
Inhibits Virulence through Suppression of Pyochelin and Pyoverdine Biosynthesis
Pseudomonas aeruginosa and Candida albicans are two medically important human pathogens that often co-infect or co-colonize the same human niches, such as the gut. In a normal healthy host, P. aeruginosa and C. albicans can colonize the gut without any significant pathologic sequelae. But in immunocompromised hosts, both pathogens can escape the gut and cause life-threatening disseminated infections. Yet the mechanisms and pathogenic consequences of interactions between these two pathogens within a living mammalian host are not well understood. Here, we use a mouse model of P. aeruginosa and C. albicans gut co-infection to better understand the mechanisms by which C. albicans inhibits P. aeruginosa infection. C. albicans inhibits the expression of P. aeruginosa genes that are vital for iron acquisition. Accordingly, deleting these iron acquisition genes in P. aeruginosa prevents infection. Understanding how microbes interact and antagonize each other may help us identify new potential therapeutic targets for preventing or treating infections.
Published in the journal:
. PLoS Pathog 11(8): e32767. doi:10.1371/journal.ppat.1005129
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1005129
Summary
Pseudomonas aeruginosa and Candida albicans are two medically important human pathogens that often co-infect or co-colonize the same human niches, such as the gut. In a normal healthy host, P. aeruginosa and C. albicans can colonize the gut without any significant pathologic sequelae. But in immunocompromised hosts, both pathogens can escape the gut and cause life-threatening disseminated infections. Yet the mechanisms and pathogenic consequences of interactions between these two pathogens within a living mammalian host are not well understood. Here, we use a mouse model of P. aeruginosa and C. albicans gut co-infection to better understand the mechanisms by which C. albicans inhibits P. aeruginosa infection. C. albicans inhibits the expression of P. aeruginosa genes that are vital for iron acquisition. Accordingly, deleting these iron acquisition genes in P. aeruginosa prevents infection. Understanding how microbes interact and antagonize each other may help us identify new potential therapeutic targets for preventing or treating infections.
Introduction
The bacterium Pseudomonas aeruginosa and the fungus Candida albicans, two medically important human pathogens, co-infect or co-colonize numerous sites on the human body, including the gut [1], lung (including respiratory ventilator) [2–4], burn wound [5], genitourinary tract [1], and skin sites (including vascular catheters) [6,7]. In vitro studies suggest that mutually antagonistic interactions take place between P. aeruginosa and C. albicans. P. aeruginosa modulates C. albicans morphology [8] and can kill C. albicans filaments [9,10]. C. albicans inhibits P. aeruginosa cellular signaling [11] and metabolite production [11]. Although many clinical studies report the observation of mixed infections with P. aeruginosa and C. albicans [1,3,12,13], the impact on bacterial and/or fungal pathogenesis is still unclear [14,15].
In cancer and stem cell transplant patients, invasive P. aeruginosa and C. albicans infections are thought to arise from initial GI colonization and subsequent translocation after medically induced immune deficits [16–20]. Three primary defense mechanisms that prevent microbial translocation from the GI tract in humans and mice include 1) a stable gut microbiota; 2) intact intestinal mucosal barriers; and 3) intact host immune defenses, particularly cellular immunity [21–23]. Bacterial-fungal interactions can significantly impact gut microbiota homeostasis and gut mucosal integrity. For instance, bacteria can inhibit C. albicans GI colonization [24–26] and conversely, C. albicans modulates bacterial repopulation in the gut [27,28]. Importantly, the risk for bacteremia in cancer patients is directly proportional to gut bacterial burden [29]. Furthermore, bacteria and fungi can damage epithelial barriers by production of cytotoxic effector molecules (e.g. Type III secretion system in P. aeruginosa) [30–32] and morphology (yeast to hyphal transition in C. albicans) [33–36]. Thus bacterial-fungal interactions that affect these virulence determinants could significantly impact the pathogenesis of invasive disease.
Another line of host defense against microbial pathogen infection is the withholding of nutrients (such as iron) to prevent microbial overgrowth [37], a strategy known as nutritional immunity [38]. Since iron is not freely available in the mammalian host, most bacterial pathogens utilize high-affinity iron uptake mechanisms that compete against host-mediated sequestration of iron [38]. Accordingly, P. aeruginosa produces low-molecular weight secreted molecules known as siderophores (pyochelin and pyoverdine) that specifically chelate iron (Fe3+). Both pyochelin and pyoverdine have been shown to be important for virulence in pulmonary and burn wound models of P. aeruginosa infection [39–42]. C. albicans possesses similar iron acquisition mechanisms [43,44]. Thus, in iron-limited environments, such as the mammalian gut, the ability of one microbe (e.g. C. albicans) to prevent a competing microbe (e.g. P. aeruginosa) from acquiring iron could provide a significant fitness advantage.
To study the impact of fungi on bacterial virulence, we created a murine model of P. aeruginosa and C. albicans GI co-colonization and neutropenia-induced P. aeruginosa virulence. While C. albicans had no effect on P. aeruginosa GI colonization, C. albicans repressed expression of P. aeruginosa pyochelin and pyoverdine biosynthesis genes. Of note, the presence of C. albicans did not increase gut iron levels. Accordingly, deletion of both pyochelin and pyoverdine genes attenuated P. aeruginosa virulence. C. albicans secreted proteins were sufficient to inhibit P. aeruginosa pyochelin and pyoverdine gene expression and decrease P. aeruginosa’s cytotoxic effect on colonocytes in vitro. Strikingly, oral administration of C. albicans secreted proteins protected mice from P. aeruginosa infection. Finally, supplementation with oral iron restored P. aeruginosa virulence in P. aeruginosa and C. albicans colonized mice. Thus, by exploring bacterial-fungal interactions in the mammalian GI tract, we can identify new strategies for preventing invasive microbial infections.
Results
C. albicans inhibits P. aeruginosa virulence in neutropenic mice
We adapted a well-established murine model using oral antibiotic treatment to promote C. albicans [24,25] and P. aeruginosa colonization [31] and monoclonal antibody induced neutropenia to promote P. aeruginosa virulence only [31]. The presence of C. albicans SC5314 in the GI tract did not significantly affect P. aeruginosa PAO1 GI colonization levels compared to mice that had been mono-colonized with PAO1 (Fig 1A). The temporal sequence of GI colonization (P. aeruginosa first, C. albicans first, or P. aeruginosa and C. albicans simultaneously) did not affect P. aeruginosa GI colonization levels (Fig 1A). Conversely, the presence of P. aeruginosa did not significantly affect C. albicans colonization levels compared to mice mono-colonized with C. albicans (Fig 1A). In contrast, when P. aeruginosa and a single bacterial species (Enterococcus faecalis, Bacteroides thetaiotaomicron, Escherichia coli, or Blautia producta) were simultaneously introduced into the GI tract, P. aeruginosa levels significantly decreased compared to mice mono-colonized with P. aeruginosa (S1 Fig).

We previously demonstrated that neutropenia is sufficient for P. aeruginosa virulence [31], whereas both neutropenia and GI mucosal damage are required for C. albicans virulence [25]. Therefore, in the setting of neutropenia, 0% of the mice mono-colonized with P. aeruginosa survived (Fig 1B), with all deceased mice exhibiting evidence of extra-intestinal dissemination of P. aeruginosa (e.g. cultured P. aeruginosa from spleen homogenates) (Fig 1C). Surprisingly, 87.5–100% of mice co-colonized with P. aeruginosa and C. albicans survived after neutropenia (p<0.001 by Fisher’s exact, Fig 1B and 1C). There was no evidence of C. albicans dissemination (e.g. cultured C. albicans from liver homogenates) in any of the mice co-colonized with C. albicans and P. aeruginosa (Fig 1D).
To eliminate the potential immunomodulatory effects of other commensal gut microbiota, we repeated these experiments in germ-free mice. Again, although co-colonization with C. albicans did not significantly change P. aeruginosa GI colonization in germ-free mice (Fig 1E), co-colonization with C. albicans did increase length of survival (p = 0.04, log-rank test) and overall survival from P. aeruginosa infection (3 of 4 mice survived in the C. albicans and P. aeruginosa co-colonized group; 0 of 4 mice survived in the P. aeruginosa group; p = 0.07, Fisher’s exact) (Fig 1F). In deceased mice, P. aeruginosa dissemination was confirmed, but no evidence of C. albicans dissemination was noted (Fig 1G and 1H).
To exclude microbial strain-specific effects, we repeated these experiments with antibiotic-treated mice (n = 8) utilizing additional P. aeruginosa (PA14, PAK) and C. albicans strains (3153A, clinical isolate, biofilm[45]; Can098, clinical isolate, bloodstream; and Can091, clinical isolate, bloodstream) in all possible combinations. Again, P. aeruginosa GI colonization was unaffected by co-colonization with C. albicans (S2 Fig). C. albicans levels were also unaffected by P. aeruginosa (with the exception of decreased C. albicans 3153A levels in the presence of either P. aeruginosa PAO1 or PA14) (S2 Fig). Regardless of the P. aeruginosa or C. albicans strain used, mice co-colonized with P. aeruginosa and C. albicans had significantly increased length of survival (S3 Fig) and significantly increased overall survival (with the only exception being mice co-colonized with P. aeruginosa PA14 and C. albicans 3153A, p = 0.10, Fisher’s exact, Table 1) compared to mice mono-colonized with P. aeruginosa. All deceased mice exhibited evidence of P. aeruginosa dissemination (S3 Fig).

In summary, we conclude that co-colonization with C. albicans does not affect P. aeruginosa’s ability to colonize the murine GI tract, regardless of the temporal sequence of colonization or the microbial strains used. Furthermore, co-colonization with C. albicans significantly increases both length of survival and overall survival from P. aeruginosa infection.
