (Fig 1C), as determined using a standard dual fluorescence assay based on flow cytometry to quantify transfer of EGFP and calcein red between donor and target cells [32]. To more precisely identify which residue(s) in the GAG74-76 and LPL80-81 regions of the p15 HP are essential for fusion, each residue was individually substituted with Ala. Substitutions of L80 or L82 had little to no effect on syncytiogenesis, while substitutions of P81, G74 or G76 essentially eliminated syncytium formation (Fig 1D). As shown by western blotting (Fig 1E) and flow cytometry analysis of cell surface localization, analyzed by percent cell fluorescence relative to wt p15 (Fig 1F) or by mean fluorescence intensity (S1 Fig), none of the triple Ala substitutions had any effect on p15 expression or trafficking to the plasma membrane. The p15 HP is therefore essential for cell-cell pore formation, and this activity is dependent on Gly and Pro residues near the center of the HP.
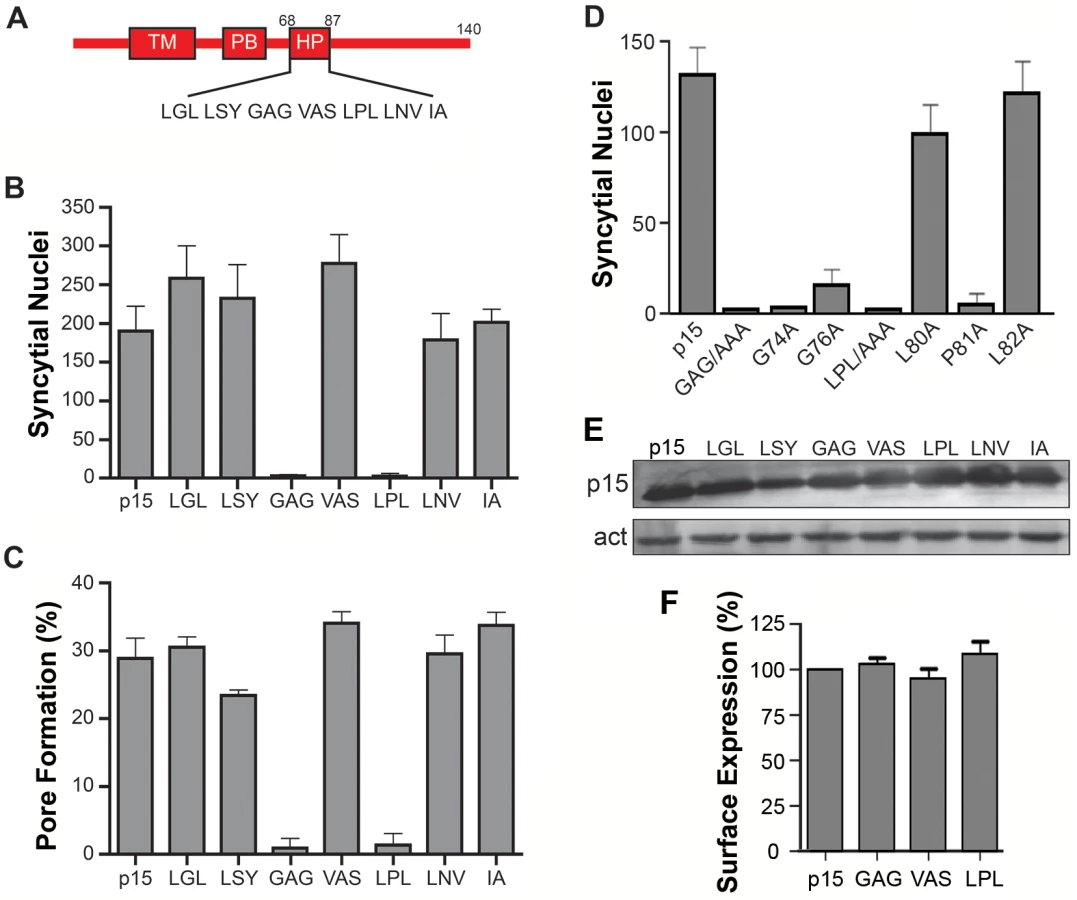
Helical properties and hydrophobicity influence the function of the p15 HP
Secondary structure predictions [44] suggested a helix-loop-helix (H-L-H) conformation for the wild type (wt) p15 HP, while the fusion-dead p15GAG74-76, G74A and p15P81A constructs were predicted to be a continuous α-helix (Fig 2A). These predictions were experimentally confirmed using circular dichroism (CD) spectroscopy of synthetic peptides corresponding to the wt p15HP (p15HPpep) and G74/76A (GAG) and G74A mutant versions of this peptide. CD spectra revealed both mutant peptides assumed more helical structure in the presence of 1-palmitoyl-2-hydroxy-sn-glycero-3-[phospho-rac-(1-glycerol)] (LPPG) micelles, as evidenced by increased maxima at 190 nm and increased minima at 208 nm and 222 nm relative to the wt peptide (Fig 2A). These shifts in the CD spectra of the peptides imply increased helicity and suggest conversion of at least part of the loop region to an α-helix, with the Pro81 residue likely preventing complete helix formation. Several attempts to synthesize and purify a peptide containing the P81A substitution were unsuccessful.

As its name suggests, the p15 HP comprises mostly hydrophobic or apolar residues (16 of 20 residues). The functional significance of hydrophobic residues in the p15 HP was supported by substitution studies in the context of the p15 protein, sequentially replacing residues in the predicted N - and C-terminal helices with the uncharged, polar residue asparagine. Asparagine replacement of the N-terminal L71 residue reduced p15-induced syncytium formation by ~20%, and when combined with a similar replacement of Y73 in the predicted N-terminal helix by ~50% (constructs HP1 and HP2 in Fig 2B). Additional replacements of V77 and L80 in the predicted loop region had no further inhibitory effect (constructs HP3 and HP4), but additional substitution of I86 in the predicted C-terminal helix reduced syncytium formation by ~80% (construct HP5 Fig 2B). These results suggest that changes in the predicted helicity and hydrophobicity of the p15 HP compromise its role in cell-cell fusion.
Helix-loop-helix conformation of p15HPpep in a membrane mimetic environment
The functional implications of a predicted H-L-H p15 HP architecture prompted determination of the atomic-level structure of a 20-residue p15HPpep synthetic peptide (L68GLLSYGAGVASLPLLNVIA87; numbers refer to the residue location in p15) using solution-state nuclear magnetic resonance (NMR) spectroscopy. High-resolution NMR spectra of p15HPpep implied a uniform, homogeneous structure in dodecylphosphocholine (DPC) micelles, as indicated by a single observed set of resonance frequencies for each spin system (S2 Fig). The 1H-1H pairwise distance restraints used in the structure calculation were generated from observed Nuclear Overhauser Effects (NOEs) (S1 Table). Approximately 99% of the expected 1H chemical shifts were determinable using sequential assignment protocols [45] based upon 2D 1H-1H TOCSY and 2D 1H-1H NOESY experiments (S2A Fig). Natural abundance 2D 1H-15N and 2D 1H-13C HSQC spectra allowed for complete amide 15N and α/β 13C chemical shift determination (S2B and S2C Fig). Following an iterative NOE distance restraint refinement protocol [46], the final structural ensemble of p15HPpep was calculated using 511 NOE-based distance restraints (S1 Table). The 50 lowest-energy structures (out of 100 calculated) were retained, showing excellent agreement with NOE data and reasonable Ramachandran plot statistics (S1 Table).
The 50-member solution NMR structural ensemble indicated almost all of the p15HPpep conformers contain two short α-helical segments at the N - and C-termini connected by an unstructured turn/loop (Fig 3C). Due to the turn/loop between helices and lack of long-range distance restraints between the helices, the orientation of the two helices relative to each other was not constrained, as clearly evident when conformers where arranged by overlaying the N - or C-terminal helices (Fig 3A and 3B). The C-terminal helix spanning residues L82-I86 was consistent throughout the ensemble; this helix extended to P81 in 38 of the 50 members. Positioning of L82, L83, V85 and I86 side chains on the same face of this C-terminal helix forms a hydrophobic face (Fig 3D). The N-terminal helix exhibited a higher degree of variability throughout the ensemble (Fig 3C), but spanned residues L71-Y73 in 37 of the 50 members; in 33 of these 37 members the helix extended to include L70 and/or G74. Notably, no ensemble member contained a continuous helix spanning residues G74, G76 and P81, the three residues where Ala substitutions in the context of the p15 protein eliminated membrane fusion (Fig 1D). Structural and functional studies therefore indicate a H-L-H conformation is a functional requirement of the p15 HP.

The p15HPpep peptide preferentially partitions into highly curved lipid bilayers containing hydrophobic defects
To examine the membrane interactive properties of the p15 HP, wt p15HPpep was incubated with liposomes of varying diameters (50, 100 or 400 nm) at a constant peptide:lipid molar ratio (1 : 500). Liposomes were separated from free peptide by flotation using sucrose density gradient ultracentrifugation, and reverse phase HPLC was used to quantify peptide concentrations in the top 0–20% sucrose interface and bottom 30% sucrose gradient fractions, corresponding to liposome-associated and free peptide, respectively. Control experiments confirmed different sized liposomes were all recovered at 90–95% efficiency from the 0–20% sucrose interface (S3A Fig), and HPLC analysis of different peptide concentrations defined a linear dose-response range for peptide quantification (S3B Fig). Results indicated a progressive, size-dependent increase in p15HPpep association with liposomes at the 0–20% sucrose interface (Fig 4B). Replicate experiments (n = 3) indicated ~5% of total p15HPpep associated with 400 nm liposomes, ~20% with 100 nm liposomes, and ~40% with 50 nm liposomes (Fig 4C). When corrected for background peptide present in 0–20% sucrose interface in the absence of liposomes (~3% of total peptide, Fig 4B), decreasing liposome size from 400 nm to 50 nm resulted in an ~4-fold increase in p15HPpep partitioning into liposomes. Thus, the p15HPpep peptide preferentially partitions into lipid bilayers with a high degree of positive curvature.

To determine whether the p15 HP might function as a “curvature sensor” to detect hydrophobic defects in highly curved lipid bilayers, the small molecule fluorescent hydrophobic probe bis-ANS (4,4'-dianilino-1,1'-binaphthyl-5,5'-disulfonic acid) was used as a partitioning competitor. Analysis of bis-ANS partitioning into liposomes (bis-ANS fluorescence increases in non-polar environments) indicated increased bis-ANS partitioning maxima as liposome size decreased from 400 nm to 50 nm (Fig 5A). These results confirmed small liposomes (<100 nm diameter) have more hydrophobic defects than larger liposomes, and they determined 10 μM bis-ANS saturates hydrophobic defects in the different sized liposomes. Pre-treatment of liposome suspensions with 10 μM bis-ANS before peptide addition essentially eliminated the size-dependent partitioning of the p15HPpep peptide (Fig 5B and 5C); p15HPpep partitioned into liposomes at levels only slightly above background (~5–10% of total peptide) and independent of liposome size. The p15HPpep peptide therefore senses membrane curvature via hydrophobic defects in highly curved lipid bilayers.

To examine whether the H-L-H conformation of the p15 HP required for p15-mediated cell-cell fusion activity is also required for membrane curvature sensing, we used the two mutant p15HPpep peptides containing the G74/76A substitutions in the GAG sequence or just the G74A substitution. Both of these substitutions abrogated cell-cell fusion activity in the context of the p15 protein (Fig 1D) and increased helicity of the respective mutant p15HPpep peptides (Fig 2A). When examined using the liposome partitioning assay, both mutant peptides lost their ability to function as a membrane curvature sensor. In marked contrast to wt p15HPpep, in replicate experiments (n = 3) the mutant peptides avidly and equally associated with both 50 nm and 400 nm liposomes. Essentially all (>95%) of the GAG mutant peptide associated with both sizes of liposomes; ~80% of the G74A peptide showed similar size-indiscriminate association with liposomes (Fig 6). Hence, loss of the p15HPpep HLH conformation correlates with both a loss of syncytium formation induced by the p15 protein and loss of membrane curvature sensing by p15HPpep peptides.

Heterologous lipid-packing sensors can functionally replace the p15 HP
Well-characterized amphipathic lipid packing sensors (ALPS) respond to membrane curvature by partitioning linear AHs into lipid packing defects present in highly curved membranes [47–49]. The p15 HP is the first example of lipid packing sensor requiring a H-L-H conformation. Calculations of the mean hydrophobic moment, an indication of the relative amphipathic nature of a helix [50], also indicates the p15 HP motif is considerably less amphipathic than classical ALPS motifs (Fig 7A). Nonetheless, the p15HPpep peptide shares the curvature sensing capability of these heterologous ALPS motif peptides. To determine whether ALPS motifs can functionally replace the p15 HP during cell-cell membrane fusion, a series of chimeric p15 constructs containing various ALPS motifs in place of the p15 HP were created and assessed for their syncytiogenic activity. These ALPS motifs were from the membrane curvature sensing modules of Golgi-localized ArfGAP1 (F199LNSAMSSLYSGWSSFTTGASKFAS223), the sterol transporter protein Kes1p (S7SSWTSFLKSIASFNGDLSSLSA29), and the vacuole protein sorting effector subunit Vps41 (T359TNIGSLLSSAASSFRGT376) (Fig 7A). Most interestingly, all three ALPS motifs could functionally substitute for the p15 HP to varying degrees, independent of their relative expression levels (Fig 7B and 7C); ArfGAP1 yielded equivalent levels of syncytium formation as wt p15, Kes1p augmented p15 fusion activity, while p15 with the Vps41 motif retained modest, but significant, levels of cell-cell fusion activity. We note that Vps41 had the lowest hydrophobic moment and smallest hydrophobic face of the three ALPS motifs, although the relative fusion levels should be interpreted with caution in the absence of quantitative cell surface expression data and kinetic analysis of syncytium formation. Substitution of residues in the hydrophobic face of ALPS motifs that renders them insensitive to membrane curvature, L200A, W211A, and F221A in the ArfGAP1 motif and W10A, F13A, and F20A in the Kes1p motif [47, 51, 52], reduced syncytium formation of these p15 constructs by >95% (Fig 7C). The loss of cell-cell fusion activity was unlikely to be due to dramatic changes in protein expression or trafficking since three unrelated, heterologous ALPS motif can functionally replace the p15 HP, indicating the p15 HP is not required for p15 expression or trafficking to the plasma membrane. ALPS motifs that detect positive membrane curvature dependent on their hydrophobic face can therefore functionally substitute for the p15 HP.