C. albicans does not inhibit Escherichia coli GI colonization or virulence in mice
To determine whether C. albicans’s protective effect was unique to P. aeruginosa, we tested Escherichia coli (a clinical bloodstream isolate recovered from a pediatric cancer patient), another bacteria from the Enterobacteriaceae family frequently isolated from bloodstream infections in cancer patients [18,46]. As with P. aeruginosa, C. albicans did not affect E. coli GI colonization levels (S4 Fig). While C. albicans partially protected mice from E. coli mortality, this protective effect was not statistically significant (S4 Fig).
The C. albicans quorum-sensing molecule farnesol does not affect P. aeruginosa GI colonization or virulence
Cross-kingdom cellular signaling has been documented between P. aeruginosa and C. albicans in vitro: the P. aeruginosa molecule 3-oxo-C12HSL affects C. albicans morphology [8], and the C. albicans metabolite farnesol inhibits P. aeruginosa quinolone signaling [11], reduces pyocyanin synthesis [11], and inhibits swarming motility [4]. Thus, we hypothesized that C. albicans farnesol might play a critical role in inhibiting P. aeruginosa virulence in our murine model. The deletion of the C. albicans dpp3 gene, which encodes a phosphatase that converts farnesyl pyrophosphate to farnesol [47], did not affect P. aeruginosa or C. albicans GI colonization levels (S5 Fig). Importantly, mice co-colonized with C. albicans dpp3Δ/Δ were still protected from P. aeruginosa (87.5% survival, 7 of 8 mice) compared to the P. aeruginosa mono-colonized group (0% survival, 0 of 8 mice) (S5 Fig). Finally, mice mono-colonized with P. aeruginosa and treated with oral farnesol [47] showed no significant difference in P. aeruginosa GI colonization levels and no significant difference in overall survival or length of survival from P. aeruginosa infection compared to untreated controls (S5 Fig). Thus, C. albicans farnesol does not inhibit P. aeruginosa virulence and is not responsible for the C. albicans protective effect from P. aeruginosa infection.
In vivo Pseudomonas aeruginosa transcriptome analysis reveals C. albicans induced suppression of pyochelin and pyoverdine biosynthetic pathways
We used RNA-Seq to examine the in vivo gene expression of P. aeruginosa PAO1 in the GI tract of neutropenic mice (n = 8), in the presence or absence of C. albicans. Our goal was to identify P. aeruginosa virulence genes normally activated during virulence from the GI tract (P. aeruginosa colonized mice) but suppressed by C. albicans in co-colonized mice (P. aeruginosa and C. albicans colonized mice) (Fig 2A). We utilized the antibiotic-treated, rather than germ-free, murine model to emulate the pathogenesis of P. aeruginosa infections in the clinical setting, realizing that we would be isolating transcripts from a complex mixed microbial population. We identified a total of 35 genes that were significantly repressed and only 2 genes whose mRNA levels were increased when C. albicans co-colonized the GI tract (Fig 2B, S1 File). 31% (11 of 35) of the down-regulated genes are known to be involved in pyochelin and pyoverdine biosynthetic pathways (Fig 2C and 2D). We performed RT qPCR of P. aeruginosa pyochelin and pyoverdine biosynthetic genes and confirmed a similar pattern of uniform repression of these genes consistent with our RNASeq results (Fig 2E). In sum, expression of P. aeruginosa pyochelin and pyoverdine genes significantly decreases in the murine GI tract when C. albicans is present.

Deletion of both pyochelin and pyoverdine genes attenuates P. aeruginosa virulence
In order to assess the relative importance of specific pyochelin and/or pyoverdine genes for P. aeruginosa GI colonization or virulence, we constructed P. aeruginosa pyochelin, pyoverdine, and pyochelin/pyoverdine deletional mutants. Since iron is essential for microbial growth and survival, the deletion of pyochelin/pyoverdine genes might significantly limit P. aeruginosa iron acquisition and thus inhibit P. aeruginosa growth and GI colonization. Interestingly, deleting either pyochelin or pyoverdine genes, or both pyochelin and pyoverdine genes, did not affect P. aeruginosa GI colonization (Fig 3A).

In terms of virulence, deleting pyochelin (Fig 3B) or pyoverdine (Fig 3C) genes did not attenuate the virulence of P. aeruginosa, but deleting both pyochelin and pyoverdine genes significantly increased survival from P. aeruginosa infection (87.5–100% survival, p = <0.0001, Fisher’s exact test) when compared to mice colonized with wild-type P. aeruginosa (0% survival) (Fig 3D). Complementation with the pyochelin genes pchBA (pME6477) in the pyochelin/pyoverdine deletional mutants was sufficient to restore P. aeruginosa virulence (S6 Fig) thus proving that the deletion phenotype was not due to unexpected “off-target” effects of deletion. P. aeruginosa dissemination was confirmed by the presence of cultured P. aeruginosa or P. aeruginosa mutants from spleen homogenates of deceased mice (S6 Fig). Thus, either P. aeruginosa pyochelin or pyoverdine is sufficient to maintain P. aeruginosa virulence.
Addition of C. albicans to a P. aeruginosa-colonized murine gut does not increase gut iron levels
Since it is well-established that iron can repress the synthesis of P. aeruginosa siderophores, we first determined whether the iron concentration in the GI tract (as determined by measuring total fecal iron content) of a mouse co-colonized with P. aeruginosa and C. albicans was higher than in mice mono-colonized with P. aeruginosa, as this would then explain the observed decrease in P. aeruginosa pyochelin and pyoverdine gene expression when C. albicans is present. Total fecal iron content (Fe2+and Fe3+ as determined by a ferrozine assay [48]) in untreated (no antibiotics, no microbes) mice was greater than in antibiotic treated mice (Fig 4A). Mouse chow did have detectable iron (albeit a very low concentration), whereas the antibiotic (penicillin-streptomycin) water did not have any detectable iron (Fig 4A). In general, introduction of P. aeruginosa and/or C. albicans into the GI tract of antibiotic-treated mice did not appreciably increase fecal iron levels compared to mice treated only with antibiotics. But most importantly, fecal iron levels in mice co-colonized with P. aeruginosa and C. albicans were not significantly increased compared to mice mono-colonized with the corresponding P. aeruginosa strain (e.g. fecal iron levels in mouse co-colonized with PAO1 and SC5314 were not significantly higher than in mice mono-colonized with PAO1) (Fig 4A). Furthermore, mice colonized with P. aeruginosa pyochelin and pyoverdine mutants did not have significantly increased fecal iron levels compared to mice colonized with wild-type PAO1 (Fig 4A). Mice colonized with the pyochelin/pyoverdine deletional mutant PAO1ΔpchBAΔpvdS, however, did have significantly lower fecal iron levels compared to PAO1, which may be due to an a unique interaction with the gut microbiota or host.
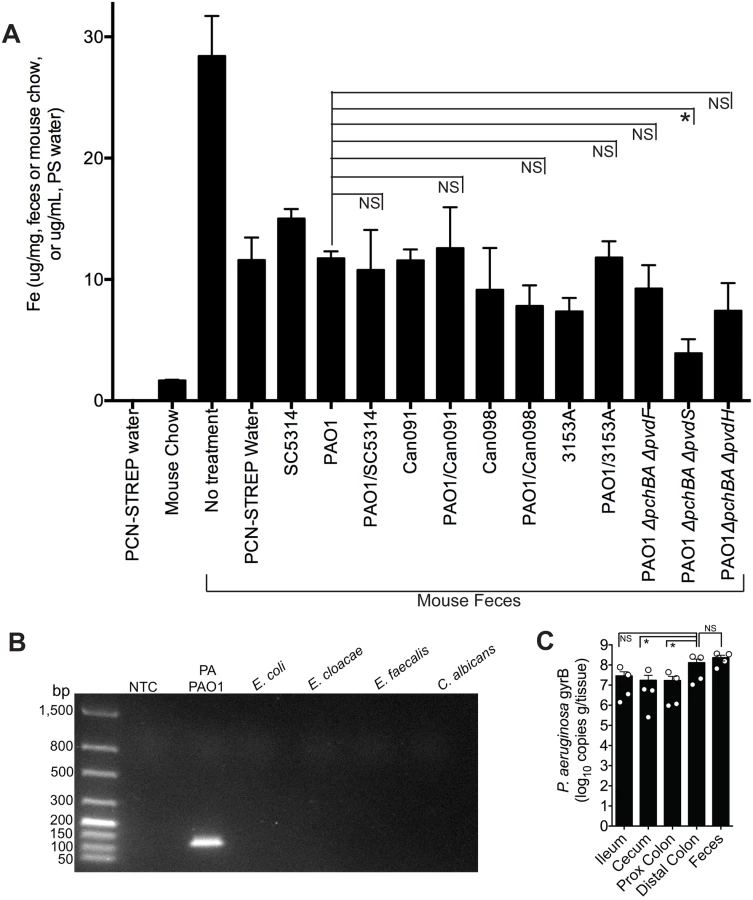
Iron is critical for P. aeruginosa biofilm initiation and maturation [49–51]. Furthermore, disruptions in P. aeruginosa iron metabolism have been shown to inhibit P. aeruginosa biofilm formation [52]. This point is particularly relevant given that there are at least two distinct populations of P. aeruginosa in the murine GI tract: P. aeruginosa in the gut lumen and P. aeruginosa adherent to the GI epithelium, a subset of which may be in biofilms. Fecal specimens are routinely used to measure gut microbial abundance or profile gut microbial populations, but these data may better represent gut lumen microbial populations and may underrepresent microbial populations adherent to GI epithelium. Therefore in order to determine whether measuring P. aeruginosa levels in the feces was an accurate surrogate for the total P. aeruginosa levels in the GI tract, we utilized a P. aeruginosa-specific qPCR assay to measure copies of the P. aeruginosa gyrB gene from gDNA extracted from feces and gastrointestinal segments (flushed and rinsed with PBS to remove the luminal contents) (Fig 4B)—rather than culturing intestinal tissue homogenates for P. aeruginosa which might miss (or underrepresent) P. aeruginosa present biofilms adherent to intestinal tissue. Total P. aeruginosa levels (as represented by copies of P. aeruginosa gyrB per gram/tissue) in the feces were not significantly different from P. aeruginosa levels in the distal colon (Fig 4C), suggesting that measuring P. aeruginosa in feces is an adequate surrogate for total P. aeruginosa in the distal colon. Of note, P. aeruginosa levels in the more proximal intestinal segments were approximately one log lower than in the distal colon (Fig 4C).