To ascertain if ALPS motifs are unique in their ability to functionally replace the p15 HP, three distinct membrane-interactive AHs were used to replace the p15 HP (Fig 8A). The H0-NBAR motif (Q16VQKKFSRAQEKVLQKLGK34) from BRAP/BIN2 has a high hydrophobic moment and strong positively charged face (Fig 8), but only partitions into membranes with negatively-charged lipid headgroups, independent of membrane curvature, and induces membrane rigidity [53]. The AH from melittin [G1IGAVLKVLTTGLPALSWI KRKRQQ25] is a lytic peptide that forms pores [54], and has a low hydrophobic moment (Fig 8). The AH of the Bacillus subtilis DivIVA protein (E21VNEFLAQVRKDYEIVLR41) has a high hydrophobic moment, but is involved in localizing DivIVA to negatively curved membranes [55]. Despite being expressed at levels similar to wt p15 (Fig 8C), none of these constructs were functional for syncytium formation (Fig 8B).

A membrane-proximal amphipathic helix in the p14 FAST protein cytoplasmic tail serves a similar function as the p15 HP
We were interested in determining whether other FAST proteins were similarly reliant on a cytosolic lipid packing sensor. The HP of the p14 FAST protein resides in the ectodomain, not the cytosolic endodomain, where it functions as a fusion peptide to promote early stages of membrane fusion [36]. However, we noted the presence of a previously unrecognized 22-residue AH (p14 residues 70–92; sequence—LTEFQKRYLRNSYR LSEIGRPIS; hydrophobic moment—0.402) located membrane-proximal and immediately downstream of the polybasic motif in the p14 endodomain (Fig 9A and 9B). A p14 construct containing an ArfGAP1 ALPS motif substitution of this AH retained ~60% cell-cell fusion activity relative to wt p14 (Fig 9C), while p15 containing the p14AH in place of the p15 HP retained ~40% fusion activity (Fig 9D). These results suggested the FAST proteins may be generally reliant on a cytosolic, membrane-proximal curvature sensor for cell-cell fusion.

Discussion
Formation and stabilization of the highly curved rim of a fusion pore is the rate limiting and most energy-dependent event in membrane fusion [26–28, 56]. How viral membrane fusion proteins overcome this barrier is unclear. We now show that a membrane-proximal hydrophobic patch present in the cytoplasmic tail of the p15 FAST protein preferentially partitions into membranes with high positive curvature. This partitioning is based on the presence of hydrophobic defects present in highly curved membranes, and requires the p15 HP to assume a H-L-H conformation. Furthermore, the p15 HP can be functionally replaced by ALPS motifs from heterologous proteins, but not by other membrane-interactive AHs. These results identify the p15 HP as a fusion-inducing lipid packing sensor (FLiPS), and they extend the known roles of membrane curvature-sensing motifs to a novel role in cell-cell pore formation and membrane fusion. Furthermore, a previously unrecognized membrane-proximal AH in the p14 FAST protein cytoplasmic tail can functionally replace the p15 HP, and can itself be functionally replaced by a heterologous ALPS motif, implying it serves a similar curvature sensing function as the p15 HP. Other FAST proteins, including Broome reovirus p13 [9] and aquareovirus p22 [7], also contain cytoplasmic, membrane-proximal HPs, suggesting FLiPSs may be a common mechanism employed by the FAST proteins to induce pore formation.
Several lines of evidence support the role of the p15 HP as the first example of a novel type of lipid packing sensor dependent on a H-L-H conformation: (1) circular dichroism studies indicate the wt p15HPpep peptide is only partially helical in a membrane environment (Fig 2A); (2) NMR analysis indicates p15HPpep in DPC micelles comprises two small, amphipathic helices connected by a highly flexible loop (Fig 3); (3) p15HPpep peptides containing mutations of key Gly and Pro residues in the loop yield CD spectra indicative of increased helicity, consistent with secondary structure predictions (Fig 2A); (4) these same HP mutations in the context of the p15 protein abrogate syncytium formation, inhibiting events occurring at, or upstream, of cell-cell pore formation (Fig 1); (5) the wt p15HPpep, but not mutant versions of this peptide, preferentially partitions into small liposomes with increased membrane curvature (Figs 4 and 6), and this partitioning is blocked by masking hydrophobic defects using bis-ANS (Fig 5). Thus, curvature sensing by the p15HPpep peptide and cell-cell fusion mediated by the p15 protein both directly correlate with a H-L-H architecture in the p15 HP.
The small size of the FAST protein ectodomains indicates their mechanism of mediating membrane fusion cannot rely on membrane stresses induced by complex refolding of large ectodomains, as occurs with enveloped virus fusion proteins [10]. The newly defined curvature-sensing properties of the p15 HP described in this report provide a means for FAST proteins to overcome curvature stresses and lower the energy barrier to pore formation (Fig 10). During the fusion reaction, curvature changes in the outer membrane leaflet are needed to initiate merger with the outer monolayer of a closely apposed target cell membrane. In the case of FAST proteins, these curvature changes are mediated by a fusion peptide motif in the ectodomain and by essential residues in the outer interfacial region of the transmembrane domain [34–36, 38]. The presumed stalk-like hemifusion intermediate that arises from these lamellar rearrangements [57] has strong negative curvature in the outer monolayer (i.e., lipid headgroups are forced together and lipid tails splay apart). Coupling of the bilayer leaflets means the inner monolayer has to assume increasing positive curvature at the base of the stalk structure (i.e., lipid headgroups are forced apart). These curvature stresses render the hemifusion intermediate a thermodynamically unstable structure, which must either revert to two planar bilayers or advance to pore formation to resolve curvature stresses. However, pore formation requires induction of further positive curvature to merge the inner monolayers, creating an energetic barrier to pore formation and making the reverse reaction a more likely outcome [58]. Anchored near the cytoplasmic side of the plasma membrane by the p15 transmembrane domain, the p15 HP is perfectly positioned to function as a fusion-inducing lipid packing sensor (FLiPS). By partitioning into hydrophobic defects near the base of an emerging hemifusion structure to stabilize positive curvature, the FLiPS would promote stalk formation and allow further positive curvature to develop resulting in a seamless transition to pore formation (Fig 10). Hence, the FLiPS may serve as both a curvature sensor and curvature inducer. This tightly coupled hemifusion-pore formation model may explain the difficulty in detecting a hemifusion intermediate during FAST protein-mediated membrane fusion [59].
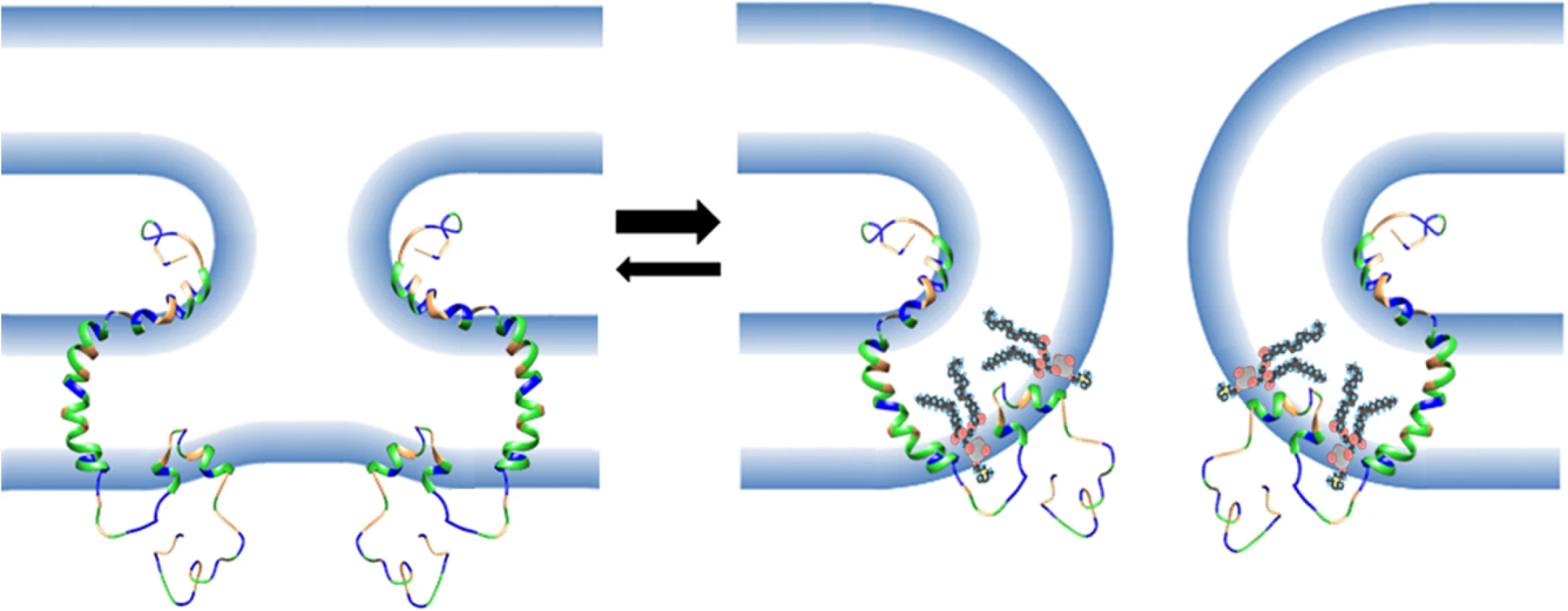
A surprising result was the ability of heterologous ALPS motifs to functionally replace the p15 HP, and this phenotype was ablated by mutations that disrupt the curvature sensing function of the ALPS motifs [47, 51, 52] (Fig 7). These observations imply linear, amphipathic ALPS motifs and the H-L-H p15 HP impart a similar curvature sensing ability to the p15 endodomain. How might these two disparate structures provide a similar function during the fusion reaction? ALPS motifs contain one or more bulky aromatic residues on their hydrophobic face and have considerably higher hydrophobic moments than the p15 HP (Fig 7), properties well-suited for interaction with curved membranes and hydrophobic defects [60]. The hydrophobic moment of AHs, while required, is not the sole determinant of curvature sensing since other AHs such as those of HO-NBAR and DivIVA have similar hydrophobic moments as the ALPS motifs but do not sense or induce positive curvature [53, 55]. Presumably, other features of these AHs influence how they interact with and insert into membranes. Unlike ALPS motifs, the p15 HP is devoid of aromatic residues and comprises predominantly hydrophobic or apolar residues resulting in limited amphipathic character when modeled as a straight helix (Fig 7). However, the two small terminal helices in the H-L-H conformation are amphipathic; residues L82, L83, V85, I86 are on one face of the C-terminal helix, and residues L70, L71 and possibly L68 in the N-terminal helix present another hydrophobic face (Fig 3). Connected by a highly flexible loop region, these two helices are free to change their relative orientation to each other; the structure of some conformers actually resembled helical hairpins, similar to conformations of the caveolin intramembrane domain [61] and the influenza virus HA fusion peptide [62]. The dynamic H-L-H structure may therefore be required to reposition the helical hydrophobic faces in the correct geometry to allow the p15 HP to optimally interact with, and insert to the correct depth into, hydrophobic defects.
The dependence of the p15 HP on a dynamic H-L-H conformation for membrane fusion activity is consistent with the phenotype of mutant p15 proteins and p15HPpep peptides. Mutations in the loop region that increased the helicity of the p15HPpep peptides resulted in loss of curvature sensing capability. Their increased retention times on reverse-phase HPLC columns (Fig 6 vs Fig 4) indicate these mutant peptides are also considerably more hydrophobic than wt p15HPpep, which presumably explains their increased association with liposomes independent of liposome size and curvature (Fig 6). In the context of the p15 protein, deeper insertion of these hydrophobic helices into the cytoplasmic leaflet would force apart lipid acyl chains and induce negative curvature, the opposite curvature needed for pore, which may explain why these HP mutations abrogate p15-induced cell-cell pore formation (Fig 1).
Membrane-proximal AHs or hydrophobic regions have also been noted in the cytoplasmic tails of many enveloped viral fusogens from diverse viruses, including retroviruses, lentiviruses, rhabdoviruses, herpesviruses and paramyxoviruses [63]. The functional significance of these AHs is not well established since tail truncations of several of these proteins retain pore forming capability [64–66]. In the case of MoMuLV, differential scanning calorimetry indicates the env protein AH induces positive curvature in membranes [67], suggesting this motif may induce or stabilize positive curvature to promote pore formation, similar to the p15 FLiPS. However, a tail truncation that deletes the MoMuLV AH has no effect on fusion pore formation, but it does partially inhibit syncytia formation [63, 66]. The MoMuLV AH may therefore promote pore expansion, not pore formation. A similar role for the cytoplasmic tails of the paramyvovirus and influenza virus fusion proteins in pore expansion has also been reported [68, 69]. Cellular curvature-generating proteins such as dynamin, ENTH domain proteins, and BAR domain proteins have also recently been shown to promote pore expansion during cell-cell fusion and vesicle exocytosis [70, 71], and the p15 HP may serve a similar role in stabilizing and propagating positive curvature in the rim of laterally expanding fusion pores. Positive curvature sensors are therefore involved in pore expansion, and as we now show for the first time, in pore formation.