Thus, the addition of C. albicans to a P. aeruginosa-colonized murine GI tract does not increase fecal iron levels. Therefore, iron is unlikely to cause the C. albicans-induced decrease in P. aeruginosa pyochelin and pyoverdine gene expression.
C. albicans secreted factors inhibit P. aeruginosa pyochelin and pyoverdine gene expression and pyoverdine production in vitro
We next assessed whether C. albicans inhibits P. aeruginosa pyochelin and pyoverdine gene expression through a direct mechanism. We grew P. aeruginosa in GGP medium (a medium with limited iron availability to enhance the production of pyochelin and pyoverdine) to mid-log phase, added either mid-log phase C. albicans grown in YPD medium or a YPD alone control, co-incubated at 37°C for 10 minutes, and then isolated bacterial RNA. Pyochelin and pyoverdine gene expression uniformly decreased following addition of C. albicans (yeast or hyphal form), compared to a P. aeruginosa only control (Fig 5A, S7 Fig). The YPD only control, however, also significantly inhibited pyochelin and pyoverdine gene expression. This was most likely due to the relatively high iron concentration of YPD compared GGP medium (Fig 5B). To confirm this hypothesis, we repeated the pyochelin and pyoverdine gene expression experiments using YPD, YPD that had been depleted of iron, using the chelating agent Chelex100, and iron-depleted YPD supplemented with iron (to iron concentrations comparable to untreated YPD) (S8 Fig). The results confirmed that the iron in YPD was responsible for the inhibition of pyochelin and pyoverdine gene expression (S8 Fig).

We were, however, unable to detect any iron in YPD in which C. albicans had been grown to mid-log phase, presumably because the iron had been taken up by the yeast cells (Fig 5B). Thus, to further investigate whether this effect was from direct C. albicans and P. aeruginosa interactions, we exposed mid-log phase P. aeruginosa grown in GGP to heat-killed (HK) C. albicans cells suspended in PBS or a PBS only control. Addition of HK C. albicans or the PBS control did not suppress pyochelin and/or pyoverdine gene expression (Fig 5C).
We then asked whether a C. albicans secreted factor might be mediating the P. aeruginosa pyochelin and pyoverdine gene expression suppression. Supernatant from a C. albicans overnight culture (YPD, 30°C) significantly decreased P. aeruginosa pyochelin and pyoverdine gene expression (Fig 5D), and iron was not detectable in the C. albicans YPD supernatant (Fig 5B). To further determine if this inhibitory C. albicans secreted factor was a protein, we isolated C. albicans supernatant proteins by ammonium sulfate precipitation, followed by desalting with Sephadex G-25 gel filtration, and then dialysis against PBS. Indeed, C. albicans supernatant proteins alone significantly inhibited P. aeruginosa pyochelin and pyoverdine gene expression in vitro (Fig 5D). Iron was detected in the C. albicans supernatant protein, albeit at very low concentrations (mean 0.1830 ug/mL), but we were able to deplete the iron in the C. albicans supernatant protein to undetectable levels (Fig 5B). YPD that had undergone the same protein purification process (ammonium sulfate precipitation, desalting, and dialysis) had no detectable iron and did not significantly decrease P. aeruginosa pyochelin and pyoverdine gene expression (Fig 5B and 5D).
To further delineate whether the C. albicans secreted factor mediating this effect was a protein, we subjected the C. albicans supernatant protein to physical (boiled for 60 minutes) or chemical (treated with protease from Streptomyces griseus for 60 minutes) denaturation. Addition of boiled or protease-treated C. albicans supernatant protein to P. aeruginosa cultures resulted in significantly higher P. aeruginosa pyochelin and pyoverdine gene expression compared to P. aeruginosa cultures given untreated C. albicans supernatant protein (Fig 5E), strongly suggesting that the inhibitory C. albicans secreted effector is a protein.
In order to corroborate our gene expression data with protein-level expression, we utilized a well-established method of quantifying pyoverdine levels, by measuring the characteristic green fluorescence attributed to pyoverdine (Fig 5G). We first demonstrated that P. aeruginosa pyoverdine production was inhibited by C. albicans secreted protein in a dose-dependent manner (Fig 5F), comparable to levels in a P. aeruginosa pyoverdine deficient mutant (Fig 5G and 5H). Consistent with our gene expression data, C. albicans supernatant proteins inhibited pyoverdine production, and physical or chemical denaturation of C. albicans secreted protein abrogated this effect (Fig 5G and 5H). C. albicans secreted proteins did not inhibit P. aeruginosa growth in GGP medium in vitro (S9 Fig). Finally, we found comparable results (S10 Fig) when repeating these experiments using the additional P. aeruginosa and C. albicans strains used in in the in vivo experiments described previously (S2 and S3 Figs).
In sum, a C. albicans secreted factor, most likely a secreted protein, suppresses P. aeruginosa pyoverdine and pyochelin gene expression and reduces pyoverdine production in vitro.
C. albicans secreted proteins inhibit P. aeruginosa mediated cytotoxicity in vitro and inhibit P. aeruginosa virulence in vivo
The relationship between iron acquisition and microbial virulence is well-established. Iron uptake mutants can be avirulent [53], and iron deficiency is associated with increased resistance to infection [54]. Suppression of iron acquisition could lead to decreased growth and colonization and thus decreased risk for virulence. In fact, high-level gut bacterial colonization significantly increases the risk for virulence from the gut in cancer patients [29]. In our study, however, inhibition or deletion of pyochelin and pyoverdine had no effect on P. aeruginosa GI colonization.
Interestingly, pyoverdine is not only a siderophore but also a signaling molecule that induces the production of two extracellular virulence factors, the protease PrpL and exotoxin A [55]. Extracellular virulence factors (including proteases, cytotoxins, and phospholipases) have been shown to contribute to P. aeruginosa virulence in various animal models [56–58]. Therefore, we postulated that C. albicans mediated protection from P. aeruginosa infection might be due to inhibition of P. aeruginosa extracellular cytotoxic molecule production (i.e. exotoxin A, PrpL, etc). To explore this hypothesis, we utilized an in vitro cytotoxicity assay using cultured human colonocytes and P. aeruginosa culture supernatant proteins (recovered from stationary-phase cultures grown in iron-limited GGP media). P. aeruginosa culture supernatant proteins had variable levels of cytotoxicity against cultured human colonocytes: wild-type P. aeruginosa (5.8 fold increase compared to untreated control) > ΔpchBA (5.0) > ΔpvdS (1.8) > wild-type P. aeruginosa + C. albicans secreted protein (1.3) > E. coli secreted protein (0.95) > C. albicans secreted protein (0.6) (Fig 6A). In fact, the cytotoxicity induced by supernatant proteins recovered from P. aeruginosa grown with C. albicans secreted proteins was not significantly different than the untreated control group. Finally, immunoblotting showed that exotoxin A was nearly 80% reduced when P. aeruginosa was grown with C. albicans secreted proteins (Fig 6B and 6C). In summary, C. albicans secreted proteins inhibit the production of P. aeruginosa extracellular cytotoxic molecules, such as exotoxin A, and thus inhibit the cytotoxic effect of P. aeruginosa extracellular proteins against cultured human colonocytes.

To determine whether live C. albicans was necessary to protect against invasive P. aeruginosa in vivo, we first established P. aeruginosa mono-colonization in antibiotic-treated mice. We then treated P. aeruginosa mono-colonized mice with heat-killed (HK) C. albicans cells by gavage; HK C. albicans suspended in drinking water; iron-depleted C. albicans supernatant protein (200 ug by oral gavage daily and 100 μg/mL suspended in drinking water, renewed every 2 days); or a no treatment control on the day prior to induction of neutropenia and maintained throughout the duration of the experiment. Mice treated with HK C. albicans were not protected from P. aeruginosa infection (Fig 6D), while mice treated with iron-depleted C. albicans supernatant protein had significantly increased length of survival (p = 0.0208, log-rank test, Fig 6E) and overall survival from P. aeruginosa infection (4 of 8 mice, p = 0.038, Fisher’s exact test). Oral delivery of protein and peptide drugs is often problematic because of the high acidity of the stomach and proteolysis by intestinal enzymes. Yet both nutritional and pharmaceutical proteins can survive passage through the GI tract, albeit in minute quantities[59,60]. Given that laboratory mice drink ~4–8 ml water/day (with C3H mice drinking ~ 5–6 ml/day) [61], the mice used in this experiment would have ingested up to 800 ug of C. albicans supernatant protein per day. Furthermore, the total GI transit time in mice is roughly 6 hours compared to between 40–50 hours in humans [62]. Therefore, this rapid transit time may help proteins better survive gastric acidity and intestinal proteolysis, and thus be delivered throughout the murine GI tract. Despite the difficulty of surviving passage through the stomach and intestine, the C. albicans supernatant proteins that survived intestinal passage still afforded some degree of protection from P. aeruginosa infection, although not to the degree seen with C. albicans co-colonization. These data suggest that a C. albicans secretable factor, most likely a secreted protein, is responsible for C. albicans-mediated protection from P. aeruginosa infection in our murine model.