Materials and Methods
Cells and antibodies
Vero cells were obtained from the American Type Culture Collection (ATCC) and QM5 cells were obtained from Charles Ordahl [72] and were cultured as previously described [4]. The production of rabbit polyclonal p14 - and p15-specific antisera has been previously described [5, 73]. Monoclonal mouse anti-FLAG or anti-actin (Sigma-Aldrich), and Alexa Fluor 647 - or horseradish peroxidase-conjugated goat-anti rabbit and goat anti-mouse conjugated secondary antibodies (Jackson ImmunoResearch) were from the indicated suppliers.
Cloning
All FAST proteins were cloned into pcDNA3 mammalian expression vector (Invitrogen) as previously described (Corcoran and Duncan, 2004; Dawe et al., 2005). The Stratagene QuikChange mutagenesis method was used to generate alanine scan mutants, using pcDNA3-p15 as a template. A triple FLAG tag was added to the N-terminus of the indicated p15 constructs by PCR amplification for use in cell-surface immunofluorescence. The pcDNA3-p15 plasmid was used as a template to perform PCR-driven overlap extension [74] to replace the hydrophobic patch with heterologous amphipathic helices. Custom oligonucleotide primers were purchased from IDT, and all constructs were confirmed by sequencing.
Transfection and syncytial indexing
Cells at ~70% confluency were used for transient transfection with polyethylenimine (Polysciences Inc.) 24 h after seeding. Cell monolayers were fixed with methanol at various timepoints and stained with Wright-Giemsa stain (Siemen’s Healthcare). Images were captured on a Nikon Diaphot TMD inverted microscope using the 20x objective. Fusogenicity of constructs was determined by counting the number of syncytial nuclei per microscopic field as previously described (Corcoran et al, 2004). Results are reported as the mean number of syncytial nuclei per field of view.
Pore formation assay
Pore formation was quantified as previously described [32]. Briefly, sparsely seeded QM5 cells were co-transfected with pEGFP and the p15 construct of interest and incubated for 9 h post-transfection. Vero target cells were loaded with CellTrace calcein red-orange AM (Molecular Probes) and incubated in growth media for 2–4 h. Target cells were then resuspended with trypsin and overlaid on the transfected donor cells. At 3 h after co-culturing, cells were resuspended into PBS by trypsin digestion and fixed in suspension at 4°C using 3.7% formaldehyde. Fixed cells were analyzed by flow cytometry (FACSCaliber, Becton Dickinson), quantifying the extent of calcein red fluorescence in 10,000 gated EGFP-positive cells, indicative of cytoplasmic transfer and pore formation between donor and target cells, using FCS Express 2.0 (DeNovo software). Donor cells transfected with pEGFP plus empty vector served as negative controls.
Cell surface expression
Transfected QM5 cells expressing N-terminally FLAG-tagged p15 constructs were incubated at 4˚C for 30 min in blocking buffer (5% normal goat serum, 1% bovine serum albumin (BSA), 0.02% NaN3 in HBSS) and stained with a 1 : 1000 dilution of anti-FLAG antibody and a 1 : 2,000 dilution of Alexa 647-conjugated goat anti-mouse secondary antibody, each for 1 h at 4˚C. Cells were resuspended in phosphate buffered saline (PBS) containing 10 mM EDTA, fixed with 3.7% formaldehyde, and 10,000 cells were analyzed by flow cytometry (Becton Dickinson FACSCalibur) using De-Novo software. The fluorescence gate was set to <5% for negative control cells transfected with empty vector, and the same gate was applied to quantify surface fluorescence of the p15 transfected cells. Background fluorescence from vector-transfected cells was subtracted before calculating mean percent surface fluorescence.
Peptides
Wild-type and mutant p15HPpep peptides were synthesized by GenScript using click peptide synthesis [75] and purified to >95% purity by reverse-phase HPLC. Sequences are as follows: wt p15HPpep, acetyl-LGLLSYGAGVASLPLLNVIA-amide; p15HPpep GAG, acetyl-LGLLSYAAAVASLPLLNVIA-amide; p15HPpep G74A, acetyl-LGLLSYAAGVASLPLLNVIA-amide. Peptides were dissolved in DMSO (10μg/μl) at room temperature and then diluted 4-fold with DMSO/500 mM Hepes buffer, pH 8.4 (70 : 30, v:v) and incubated at 37°C overnight to quantitatively convert the O-acyl isopeptide bond to an N-acyl peptide bond [75]. For CD experiments, the native peptides were further concentrated using preparative reverse-phase HPLC and lyophilisation.
Circular dichroism spectroscopy
Far-ultraviolet CD spectra of wt and mutant p15HPpep (200 μM) dissolved in 76 mM 1-palmitoyl-2-hydroxy-sn-glycero-3-[phospho-rac-(1-glycerol)] (LPPG) micelles (2 : 1 micell:peptide ratio) were obtained using a Jasco J-810 spectropolarimeter with an optical path of 0.1 mm. Spectra were acquired from 260 to 185 nm in 0.1 nm steps at a temperature of 37C. Measurements were collected from three scans in each of two independent experiments. Data were converted to the mean residue ellipticity, [θ], averaged, and blank.
Liposome flotation assay
Liposomes were prepared in HEPES-buffered saline (HBS; 10mM HEPES, 150mM NaCl, pH 7.4) as previously described [36], using a 1 : 1:1 molar ratio of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), and cholesterol (Avanti Polar Lipids) at a final total lipid concentration of 25.2 mM, and the lipid mixture was sequentially extruded through 400 nm, 100 nm and 50 nm polycarbonate filters. Electron microscopy confirmed a fairly uniform, normal distribution of liposome sizes around the mean liposome diameter expected from the filter pore size [42]. Peptides and liposomes were mixed at 1 : 500 peptide:lipid molar ratio (15.8 μg of peptide [75.6 nmoles] and 3.78 μmoles of lipid in a final volume of 150 μl) and incubated for 30 minutes at room temperature. The suspension was adjusted to 30% (w/v) sucrose by adding 100 L of a 75% (w/v) sucrose solution in HBS. The resulting high-sucrose suspension was overlaid with 200 μL of 25% (w/v) sucrose in HBS and 200 L of sucrose-free HBS. The sample was centrifuged at 55,000 rpm in a S55S swinging bucket rotor (206,000 x g) for 3 h at 4°C using a Sorval MTX150 benchtop ultracentrifuge. The bottom (250 μL), middle (200 μl) and top (200 μL) fractions were harvested from the bottom using a Hamilton syringe and 100 μl of the top and bottom fractions were analyzed by reverse-phase HPLC. The percent total peptide in the liposome fraction was determined by integrating the area under the HPLC peptide peaks and normalizing to the relative sample volumes.
HPLC
HPLC was performed on a Waters 2690 Separation Module with a Vydac 214MS C4 reverse phase column (5 μm particle, 250mm × 4.6mm; Grace Davison Discovery Sciences) using a water + 0.1% trifluoroacetic acid (TFA) (Solvent A) and acetonitrile + 0.085% acetonitrile (Solvent B) mobile phase gradient (% Solvent A/B, time: [80/20, 0], [40/60, 20], [20/80, 25]) with a flow rate of 1mL/min. Peptide elution was monitored at 215nm with a Waters 2487 dual wavelength detection unit.
Bis-ANS competition assay
Binding of 4,4′-dianilino-1,1′-binaphthyl-5,5′-disulfonic acid dipotassium salt (bis-ANS; Invitrogen) to liposomes was monitored by measuring the fluorescence after addition of a 10 mM stock solution (0.1 to 15 M final concentration) to 100 M liposomes. Fluorescence was monitored on a Varian Cary Eclipse fluorescence spectrophotometer with excitation and emission wavelengths of 395 nm and 500 nm, and a slit width of 5 nm. Liposomes were blocked with 10 M bis-ANS, pelleted by ultracentrifugation and resuspended in buffer for use in peptide-binding competition assays. Blocked liposomes were incubated with peptide under conditions identical to the liposome flotation assay described above.
Solution NMR data acquisition and structure calculation
Atomic resolution structure of the p15HPpep (LGLLSYGAGVASLPLLNVIA; 1 mM), representing residues 68–87 of p15 FAST protein, was determined in the presence of dodecylphosphocholine (DPC-d38; 150 mM) micelles in 20 mM sodium acetate, 90% H2O/10% D2O at pH 5.0. 1D 1H, 2D 15N-1H HSQC (natural abundance), 2D 13C-1H HSQC (natural abundance), 2D 1H-1H TOCSY (80 ms mixing time), and 2D 1H-1H NOESY (200–400 ms mixing times) experiments were acquired at 37°C using a Bruker Avance III 700 MHz spectrometer equipped with a 5 mm TCI cryoprobe. Following NOE buildup analysis, distance restraints were determined to be unaffected by spin diffusion in the NOESY at 200 ms mixing time. The NOE restraints were therefore produced from NOESY cross-peak analysis at 200 ms. Following iterative refinement [46], 511 nonredundant NOEs were retained, 173 of which corresponded to intraresidue contacts, 294 to interresidue contacts, and 44 to ambiguous contacts (see S1 Table). Structure calculations were performed using the simulated annealing algorithm within the python scripting interface of XPLOR-NIH 2.34 [76] incorporating the RAMA torsion angle database potential term [77]. The 50 p15HPpep lowest energy structures out of 100 calculated were retained for analysis. Atomic coordinates are deposited in the Research Collaboratory for Structural Bioinformatics (www.rcsb.org), with PDB ID 2MNS. Chemical shifts are deposited in the Biological Magnetic Resonance Bank (www.bmrb.wisc.edu) with entry ID 19902.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Boutilier J, Duncan R, editors. The reovirus fusion-associated small transmembrane (FAST) proteins: virus-encoded cellular fusogens. USA: Elsevier; 2011.
2. Ciechonska M, Duncan R. Reovirus FAST proteins: virus-encoded cellular fusogens. Trends Microbiol. 2014;22(12):715–24. doi: 10.1016/j.tim.2014.08.005 25245455
3. White JM, Delos SE, Brecher M, Schornberg K. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit Rev Biochem Mol Biol. 2008;43(3):189–219. doi: 10.1080/10409230802058320 18568847
4. Corcoran JA, Duncan R. Reptilian reovirus utilizes a small type III protein with an external myristylated amino terminus to mediate cell-cell fusion. J Virol. 2004;78(8):4342–51. 15047847
5. Dawe S, Corcoran JA, Clancy EK, Salsman J, Duncan R. Unusual topological arrangement of structural motifs in the baboon reovirus fusion-associated small transmembrane protein. J Virol. 2005;79(10):6216–26. 15858006
6. Guo H, Sun X, Yan L, Shao L, Fang Q. The NS16 protein of aquareovirus-C is a fusion-associated small transmembrane (FAST) protein, and its activity can be enhanced by the nonstructural protein NS26. Virus Res. 2013;171(1):129–37. doi: 10.1016/j.virusres.2012.11.011 23201583
7. Racine T, Hurst T, Barry C, Shou J, Kibenge F, Duncan R. Aquareovirus effects syncytiogenesis by using a novel member of the FAST protein family translated from a noncanonical translation start site. J Virol. 2009;83(11):5951–5. doi: 10.1128/JVI.00171-09 19297495
8. Shmulevitz M, Duncan R. A new class of fusion-associated small transmembrane (FAST) proteins encoded by the non-enveloped fusogenic reoviruses. Embo J. 2000;19(5):902–12. 10698932
9. Thalmann CM, Cummins DM, Yu M, Lunt R, Pritchard LI, Hansson E, et al. Broome virus, a new fusogenic Orthoreovirus species isolated from an Australian fruit bat. Virology. 2010;402(1):26–40. doi: 10.1016/j.virol.2009.11.048 20350736
10. Albertini A, Bressanelli S, Lepault J, Gaudin Y. Structure and working of viral fusion machinery. Current topics in membranes. 2011;68 : 49–80. doi: 10.1016/B978-0-12-385891-7.00003-9 21771495
11. Brown CW, Stephenson KB, Hanson S, Kucharczyk M, Duncan R, Bell JC, et al. The p14 FAST protein of reptilian reovirus increases vesicular stomatitis virus neuropathogenesis. J Virol. 2009;83(2):552–61. doi: 10.1128/JVI.01921-08 18971262
12. Duncan R, Sullivan K. Characterization of two avian reoviruses that exhibit strain-specific quantitative differences in their syncytium-inducing and pathogenic capabilities. Virol. 1998;250 : 263–72.