Iron supplementation restores P. aeruginosa virulence in P. aeruginosa-C. albicans co-colonized mice
P. aeruginosa has an alternative iron uptake system, the FeoABC system, that allows uptake of Fe2+ which is soluble and present in anaerobic conditions or in microaerobic environments at lower pH [63]. The FeoABC system is active during infections in patients with cystic fibrosis [64,65]. Since the distal gut is an anaerobic/ microaerobic environment with lower pH [66], we hypothesized that if supplemental oral iron were introduced into the GI tract of P. aeruginosa and C. albicans colonized mice, P. aeruginosa might utilize the FeoABC system to acquire iron and thus restore P. aeruginosa virulence. Consistent with our prior findings (Fig 4), total fecal iron content (Fe2+and Fe3+ as determined by a ferrozine assay [48]) in antibiotic treated mice was significantly lower than in untreated mice (Fig 7A). Introduction of P. aeruginosa and C. albicans into the GI tract of antibiotic-treated mice did not appreciably increase fecal iron levels. Oral administration of FeSO4, however, significantly increased overall fecal iron levels in P. aeruginosa—C. albicans co-colonized mice (Fig 7A) but did not restore Fe levels to those found in untreated mice. FeSO4 supplementation did not change P. aeruginosa or C. albicans GI colonization levels when compared to the untreated control group (Fig 7B). Importantly, iron supplementation restored P. aeruginosa virulence: 25% survival from P. aeruginosa infection in P. aeruginosa and C. albicans co-colonized mice treated with FeSO4 compared to 100% survival in untreated P. aeruginosa and C. albicans co-colonized mice (overall survival: p = 0.003, Fisher’s exact test; length of survival: p = 0.0024, log-rank test) (Fig 7C).

While iron supplementation clearly restored P. aeruginosa virulence, we were still uncertain as to why. To best answer this question, we turned our focus to the three primary defense mechanisms that prevent microbial translocation from the GI tract in humans and mice: 1) gut microbiota homeostasis, preventing the overgrowth or expansion of pathogenic microbes; 2) intact cellular immunity, particularly neutrophils; and 3) intact intestinal mucosal barriers [21–23]. Iron supplementation did not promote P. aeruginosa expansion in the gut (Fig 7B), so increased bacterial burden was not the cause of increased virulence. Excess iron may inhibit host phagocytic cells, repressing both chemotaxis and phagocytosis [67,68]. However, in our murine model, absolute neutrophil and macrophage counts are already < 100 cells/mm3 [31], so it is unlikely that further qualitative immune deficits (chemotactic or phagocytic) would have significant consequences. The final possibility was that iron may have some role in modulating gut epithelial integrity—either directly through the host or indirectly through its effect on P. aeruginosa. To investigate this possibility, we used an intestinal permeability assay in which we orally gavaged mice with fluorescent-labeled dextran (FITC-dextran) and then measured FITC-dextran levels in the serum[69]. Low levels of serum FITC-dextran would indicate low intestinal permeability (strong gut epithelial integrity) and thus low absorption from the gut. Mono-colonization with P. aeruginosa, or mono-colonization with C. albicans did not significantly increase intestinal permeability compared to mice treated with antibiotics alone (Fig 7C). When P. aeruginosa colonized mice were made neutropenic, intestinal permeability significantly increased. This effect was not seen in neutropenic C. albicans colonized mice. Neutropenic mice co-colonized with C. albicans and P. aeruginosa exhibited low intestinal permeability (< 1 ng/uL), and iron supplementation in the co-colonized group significantly increased intestinal permeability (Fig 7C). Finally, neither 1) iron supplementation alone or 2) iron supplementation and C. albicans colonization in the setting of neutropenia had any significant effect on increasing intestinal permeability. Collectively these results suggest that the iron-induced increase in gut permeability in co-colonized mice was due to P. aeruginosa. Of note, mice with the highest intestinal permeability (serum-FITC dextran concentrations > 3 ng/uL) were ill-appearing, with signs consistent with acute infection (hunched backs, cold to touch, raised fur). Thus, these findings suggest that iron supplementation restores P. aeruginosa virulence in P. aeruginosa and C. albicans co-colonized mice by modulating gut epithelial integrity (increasing gut permeability) indirectly through P. aeruginosa and not directly on host intestinal tissue.
Discussion
Since P. aeruginosa and C. albicans co-infect or co-colonize numerous human body niches, we developed a murine model of P. aeruginosa and C. albicans GI co-infection that would allow us to better study bacterial-fungal interactions and determine the pathophysiological impact of these interactions. Using this model system, we found that co-colonization with C. albicans significantly inhibited P. aeruginosa virulence in the setting of neutropenia—while having no impact on P. aeruginosa or C. albicans GI colonization levels. P. aeruginosa pyoverdine and pyochelin gene expression uniformly decreased when C. albicans was present in the GI tract. Accordingly, deletion of both pyochelin and pyoverdine genes was sufficient to attenuate P. aeruginosa virulence. A C. albicans secreted factor, most likely a secreted protein and not the quorum-sensing molecule farnesol, suppressed P. aeruginosa pyochelin and pyoverdine gene expression and inhibited pyoverdine production. C. albicans secreted proteins inhibited P. aeruginosa-mediated cytotoxic effects (via extracellular effector molecules) against cultured human colonocytes and were sufficient to significantly reduce P. aeruginosa virulence. Oral iron supplementation restored P. aeruginosa virulence in C. albicans and P. aeruginosa co-colonized mice. Here we describe a novel observation of fungal-inhibition of bacterial effectors critical for virulence but not important for colonization. These findings validate the use of a mammalian model system to investigate complex prokaryotic-eukaryotic interactions in the GI tract.
The antagonistic relationship between P. aeruginosa and C. albicans has been well-documented, but largely described with in vitro studies. P. aeruginosa [70,71] or P. aeruginosa products (notably pyocyanin and 1-hydroxyphenazine) [72] inhibit the growth of Candida spp. in vitro. Furthermore, P. aeruginosa forms dense biofilms on C. albicans hyphae and directly kills the fungus in vitro [10]. In fact, both P. aeruginosa contact-mediated and soluble factors can kill C. albicans filaments in vitro [9]. Interestingly, the yeast form of C. albicans is resistant to P. aeruginosa killing [9,10]. In fact, P. aeruginosa pyocyanin and the quorum-sensing molecule, 3-oxo-C12 homoserine lactone, can promote growth of the yeast form of C. albicans in vitro [8,72]. In this study, however, co-colonization surprisingly had no effect on C. albicans or P. aeruginosa’s individual ability to colonize the GI tract. In terms of rectifying these discrepant findings, the most plausible explanation is that C. albicans in the murine gut may be predominantly in the yeast form and therefore largely resistant to any P. aeruginosa-mediated growth inhibition or killing. C. albicans that colonizes human cutaneous epithelial cell surfaces is predominantly in the yeast form [73,74], and this may also hold true with C. albicans colonizing GI epithelial surfaces. Future studies that can determine C. albicans morphology and state (planktonic vs biofilm) in the mammalian GI tract would be immensely informative.
Interestingly, when P. aeruginosa and C. albicans form mixed biofilms in vitro, P. aeruginosa pyoverdine, pyochelin, and exotoxin A secretion is significantly increased (thus providing improved iron sequestration); C. albicans iron acquisition proteins are reduced, and C. albicans metabolism decreased [75]. Our results differ from these in vitro findings for two likely reasons: 1) the microenvironments of P. aeruginosa and C. albicans biofilms on plastic versus P. aeruginosa and C. albicans in the murine gut are markedly different and may result in significant differences in transcriptional changes and phenotype and 2) P. aeruginosa and C. albicans in the murine gut is likely a heterogeneous mix of planktonic, mono biofilm and mixed biofilm microbial populations.
P. aeruginosa and C. albicans interactions in in vivo models have also been described. For instance, A. baumanii reduced C. albicans virulence, and C. albicans was able to inhibit A. baumanii growth via farnesol production in a C. elegans model of co-infection [76]. In contrast, several murine models have demonstrated a synergistic effect of C. albicans-bacterial co-infection: C. albicans/E. coli intravenous bloodstream infection [77,78], P. aeruginosa/C. albicans burn wound [79], and C. albicans/S. aureus peritonitis [80] models. To the best of our knowledge, our study is the first in vivo system to describe a C. albicans-induced inhibition of P. aeruginosa iron acquisition proteins resulting in a loss of P. aeruginosa virulence. Taken together, these prior studies and the data presented here demonstrate the complexity of bacterial-fungal interactions and how significantly they can vary depending on experimental procedures (in vitro vs in vivo) and body niche/disease model.
A stable gut microbiota promotes pathogenic bacteria colonization resistance, preventing pathogenic microbes from overgrowing and thus decreasing the risk of pathogen virulence. High gut bacterial burden significantly increases the risk of bacteremia (with the same gut bacteria) in immunocompromised patients [29]. P. aeruginosa is not a normal member of the human commensal microbiota [17,81]. In contrast, the prevalence of C. albicans GI colonization in humans ranges from less than 10% in remote and traditional societies [82–84] to 40–80% in modern, developed countries [85–87]. Interestingly, adult mice with intact gut microbiota (no antibiotic treatment) are resistant to P. aeruginosa and C. albicans [24–26] GI colonization. As with other P. aeruginosa infections (such as burn wounds, ophthalmic infections, and ventilator associated pneumonia), P. aeruginosa gut colonization appears to require a deficit in host immune defenses, in this case a disturbance in gut microbial homeostasis. Thus, depletion of anaerobic gut commensals (e.g via antibiotics) is critical for establishing both sustained and high levels of P. aeruginosa [31] and C. albicans [24,25] GI colonization in mice.