13. Salsman J, Top D, Boutilier J, Duncan R. Extensive syncytium formation mediated by the reovirus FAST proteins triggers apoptosis-induced membrane instability J Virol. 2005;79(13):8090–100. 15956554
14. Zaitseva E, Mittal A, Griffin DE, Chernomordik LV. Class II fusion protein of alphaviruses drives membrane fusion through the same pathway as class I proteins. J Cell Biol. 2005;169(1):167–77. 15809312
15. Harrison SC. Viral membrane fusion. Nat Struct Mol Biol. 2008;15 : 690–8. doi: 10.1038/nsmb.1456 18596815
16. Melikyan GB. Common principles and intermediates of viral protein-mediated fusion: the HIV-1 paradigm. Retrovirology. 2008;5 : 111. doi: 10.1186/1742-4690-5-111 19077194
17. Chernomordik LV, Kozlov MM. Mechanics of membrane fusion. Nat Struct Mol Biol. 2008;15(7):675–83. doi: 10.1038/nsmb.1455 18596814
18. Kozlov MM, McMahon HT, Chernomordik LV. Protein-driven membrane stresses in fusion and fission. Trends Biochem Sci. 2010;35(12):699–706. 20638285. doi: 10.1016/j.tibs.2010.06.003
19. Ford MG, Mills IG, Peter BJ, Vallis Y, Praefcke GJ, Evans PR, et al. Curvature of clathrin-coated pits driven by epsin. Nature. 2002;419(6905):361–6. 12353027
20. Liu TY, Bian X, Sun S, Hu X, Klemm RW, Prinz WA, et al. Lipid interaction of the C terminus and association of the transmembrane segments facilitate atlastin-mediated homotypic endoplasmic reticulum fusion. Proc Natl Acad Sci U S A. 2012;109(32):E2146–54. doi: 10.1073/pnas.1208385109 22802620
21. Groffen AJ, Martens S, Diez Arazola R, Cornelisse LN, Lozovaya N, de Jong AP, et al. Doc2b is a high-affinity Ca2+ sensor for spontaneous neurotransmitter release. Science. 2010;327(5973):1614–8. doi: 10.1126/science.1183765 20150444
22. Martens S, Kozlov MM, McMahon HT. How synaptotagmin promotes membrane fusion. Science. 2007;316(5828):1205–8. 17478680
23. Vamparys L, Gautier R, Vanni S, Bennett WF, Tieleman DP, Antonny B, et al. Conical lipids in flat bilayers induce packing defects similar to that induced by positive curvature. Biophys J. 2013;104(3):585–93. doi: 10.1016/j.bpj.2012.11.3836 23442909
24. McMahon HT, Kozlov MM, Martens S. Membrane curvature in synaptic vesicle fusion and beyond. Cell. 2010;140(5):601–5. doi: 10.1016/j.cell.2010.02.017 20211126
25. Chernomordik LV, Zimmerberg J, Kozlov MM. Membranes of the world unite! J Cell Biol. 2006;175(2):201–7. 17043140
26. Cohen FS, Melikyan GB. The energetics of membrane fusion from binding, through hemifusion, pore formation, and pore enlargement. J Membr Biol. 2004;199(1):1–14. 15366419
27. Chernomordik LV, Kozlov MM. Protein-lipid interplay in fusion and fission of biological membranes. Annu Rev Biochem. 2003;72 : 175–207. 14527322
28. Reese C, Mayer A. Transition from hemifusion to pore opening is rate limiting for vacuole membrane fusion. J Cell Biol. 2005;171(6):981–90. 16365164
29. Parmar HB, Barry C, Kai F, Duncan R. Golgi complex-plasma membrane trafficking directed by an autonomous, tribasic Golgi export signal. Mol Biol Cell. 2014;25(6):866–78. doi: 10.1091/mbc.E13-07-0364 24451258
30. Dawe S, and R. Duncan. The S4 genome segment of baboon reovirus is bicistronic and encodes a novel fusion-associated small transmembrane protein. J Virol. 2002;76 : 2131–40. 11836390
31. Shmulevitz M, Salsman J, Duncan R. Palmitoylation, membrane-proximal basic residues, and transmembrane glycine residues in the reovirus p10 protein are essential for syncytium formation. J Virol. 2003;77(18):9769–79. 12941885
32. Barry C, Duncan R. Multifaceted sequence-dependent and-independent roles for reovirus FAST protein cytoplasmic tails in fusion pore formation and syncytiogenesis. J Virol. 2009;83(23):12185–95. 19759162. doi: 10.1128/JVI.01667-09
33. Clancy EK, Duncan R. Reovirus FAST protein transmembrane domains function in a modular, primary sequence-independent manner to mediate cell-cell membrane fusion. J Virol. 2009;83(7):2941–50. doi: 10.1128/JVI.01869-08 19129451
34. Clancy EK, Duncan R. Helix-destabilizing, beta-branched, and polar residues in the baboon reovirus p15 transmembrane domain influence the modularity of FAST proteins. J Virol. 2011;85(10):4707–19. doi: 10.1128/JVI.02223-10 21367887
35. Barry C, Key T, Haddad R, Duncan R. Features of a spatially constrained cystine loop in the p10 FAST protein ectodomain define a new class of viral fusion peptide. J Biol Chem. 2010;285(22):16424–33. doi: 10.1074/jbc.M110.118232 20363742
36. Corcoran JA, Syvitski R, Top D, Epand RM, Epand RF, Jakeman D, et al. Myristoylation, a protruding loop, and structural plasticity are essential features of a nonenveloped virus fusion peptide motif. J Biol Chem. 2004;279(49):51386–94. 15448165
37. Key T, Duncan R. A compact, multifunctional fusion module directs cholesterol-dependent homomultimerization and syncytiogenic efficiency of reovirus p10 FAST proteins. PLoS Pathog. 2014;10:e1004023. doi: 10.1371/journal.ppat.1004023 24651689
38. Top D, Read JA, Dawe SJ, Syvitski RT, Duncan R. Cell-cell membrane fusion induced by p15 fusion-associated small transmembrane (FAST) protein requires a novel fusion peptide motif containing a myristoylated polyproline type II helix. J Biol Chem. 2012;287(5):3403–14. doi: 10.1074/jbc.M111.305268 22170056
39. Cortese MS, Uversky VN, Dunker AK. Intrinsic disorder in scaffold proteins: getting more from less. Prog Biophys Mol Biol. 2008;98(1):85–106. doi: 10.1016/j.pbiomolbio.2008.05.007 18619997
40. Ciechonska M, Duncan R. Efficient reovirus - and measles virus-mediated pore expansion during syncytium formation is dependent on annexin A1 and intracellular calcium. J Virol. 2014;88 : 6137–47. doi: 10.1128/JVI.00121-14 24648446
41. Top D, Barry C, Racine T, Ellis CL, Duncan R. Enhanced fusion pore expansion mediated by the trans-acting Endodomain of the reovirus FAST proteins. PLoS Pathog. 2009;5(3):e1000331. doi: 10.1371/journal.ppat.1000331 19266079
42. Key T, Sarker M, de Antueno R, Rainey JK, Duncan R. The p10 FAST protein fusion peptide functions as a cystine noose to induce cholesterol-dependent liposome fusion without liposome tubulation. Biochim Biophys Acta. 2015;1848(2):408–16. doi: 10.1016/j.bbamem.2014.10.020 25450808
43. Shmulevitz M, Epand RF, Epand RM, Duncan R. Structural and functional properties of an unusual internal fusion peptide in a nonenveloped virus membrane fusion protein. J Virol. 2004;78 : 2808–18. 14990700
44. Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4(3):363–71. doi: 10.1038/nprot.2009.2 19247286
45. Wuthrich K. NMR of proteins and nucleic acids. New York: John Wiley & Sons; 1986.
46. Ding J, Rainey JK, Xu C, Sykes BD, Fliegel L. Structural and functional characterization of transmembrane segment VII of the Na+/H+ exchanger isoform 1. J Biol Chem. 2006;281(40):29817–29. 16861220
47. Bigay J, Casella JF, Drin G, Mesmin B, Antonny B. ArfGAP1 responds to membrane curvature through the folding of a lipid packing sensor motif. Embo J. 2005;24(13):2244–53. 15944734
48. Drin G, Antonny B. Amphipathic helices and membrane curvature. FEBS Lett. 2010;584(9):1840–7. doi: 10.1016/j.febslet.2009.10.022 19837069
49. Antonny B. Mechanisms of membrane curvature sensing. Annu Rev Biochem. 2011;80 : 101–23. doi: 10.1146/annurev-biochem-052809-155121 21438688
50. Eisenberg D, Weiss RM, Terwilliger TC. The hydrophobic moment detects periodicity in protein hydrophobicity. Proc Natl Acad Sci U S A. 1984;81(1):140–4. 6582470
51. Levi S, Rawet M, Kliouchnikov L, Parnis A, Cassel D. Topology of amphipathic motifs mediating Golgi localization in ArfGAP1 and its splice isoforms. J Biol Chem. 2008;283(13):8564–72. doi: 10.1074/jbc.M709738200 18195007
52. Gonzalez-Rubio P, Gautier R, Etchebest C, Fuchs PF. Amphipathic-Lipid-Packing-Sensor interactions with lipids assessed by atomistic molecular dynamics. Biochim Biophys Acta. 2011;1808(9):2119–27. doi: 10.1016/j.bbamem.2011.05.006 21600869
53. Fernandes F, Loura LM, Chichon FJ, Carrascosa JL, Fedorov A, Prieto M. Role of helix 0 of the N-BAR domain in membrane curvature generation. Biophys J. 2008;94(8):3065–73. doi: 10.1529/biophysj.107.113118 18199667
54. Lee MT, Hung WC, Chen FY, Huang HW. Mechanism and kinetics of pore formation in membranes by water-soluble amphipathic peptides. Proc Natl Acad Sci U S A. 2008;105(13):5087–92. doi: 10.1073/pnas.0710625105 18375755
55. Lenarcic R, Halbedel S, Visser L, Shaw M, Wu LJ, Errington J, et al. Localisation of DivIVA by targeting to negatively curved membranes. EMBO J. 2009;28(15):2272–82. doi: 10.1038/emboj.2009.129 19478798
56. Leikina E, LeDuc DL, Macosko JC, Epand R, Shin YK, Chernomordik LV. The 1–127 HA2 construct of influenza virus hemagglutinin induces cell-cell hemifusion. Biochemistry. 2001;40(28):8378–86. 11444985
57. Chernomordik LV, Zimmerberg J. Bending membranes to the task: structural intermediates in bilayer fusion. Curr Opin Struct Biol. 1995;5(4):541–7. 8528771
58. Leikina E, Chernomordik LV. Reversible merger of membranes at the early stage of influenza hemagglutinin-mediated fusion. Mol Biol Cell. 2000;11(7):2359–71. 10888674
59. Clancy EK, Barry C, Ciechonska M, Duncan R. Different activities of the reovirus FAST proteins and influenza hemagglutinin in cell-cell fusion assays and in response to membrane curvature agents. Virology. 2010;397(1):119–29. doi: 10.1016/j.virol.2009.10.039 19931884
60. Vanni S, Vamparys L, Gautier R, Drin G, Etchebest C, Fuchs PF, et al. Amphipathic lipid packing sensor motifs: probing bilayer defects with hydrophobic residues. Biophys J. 2013;104(3):575–84. doi: 10.1016/j.bpj.2012.11.3837 23442908
61. Parton RG, Hanzal-Bayer M, Hancock JF. Biogenesis of caveolae: a structural model for caveolin-induced domain formation. J Cell Sci. 2006;119(Pt 5):787–96.
62. Lorieau JL, Louis JM, Bax A. The complete influenza hemagglutinin fusion domain adopts a tight helical hairpin arrangement at the lipid:water interface. Proc Natl Acad Sci U S A. 2010;107(25):11341–6. doi: 10.1073/pnas.1006142107 20534508
63. Rozenberg-Adler Y, Conner J, Aguilar-Carreno H, Chakraborti S, Dimitrov DS, Anderson WF. Membrane-proximal cytoplasmic domain of Moloney murine leukemia virus envelope tail facilitates fusion. Exp Mol Pathol. 2008;84(1):18–30. doi: 10.1016/j.yexmp.2007.11.001 18222422
64. Cathomen T, Naim HY, Cattaneo R. Measles viruses with altered envelope protein cytoplasmic tails gain cell fusion competence. J Virol. 1998;72(2):1224–34. 9445022
65. Lin X, Derdeyn CA, Blumenthal R, West J, Hunter E. Progressive truncations C terminal to the membrane-spanning domain of simian immunodeficiency virus Env reduce fusogenicity and increase concentration dependence of Env for fusion. J Virol. 2003;77(12):7067–77. 12768026
66. Melikyan GB, Markosyan RM, Brener SA, Rozenberg Y, Cohen FS. Role of the cytoplasmic tail of ecotropic moloney murine leukemia virus Env protein in fusion pore formation. J Virol. 2000;74(1):447–55. 10590134
67. Epand RF, Zhang YL, Mirzabekov T, Kagan B, Silberstein A, Hubbell WL, et al. Membrane activity of an amphiphilic alpha-helical membrane-proximal cytoplasmic domain of the MoMuLV envelope glycoprotein. Exp Mol Pathol. 2008;84(1):9–17. doi: 10.1016/j.yexmp.2007.11.003 18206141
68. Dutch RE, Lamb RA. Deletion of the cytoplasmic tail of the fusion protein of the paramyxovirus simian virus 5 affects fusion pore enlargement. J Virol. 2001;75(11):5363–9. 11333918
69. Kozerski C, Ponimaskin E, Schroth-Diez B, Schmidt MF, Herrmann A. Modification of the cytoplasmic domain of influenza virus hemagglutinin affects enlargement of the fusion pore. J Virol. 2000;74(16):7529–37. 10906206
70. Richard JP, Leikina E, Langen R, Henne WM, Popova M, Balla T, et al. Intracellular curvature-generating proteins in cell-to-cell fusion. Biochem J. 2011;440(2):185–93. doi: 10.1042/BJ20111243 21895608
71. Trouillon R, Ewing AG. Amperometric measurements at cells support a role for dynamin in the dilation of the fusion pore during exocytosis. Chemphyschem: a European journal of chemical physics and physical chemistry. 2013;14(10):2295–301. doi: 10.1002/cphc.201300319 23824748.
72. Antin PB, Ordahl CP. Isolation and characterization of an avian myogenic cell line. Dev Biol. 1991;143(1):111–21. 1985013
73. Top D, de Antueno R, Salsman J, Corcoran J, Mader J, Hoskin D, et al. Liposome reconstitution of a minimal protein-mediated membrane fusion machine. EMBO J. 2005;24(17):2980–8. 16079913
74. Heckman KL, Pease LR. Gene splicing and mutagenesis by PCR-driven overlap extension. Nat Protoc. 2007;2(4):924–32. 17446874
75. Sohma Y, Taniguchi A, Yoshiya T, Chiyomori Y, Fukao F, Nakamura S, et al. 'Click peptide': a novel 'O-acyl isopeptide method' for peptide synthesis and chemical biology-oriented synthesis of amyloid beta peptide analogues. J Pept Sci. 2006;12(12):823–8. 17131295
76. Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160(1):65–73. 12565051
77. Kuszewski J, Gronenborn AM, Clore GM. Improvements and extensions in the conformational database potential for the refinement of NMR and X-ray structures of proteins and nucleic acids. J Magn Reson. 1997;125(1):171–7. 9245376
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 6
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- HIV Latency Is Established Directly and Early in Both Resting and Activated Primary CD4 T Cells
- Battling Phages: How Bacteria Defend against Viral Attack
- A 21st Century Perspective of Poliovirus Replication
- Adenovirus Tales: From the Cell Surface to the Nuclear Pore Complex