Perhaps with the depletion of anaerobic commensals (which comprise >99% of the gut microbiota), competition for carbon sources and other nutrients is significantly decreased, creating a more permissive gut environment for colonization. But the question remains: how does C. albicans benefit from suppressing P. aeruginosa pyochelin and pyoverdine expression? One possible answer is a competitive advantage for iron. As noted earlier, the host withholds nutrients, such as iron [37], to prevent microbial overgrowth in the gut [38]. Most bacterial pathogens, including P. aeruginosa, utilize high-affinity iron uptake mechanisms that compete against host-mediated sequestration. C. albicans possesses similar iron acquisition mechanisms [43,44]. For instance, Als3, a hyphal-associated adhesin and invasin, is also essential in mediating iron acquisition from host ferritin [88]. Thus, in an effort to better secure iron from the host, C. albicans may secrete a protein that inhibits P. aeruginosa (and perhaps other microbes) from acquiring iron. Yet, despite inhibition, and even deletion, of pyochelin and pyoverdine genes, P. aeruginosa is able to grow and colonize the GI tract. As a testament to the importance of iron, P. aeruginosa employs additional strategies to acquire iron: uptake of xenosiderophores (produced by other organisms)[89–91]; uptake of host heme molecules [92]; FeoABC system (Feo system)[63,93], and extracellular reduction of Fe3+ to Fe2+ with phenazine compounds [94]. Thus, despite inhibition of pyoverdine and pyochelin by C. albicans, P. aeruginosa is probably able to utilize alternative iron acquisition pathways to sustain growth and colonize the GI tract.
While much has been written about the importance of neutropenia [25,31,95,96] and increased bacterial burden [29,62,97] in promoting acute microbial infection, our findings indicate that a rate-limiting step to pathogen virulence can be the ability of the pathogen to breach intact gut mucosal barriers. Intact mucosal barriers are a critical component of innate immunity. Newborn human and mice infants have “leaky” gut epithelium, making them more prone to microbial gut translocation [98–100]. Medications, including cytotoxic chemotherapy, can damage gut epithelium, increasing the risk of bacterial infection. And as noted before, microbial pathogens can produce extracellular molecules that damage host epithelial barriers. In P. aeruginosa, extracellular proteins exported by the type III secretion system (T3SS) have toxic effects on cultured cells [32,101–103] and can enhance virulence in animal models of P. aeruginosa pneumonia [104] and gut colonization [30]. We previously demonstrated that disruption of P. aeruginosa cytotoxic virulence effector genes (e.g. Type III secretion system ExoU) results in a loss of cytotoxicity and significantly reduces virulence in this P. aeruginosa gut model [30], without affecting P. aeruginosa GI colonization. In the current study, deletion of either pyochelin or pyoverdine decreased P. aeruginosa cytotoxic activity in vitro but virulence was still maintained in vivo. However there appears to be an in vitro cytotoxic threshold that predicts in vivo virulence: wild-type P. aeruginosa PAO1 (5.8 fold increased cytotoxicity compared to untreated control), PAO1 ΔpchBA (5.0), and PAO1 ΔpvdS (1.8) all maintained full virulence, whereas wild-type P. aeruginosa + C. albicans secreted protein (1.3) virulence was attenuated (Fig 6A).
Hence, we postulate that in P. aeruginosa and C. albicans co-colonized mice: C. albicans secretes one or more factors that inhibit P. aeruginosa pyochelin and pyoverdine expression. P. aeruginosa virulence and production of extracellular virulence effectors, such as PrpL and exotoxin A, is decreased, but host gut mucosal integrity remains intact. We further hypothesize that in P. aeruginosa and C. albicans co-colonized mice, iron supplementation restores P. aeruginosa virulence by inducing pyochelin/pyoverdine-independent P. aeruginosa cytotoxic effector molecules; host gut permeability increases (gut mucosal barriers are damaged); and P. aeruginosa virulence is restored.
While our data strongly suggests that one or more Candida albicans secreted protein(s) mediates the inhibition of P. aeruginosa virulence, the exact mechanism of pyochelin and pyoverdine gene suppression is unclear. One possible mechanism is microbial-induced modulations in the host oxidative stress response lead to changes in P. aeruginosa pyochelin and pyoverdine gene expression [105] with reduced production of pyoverdine leading to reduced synthesis of exotoxin A and PrpL protease [55]. Another possibility involves the direct regulation of P. aeruginosa exotoxin A synthesis. P. aeruginosa exotoxin A regulator protein, PtxS, inhibits the synthesis of exotoxin A and also autoregulates its synthesis [106,107]. A C. albicans secreted protein could potentially interfere with the autoregulation of ptxS or other regulatory genes involved with exotoxin A synthesis. We have ongoing studies in which we are trying to identify the C. albicans secreted protein that mediates the effect presented in this study, and we hope to be able to elucidate the specific mechanism in the future.
With advances in medical technology, artificial niches (e.g. the antibiotic-treated gut) are being created where non-commensal microbes (i.e. P. aeruginosa, C. albicans) can now establish colonization and in the right setting (e.g. deficits in the immune system) cause invasive disease. Therefore, understanding the synergistic, symbiotic, or antagonistic interactions between diverse microorganisms within clinically relevant environments may be critical for understanding their pathogenesis toward the host. By exploiting specific evolutionary defense mechanisms or survival responses used by competing pathogens, we may gain key insight into novel therapeutic targets for increasingly difficult to treat pathogens such as P. aeruginosa and C. albicans.
Methods
Bacterial and fungal strains
The Candida albicans strains, Pseudomonas aeruginosa strains, and other bacteria strains used are listed in S1 Table. Unless otherwise noted, C. albicans refers to strain SC5314 and P. aeruginosa refers to strain PAO1. Unless specified otherwise, aerobic bacteria, including P. aeruginosa, were grown overnight in Luria-Bertani (LB) broth at 37°C; anaerobic bacteria overnight in TYG [108] broth at 37°C anaerobically; and C. albicans in yeast peptone dextrose (YPD) broth at 30°C.
Ethics statement
All animal experiments were done in accordance with NIH guidelines, the Animal Welfare Act and US federal law. The University of Texas Southwestern Medical Center’s Institutional Animal Care and Use Committee approved the experimental protocol “2009–0243” that was used for this study. All animals were housed in a centralized and AAALAC-accredited research animal facility that is fully staffed with trained husbandry, technical and veterinary personnel.
P. aeruginosa and C. albicans GI co-colonization model
The P. aeruginosa and C. albicans co-colonization and virulence murine model was used as previously described [25,31], with some modifications. Unless otherwise noted, mice used for experiments were C3H/HeN (Harlan), sex-matched, 6–8 weeks of age. Mice within an experiment were littermates that remained co-housed in the same cage to ensure a shared microbiota. Mice were fed sterile water with 2 mg streptomycin /mL (STREP) and 1500 U penicillin G /mL (PCN) for 5 days. P. aeruginosa and C. albicans for oral gavage was prepared by harvesting overnight cultures (P. aeruginosa in LB, 37°C; C. albicans in YPD, 30°C); washing in PBS x 2; enumerating concentration by spectrophotometry (for P. aeruginosa) or hemacytometer (C. albicans); and resuspending the final P. aeruginosa or C. albicans oral gavage preparation in endotoxin-free sterile PBS (Gibco). P. aeruginosa and/or C. albicans was administered by oral gavage (5 x 108 cfu in 0.2 mL) after 5 days of antibiotic water treatment. Mice were then transitioned to PCN water for the remainder of the experiment.
C. albicans and P. aeruginosa GI colonization were enumerated by first weighing fecal specimens, suspending fecal pellets in 1% proteose peptone, homogenizing samples, serially diluting and culturing homogenized fecal contents on YVG agar (yeast-peptone-dextrose agar with 0.010 mg/mL vancomycin and 0.100 mg/mL gentamicin to suppress bacterial growth) [25] and cetrimide agar, respectively. For experiments utilizing bacteria, obligate anaerobic strains (B. theta, B. producta) were cultured in TYG [108] or BHI/Blood agar under anaerobic conditions (Coy anaerobic chamber) at 37°C; E. coli was grown in LB and MacConkey media under aerobic conditions at 37°C; and E. faecalis was grown on CNA media under aerobic conditions at 37°C. Bacteria were washed with PBS (aerobic or anaerobic as indicated). Bacterial concentration was determined by spectrophotometry. Bacteria were then administered by oral gavage (5 x 108 cfu). Bacterial colonization levels were enumerated by growth on the appropriate selective media and identity confirmed by gram-stain and enzymatic analysis (Rapid One for Enterobacteriaceae and RapID ANA II for anaerobes, Remel).
For virulence experiments, anti-mouse Ly-6G, Ly-6C (Gr1) monoclonal antibody RB6-8C5 was produced and administered as previously described [25,31]. Mice were monitored for mortality for 7 days. Moribund mice were euthanized with CO2 administration. Livers and spleens were resected, homogenized and plated on YVG, MacConkey, TSA, Cetrimide, and BHI/Blood agars. Livers were previously found to be the most reliable organ to confirm C. albicans extra-intestinal dissemination [25] and spleens the most reliable organ for confirming P. aeruginosa extra-intestinal dissemination [31] when using this murine model. The presence of a homogeneous population of green, oxidase-positive colonies on cetrimide agar and an absence of other bacterial growth on the MacConkey (aerobic), TSA (aerobic), and BHI/Blood (anaerobic) plates was used for confirmation of P. aeruginosa dissemination. The presence of a homogeneous population of creamy white colonies on YVG agar was used for evidence of C. albicans dissemination.
Intestinal permeability assay
Intestinal barrier function was evaluated by measuring in vivo paracellular permeability to fluorescent-labeled dextran as previously described [69]. Mice were fasted for 4.5 hours and then orally gavaged with 50 mg/100g body weight of FITC-dextran (MW 3,000–5000, FD4, Sigma). Serum was obtained 3 hours after gavage administration by terminal cardiac puncture. For neutropenic mice, serum was obtained 84 hours after RB6-8CB monoclonal antibody injection. The serum FITC-dextran concentration was calculated by comparing samples with serial dilutions of known standards using the Synergy HT Fluorimeter (BioTek, Winooski, VT) with excitation at 485 nm and emission at 530 nm.
In vivo P. aeruginosa transcriptome profiling
To obtain transcriptome information from P. aeruginosa colonizing the murine GI tract, we colonized mice with P. aeruginosa alone or P. aeruginosa and C. albicans and then administered RB6-8C5 mAb to induce virulence. When a P. aeruginosa mouse became moribund, we euthanized this mouse and an accompanying P. aeruginosa and C. albicans mouse. Cecal contents were flushed and immediately flash frozen. Cecal contents from 8 mice (1 biological replicate) of the same group were combined, and total RNA was extracted as previously described [109]. Prokaryotic total RNA was further isolated (MICROBEnrich Kit, Ambion). The quality of the resultant RNA was determined using an Agilent Bioanalyzer. For samples with RNA Integrity Numbers of greater than 7.5, the RiboMinus Kit (Life Technologies) was used to deplete rRNA from the total RNA samples. 500 ng of mRNA was used to create cDNA libraries (UTSW Microarray Core). Paired-end libraries for Illumina sequencing were prepared from purified cDNA (TruSeq RNA Sample preparation kit, Illumina). Reads were aligned to P. aeruginosa PAO1 genome (GenBank: NC_022516.2) using CLC-Biosystems RNA-Seq module. Even after removal of eukaryotic RNA (murine and C. albicans), <10% of sequence reads properly mapped to the P. aeruginosa PAO1 genome (S2 Table). The number of overlapping mapped reads was counted for every ORF in each sample of whole-transcriptome sequencing (2 biological replicates for the P. aeruginosa and P. aeruginosa and C. albicans groups). For each ORF the number of reads per kilobase of gene model per million mapped reads (RPKM) was calculated [110]. The raw read counts for each gene was processed using DESeq software (utilizing negative binomial distribution analysis), and differentially expressed genes were identified using adjusted p-values ≤ 0.05 and fold-change ≥ 2 (Benjamini-Hochberg procedure) [111].
RNA extraction and cDNA synthesis
Total RNA from P. aeruginosa cells was isolated using phenol-chloroform and bead-beating, followed by ethanol precipitation [112]. Crude RNA extracts were treated with DNaseI (Qiagen) and column purified (RNEasy Kit, Qiagen). In mixed P. aeruginosa and C. albicans experiments, prokaryotic total RNA was further isolated (MICROBEnrich Kit, Ambion). RNA concentrations were quantified by spectrophotometry (Nanodrop). Total RNA was used to synthesize cDNA (iScript, BioRad).
RT-qPCR
Total RNA was used to synthesize cDNA (iScript, BioRad). qPCR analysis was performed using the SsoAdvanced SYBR Green Supermix (Bio-Rad) and specific primers (S3 Table). Signals were normalized to P. aeruginosa rpoD [113] levels within each sample and normalized data were used to calculate relative levels of gene expression using ΔΔCt analysis.
Pseudomonas aeruginosa gyrB qPCR
The abundance of the P. aeruginosa gyrB gene was determined by qPCR analysis (SsoAdvanced Supermix, Bio-Rad) using P. aeruginosa gyrB specific gene primers and specific probe (S3 Table). To confirm specificity, P. aeruginosa gyrB qPCR was performed using gDNA from Escherichia coli ATC10798, Enterobacter cloacae (clinical isolate), Enterococcus faecalis (clinical isolate), and Candida albicans SC5314, and no false positives were noted (Fig 4B). P. aeruginosa abundance was determined using standard curves constructed with reference to cloned DNA corresponding to a short segment of P. aeruginosa gyrB gene (S3 Table). Note that qPCR measures gene copies/g tissue, not actual bacterial/fungal numbers or colony forming units.
To test P. aeruginosa gyrB levels in murine feces and intestinal tissue, fecal specimens and intestinal tract segments (2 cm; ileum, cecum, proximal and distal colon) were collected. Intestinal segments were cut open longitudinally and flushed with ice-cold sterile PBS x 2. Tissue was then flash frozen with liquid nitrogen, weighed, and immediately suspended in extraction buffer (200 mM NaCl, 200mM Tris, 20 mM EDTA, 6% SDS) and 0.5 ml of phenol/chloroform/isoamyl alcohol, pH 7.9 (Ambion) [108]. Tissue was lysed by bead-beating (0.1 mm zirconia/silica beads, BioSpec), and subjected to additional phenol/chloroform extractions. Crude DNA extracts were treated with RNAseA (Qiagen) and column-purified (PCR Purification Kit, Qiagen). DNA concentrations were quantified by fluorescence-based assay (Quant-iT PicoGreen dsDNA, Life Technologies).
In vitro P. aeruginosa and C. albicans co-culture P. aeruginosa gene expression experiments
P. aeruginosa and C. albicans were individually grown to mid-log phase. P. aeruginosa was grown in GGP media (3% glycerol, 1% proteose peptone, 2.9 mM K2HPO4, 2.0 mM MgSO4⚫7H20) at 37°C. C. albicans was grown in YPD media at 30°C. P. aeruginosa and C. albicans cultures were combined in a 1 : 1 ratio. The P. aeruginosa/C. albicans culture was grown at 37°C, and cells harvested after 10 minutes of co-incubation. Total RNA was extracted, and prokaryotic RNA was isolated (MICROBEnrich Kit, Ambion) for RT qPCR of P. aeruginosa pyochelin and pyoverdine gene expression.
P. aeruginosa pyoverdine and/or pyochelin mutant construction
P. aeruginosa mutant strains and plasmids used are listed in S4 Table. Methods for construction of unmarked deletions in pchBA and pvdH have been described previously[114,115]. Fragments of DNA flanking each deletion site were amplified by PCR, ligated together, cloned into the allele replacement vectors pEX18Gm [114] and used to replace the wild-type genes in P. aeruginosa PAO1.
To generate PAO6901 and PAO6902, the Gmr cassette from PAO1ΔpvdS::Gmr [92] was transduced via E79tv-2 [116] into PAO1 and PAO6297, respectively.
Complementation of P. aeruginosa mutants
To generate plasmid pME6477 for complementation experiments, the pchDCBA operon was fused at the ATG start codon of pchD to the lac promoter (Plac) of pME6000 by using a linker region containing a ribosome binding site (AAGCTTGATATCGAATTGTGAGCGGATAACAATTTCACACAGAATTGATTAAAGAGGAGAAATTAAGCATG; the HindIII site used for cloning into pME6000 is italicized; the ATG start codon of pchD is underlined).
For complementation of pyochelin/pyoverdine double knockout mutants, PAO1 cells were grown in LB broth to stationary phase, harvested, washed 0.1 M MgCl2, and finally suspended in ice-cold transformation buffer (75 mM CaCl2, 6mM MgCl2, 15% glycerol) [117]. 100 μL of the PAO1 competent cells were then transferred to thin-walled 13 x 100 mm borosilicate glass tubes that were pre-chilled on ice. 2–5 μL aliquots containing 100–1000 ng pME6477 (pME6000 derivative carrying pchDCBA) that had been purified using the PureYield Plasmid Midiprep kit (Promega) was then added. DNA-cell mixture was incubated on ice for 15 min and then heat-shocked at 37°C for 2 min. 500 μL LB broth was immediately added, and tubes were incubated at 37°C for 1 h in a shaking incubator. After incubation, a 200-μ L aliquot of each undiluted and diluted (1 : 10) suspension was plated on LB agar containing 100 μg/mL tetracycline. Plates were incubated at 37°C for 24 h. Select colonies were seeded into LB/Tetracycline media and grown at 37°C. Plasmids were isolated (PureYield Plasmid Miniprep Kit, Promega). The identity of plasmids were confirmed by both gel electrophoresis and sequencing.
Quantitative iron assay
Fecal iron content was measured using a colorimetric ferrozine-based assay [118,119]. Mouse fecal pellets, mouse chow (Teklad Global 16% Protein Rodent Diet, Harlan; 1 gram), liquid media (1 ml), and antibiotic water (1 ml) were collected, weighed, homogenized in 6M HCl, and stored under anaerobic conditions for 24 hours. 0.1 ml of filtered fecal homogenate was then added to 0.9 ml of hydroxylamine hydrochloride (HAHC) (10%, w/v, in 1M HCl). 1 ml of ferrozine solution was added and absorbance at 562nm was measured. Total Fe was calculated in reference to standard curves and reported as mmol/mg feces
Iron depletion
YPD and C. albicans supernatant proteins were passed over a column with Chelex100 50–100 mesh[69] (Sigma) resin beds as per the manufacturer’s instructions. Iron concentrations were measured using the ferrozine assay described above.
Pyoverdine assay
P. aeruginosa cells were grown in GGP medium[120], in which limited iron availability enhances the expression of pyochelin and pyoverdine, in black, clear bottom 96-well plates (Corning Incorporated, Costar 3603). Pyoverdine was measured by fluorescence at 400±10/460±40 nm excitation/emission, using a 96-well Microplate Fluorimeter Plate Reader (Synergy HT, Biotek Inc.), and measurements of relative fluorescence units (RFU) were normalized to cell density measured at 600 nm. Measurements were recorded dynamically up to 24 h. Between measurements, plates were incubated at 37°C and 100 rpm. The specificity of fluorescence for pyoverdine was verified using PAO1ΔpvdS mutant deficient in pyoverdin production, in which no fluorescence was found.
Heat-killed C. albicans
For heat inactivation and killing, C. albicans (yeast or hyphae) were resuspended in phosphate buffered saline (PBS) (1 x 107 cells/ml) and treated at 100°C for 1 h [45]. Killing was confirmed by plating the heat-killed on C. albicans on YPD agar and confirming lack of growth.
Microbial extracellular protein isolation
Microbial extracellular proteins were isolated as previously described [31]. P. aeruginosa strains, C. albicans (SC5314), and E. coli (ATCC 10798) were grown to stationary phase in the following media and conditions: GGP at 37°C for P. aeruginosa; YPD at 30°C for C. albicans; and LB at 37°C for E. coli. Supernatants were prepared from the cultures by centrifugation at 10,000 x g at 4°C for 30 min. C. albicans culture supernatant was then sterile filtered (0.22 μm, low-protein binding Steritop filter; Millipore). Proteins present in the supernatants were precipitated by the addition of ammonium sulfate (50% for bacterial supernatants, 80% for C. albicans supernatant) and left overnight at 4°C. Precipitated material was isolated by centrifugation at 10,000 x g at 4°C for 30 min. Protein pellets were resuspended in distilled H2O and run through PD-10 desalting columns containing Sephadex G-25 gel (Amersham Biosciences). Protein preparations then underwent two rounds of dialysis in PBS buffer (Slide-A-Lyzer Dialysis cassette, 10K MWCO; Thermo Scientific). Protein concentrations were determined by BCA protein assay (Pierce).
Physical and chemical denaturation of C. albicans supernatant proteins
For physical denaturation, C. albicans supernatant protein preparations were boiled for 60 minutes. For chemical denaturation, C. albicans supernatant protein preparations were treated with protease from Streptomyces griseus (a mixture of at least three proteolytic activities including an extracellular serine protease; Sigma P5147) at a final concentration of 5mg/mL for 60 minutes at 37°C, then heat-inactivated at 80°C for 15 minutes. Equivalent volumes (the same volume of the corresponding untreated C. albicans supernatant protein) of boiled or protease-treated C. albicans supernatant proteins were added to P. aeruginosa cultures.
In vitro P. aeruginosa and C. albicans supernatant/supernatant protein P. aeruginosa gene expression experiments
For experiments utilizing live P. aeruginosa and C. albicans supernatant (Fig 5D), C. albicans culture supernatant was prepared from an overnight C. albicans culture (grown in YPD, 30°C) by centrifugation at 10,000 x g at 4°C for 30 min. C. albicans culture supernatant was then sterile filtered (0.22 μm, low-protein binding Steritop filter; Millipore) and then dialyzed against PBS (Slide-A-Lyzer Dialysis cassette, 10K MWCO; Thermo Scientific). P. aeruginosa was grown to mid-log phase in GGP media at 37°C. P. aeruginosa and C. albicans supernatant was combined in a 1 : 1 ratio. The P. aeruginosa/C. albicans supernatant culture was grown at 37°C, and cells harvested after 10 minutes of co-incubation for RNA extraction (as described above).
For experiments utilizing live P. aeruginosa and C. albicans supernatant protein (Fig 5D and 5E), C. albicans culture supernatant proteins were prepared as described above. P. aeruginosa was grown to mid-log phase in GGP media at 37°C. C. albicans supernatant protein was added to a final concentration of 100 ug/mL. The P. aeruginosa and C. albicans supernatant protein culture was grown at 37°C, and cells harvested after 10 minutes of co-incubation for RNA extraction (as described above).
Exotoxin A immunoblot
For detection of P. aeruginosa exotoxin A protein, nitrocellulose membranes were activated by soaking in TBS-0.05% Tween. A slot blotter was used to load samples of 250 ug of total extracellular protein material. The membrane was blocked with TBS containing 5% BSA at room temperature for 1 h. The primary antibody used was a polyclonal rabbit IgG to P. aeruginosa exotoxin A (Sigma, P2318) diluted 1 : 10,000, and the blot was rotated at room temperature for 90 min. The secondary antibody was an anti-rabbit whole-molecule IgG conjugated to alkaline phosphatase diluted 1 : 50,000, which was left on the blot for 90 min at 37°C. After washing, a phosphatase substratedeveloper (KPL) was then added. Relative density of exotoxin A immunoblot was measured by ImageJ Software.
In vitro cytotoxicity assays
Human colonocyte cell lines, HT-29, were exposed to microbial culture supernatant proteins (1 μg/uL) and incubated for 3 hours at 37°C with 5% CO2. Cell toxicity was measured by CytoTox-Glo assay (Promega).
Accession numbers
Accession numbers for the genes described in this study in NCBI are pchD (880566), fptA (880419), ampP PA4218 (880532), pchF (88083), pchH (880074), pchA (881821), pchE (880048), pchG (880082), pchB (881846), pchC (881845), pvdL (882838), pvdH (882857), pvdS (882839), fpvA (878605), pvdD (879750), pvdF (878624), pvdE (877645), gbuA (881747), PA4280.1 (3240240), PA1412 (879323), PA1387 (879767), hcpB (879579), PA1316 (881564), PA4722 (881593), hsiA2 (882166), aprA (881248), PA2693 (882826), hsiC2 (879592), hcp1 (879446), oprH (878005), PA3190 (882901), pqsH (879540), hsiB2 (879559), clpV1 (879553), tssB1 (879476), cbpD (877771), PA2463 (882955), pvdP (882224), pvdA (882167), fumC1 (881033), lasI 881777), fpvA (878605), PA0572 (878237), PA2402 (878257), Exotoxin A (877850), prpL (880208)
Raw RNA-Seq data is deposited at the NCBI Sequence Read Archive (SRA): SRP061622.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Hermann C, Hermann J, Munzel U, Ruchel R (1999) Bacterial flora accompanying Candida yeasts in clinical specimens. Mycoses 42 : 619–627. 10680438
2. Azoulay E, Timsit JF, Tafflet M, de Lassence A, Darmon M, et al. (2006) Candida colonization of the respiratory tract and subsequent pseudomonas ventilator-associated pneumonia. Chest 129 : 110–117. 16424420
3. Bauernfeind A, Bertele RM, Harms K, Horl G, Jungwirth R, et al. (1987) Qualitative and quantitative microbiological analysis of sputa of 102 patients with cystic fibrosis. Infection 15 : 270–277. 3117700
4. McAlester G, O'Gara F, Morrissey JP (2008) Signal-mediated interactions between Pseudomonas aeruginosa and Candida albicans. J Med Microbiol 57 : 563–569. doi: 10.1099/jmm.0.47705-0 18436588
5. Gupta N, Haque A, Mukhopadhyay G, Narayan RP, Prasad R (2005) Interactions between bacteria and Candida in the burn wound. Burns 31 : 375–378. 15774298
6. Marrie TJ, Costerton JW (1984) Scanning and transmission electron microscopy of in situ bacterial colonization of intravenous and intraarterial catheters. J Clin Microbiol 19 : 687–693. 6429190
7. Tchekmedyian NS, Newman K, Moody MR, Costerton JW, Aisner J, et al. (1986) Special studies of the Hickman catheter of a patient with recurrent bacteremia and candidemia. Am J Med Sci 291 : 419–424. 3717200
8. Hogan DA, Vik A, Kolter R (2004) A Pseudomonas aeruginosa quorum-sensing molecule influences Candida albicans morphology. Mol Microbiol 54 : 1212–1223. 15554963
9. Brand A, Barnes JD, Mackenzie KS, Odds FC, Gow NA (2008) Cell wall glycans and soluble factors determine the interactions between the hyphae of Candida albicans and Pseudomonas aeruginosa. FEMS Microbiol Lett 287 : 48–55. doi: 10.1111/j.1574-6968.2008.01301.x 18680523
10. Hogan DA, Kolter R (2002) Pseudomonas-Candida interactions: an ecological role for virulence factors. Science 296 : 2229–2232. 12077418
11. Cugini C, Calfee MW, Farrow JM 3rd, Morales DK, Pesci EC, et al. (2007) Farnesol, a common sesquiterpene, inhibits PQS production in Pseudomonas aeruginosa. Mol Microbiol 65 : 896–906. 17640272
12. de Macedo JL, Santos JB (2005) Bacterial and fungal colonization of burn wounds. Mem Inst Oswaldo Cruz 100 : 535–539. 16184232
13. Hughes WT, Kim HK (1973) Mycoflora in cystic fibrosis: some ecologic aspects of Pseudomonas aeruginosa and Candida albicans. Mycopathol Mycol Appl 50 : 261–269. 4199669
14. Burns JL, Van Dalfsen JM, Shawar RM, Otto KL, Garber RL, et al. (1999) Effect of chronic intermittent administration of inhaled tobramycin on respiratory microbial flora in patients with cystic fibrosis. J Infect Dis 179 : 1190–1196. 10191222
15. Nseir S, Jozefowicz E, Cavestri B, Sendid B, Di Pompeo C, et al. (2007) Impact of antifungal treatment on Candida-Pseudomonas interaction: a preliminary retrospective case-control study. Intensive Care Med 33 : 137–142. 17115135
16. Pizzo PA, Poplack D, editors (2011) Principles and Practice of Pediatric Oncology, 6th Edition. Philadelphia: Lippincott Williams & Wilkins. 1531
17. Fanci R, Paci C, Anichini P, Pecile P, Marra G, et al. (2003) Incidence and molecular epidemiology of Pseudomonas aeruginosa bacteremias in patients with acute leukemia: analysis by pulsed-field gel electrophoresis. New Microbiol 26 : 353–361. 14596346
18. Tancrede CH, Andremont AO (1985) Bacterial translocation and gram-negative bacteremia in patients with hematological malignancies. J Infect Dis 152 : 99–103. 3925032
19. Miranda LN, van der Heijden IM, Costa SF, Sousa AP, Sienra RA, et al. (2009) Candida colonisation as a source for candidaemia. J Hosp Infect 72 : 9–16. doi: 10.1016/j.jhin.2009.02.009 19303662
20. Nucci M, Anaissie E (2001) Revisiting the source of candidemia: skin or gut? Clin Infect Dis 33 : 1959–1967. 11702290
21. Berg RD (1999) Bacterial translocation from the gastrointestinal tract. Adv Exp Med Biol 473 : 11–30. 10659341
22. Pasqualotto AC, Nedel WL, Machado TS, Severo LC (2006) Risk factors and outcome for nosocomial breakthrough candidaemia. J Infect 52 : 216–222. 15936825
23. Rosen GP, Nielsen K, Glenn S, Abelson J, Deville J, et al. (2005) Invasive fungal infections in pediatric oncology patients: 11-year experience at a single institution. J Pediatr Hematol Oncol 27 : 135–140. 15750444
24. Fan D, Coughlin LA, Neubauer MM, Kim J, Kim MS, et al. (2015) Activation of HIF-1alpha and LL-37 by commensal bacteria inhibits Candida albicans colonization. Nat Med.
25. Koh AY, Kohler JR, Coggshall KT, Van Rooijen N, Pier GB (2008) Mucosal damage and neutropenia are required for Candida albicans dissemination. PLoS Pathog 4: e35. doi: 10.1371/journal.ppat.0040035 18282097
26. White SJ, Rosenbach A, Lephart P, Nguyen D, Benjamin A, et al. (2007) Self-regulation of Candida albicans population size during GI colonization. PLoS Pathog 3: e184. 18069889
27. Noverr MC, Huffnagle GB (2004) Regulation of Candida albicans morphogenesis by fatty acid metabolites. Infect Immun 72 : 6206–6210. 15501745
28. Mason KL, Erb Downward JR, Mason KD, Falkowski NR, Eaton KA, et al. (2012) Candida albicans and bacterial microbiota interactions in the cecum during recolonization following broad-spectrum antibiotic therapy. Infect Immun 80 : 3371–3380. doi: 10.1128/IAI.00449-12 22778094
29. Taur Y, Xavier JB, Lipuma L, Ubeda C, Goldberg J, et al. (2012) Intestinal domination and the risk of bacteremia in patients undergoing allogeneic hematopoietic stem cell transplantation. Clin Infect Dis 55 : 905–914. doi: 10.1093/cid/cis580 22718773
30. Koh AY, Mikkelsen PJ, Smith RS, Coggshall KT, Kamei A, et al. (2010) Utility of in vivo transcription profiling for identifying Pseudomonas aeruginosa genes needed for gastrointestinal colonization and dissemination. PLoS One 5: e15131. doi: 10.1371/journal.pone.0015131 21170272
31. Koh AY, Priebe GP, Pier GB (2005) Virulence of Pseudomonas aeruginosa in a murine model of gastrointestinal colonization and dissemination in neutropenia. Infect Immun 73 : 2262–2272. 15784570
32. Sawa T, Ohara M, Kurahashi K, Twining SS, Frank DW, et al. (1998) In vitro cellular toxicity predicts Pseudomonas aeruginosa virulence in lung infections. Infect Immun 66 : 3242–3249. 9632591
33. Falgier C, Kegley S, Podgorski H, Heisel T, Storey K, et al. (2011) Candida species differ in their interactions with immature human gastrointestinal epithelial cells. Pediatr Res 69 : 384–389. doi: 10.1203/PDR.0b013e31821269d5 21283049
34. Lo HJ, Kohler JR, DiDomenico B, Loebenberg D, Cacciapuoti A, et al. (1997) Nonfilamentous C. albicans mutants are avirulent. Cell 90 : 939–949. 9298905
35. Kong EF, Kucharikova S, Van Dijck P, Peters BM, Shirtliff ME, et al. (2015) Clinical implications of oral candidiasis: host tissue damage and disseminated bacterial disease. Infect Immun 83 : 604–613. doi: 10.1128/IAI.02843-14 25422264
36. Schlecht LM, Peters BM, Krom BP, Freiberg JA, Hansch GM, et al. (2015) Systemic Staphylococcus aureus infection mediated by Candida albicans hyphal invasion of mucosal tissue. Microbiology 161 : 168–181. doi: 10.1099/mic.0.083485-0 25332378
37. Ganz T, Nemeth E (2006) Regulation of iron acquisition and iron distribution in mammals. Biochim Biophys Acta 1763 : 690–699. 16790283
38. Skaar EP (2010) The battle for iron between bacterial pathogens and their vertebrate hosts. PLoS Pathog 6: e1000949. doi: 10.1371/journal.ppat.1000949 20711357
39. Takase H, Nitanai H, Hoshino K, Otani T (2000) Impact of siderophore production on Pseudomonas aeruginosa infections in immunosuppressed mice. Infect Immun 68 : 1834–1839. 10722571
40. Takase H, Nitanai H, Hoshino K, Otani T (2000) Requirement of the Pseudomonas aeruginosa tonB gene for high-affinity iron acquisition and infection. Infect Immun 68 : 4498–4504. 10899848
41. Meyer JM, Neely A, Stintzi A, Georges C, Holder IA (1996) Pyoverdin is essential for virulence of Pseudomonas aeruginosa. Infect Immun 64 : 518–523. 8550201
42. Imperi F, Massai F, Facchini M, Frangipani E, Visaggio D, et al. (2013) Repurposing the antimycotic drug flucytosine for suppression of Pseudomonas aeruginosa pathogenicity. Proc Natl Acad Sci U S A 110 : 7458–7463. doi: 10.1073/pnas.1222706110 23569238
43. Almeida RS, Wilson D, Hube B (2009) Candida albicans iron acquisition within the host. FEMS Yeast Res 9 : 1000–1012. doi: 10.1111/j.1567-1364.2009.00570.x 19788558
44. Kronstad JW, Cadieux B, Jung WH (2013) Pathogenic yeasts deploy cell surface receptors to acquire iron in vertebrate hosts. PLoS Pathog 9: e1003498. doi: 10.1371/journal.ppat.1003498 24009498
45. Murciano C, Villamon E, O'Connor JE, Gozalbo D, Gil ML (2006) Killed Candida albicans yeasts and hyphae inhibit gamma interferon release by murine natural killer cells. Infect Immun 74 : 1403–1406. 16428793
46. Paulus SC, van Saene HK, Hemsworth S, Hughes J, Ng A, et al. (2005) A prospective study of septicaemia on a paediatric oncology unit: a three-year experience at The Royal Liverpool Children's Hospital, Alder Hey, UK. Eur J Cancer 41 : 2132–2140. 16129600
47. Navarathna DH, Hornby JM, Krishnan N, Parkhurst A, Duhamel GE, et al. (2007) Effect of farnesol on a mouse model of systemic candidiasis, determined by use of a DPP3 knockout mutant of Candida albicans. Infect Immun 75 : 1609–1618. 17283095
48. Stookey LL (1970) Ferrozine—a new spectrophotometric reagent for iron. Anal Chem 42 : 779–781.
49. Banin E, Vasil ML, Greenberg EP (2005) Iron and Pseudomonas aeruginosa biofilm formation. Proc Natl Acad Sci U S A 102 : 11076–11081. 16043697
50. Rogan MP, Taggart CC, Greene CM, Murphy PG, O'Neill SJ, et al. (2004) Loss of microbicidal activity and increased formation of biofilm due to decreased lactoferrin activity in patients with cystic fibrosis. J Infect Dis 190 : 1245–1253. 15346334
51. Singh PK, Parsek MR, Greenberg EP, Welsh MJ (2002) A component of innate immunity prevents bacterial biofilm development. Nature 417 : 552–555. 12037568
52. Kaneko Y, Thoendel M, Olakanmi O, Britigan BE, Singh PK (2007) The transition metal gallium disrupts Pseudomonas aeruginosa iron metabolism and has antimicrobial and antibiofilm activity. J Clin Invest 117 : 877–888. 17364024
53. Genco CA, Chen CY, Arko RJ, Kapczynski DR, Morse SA (1991) Isolation and characterization of a mutant of Neisseria gonorrhoeae that is defective in the uptake of iron from transferrin and haemoglobin and is avirulent in mouse subcutaneous chambers. J Gen Microbiol 137 : 1313–1321. 1919507
54. Litwin CM, Calderwood SB (1993) Role of iron in regulation of virulence genes. Clin Microbiol Rev 6 : 137–149. 8472246
55. Lamont IL, Beare PA, Ochsner U, Vasil AI, Vasil ML (2002) Siderophore-mediated signaling regulates virulence factor production in Pseudomonasaeruginosa. Proc Natl Acad Sci U S A 99 : 7072–7077. 11997446
56. Nicas TI, Bradley J, Lochner JE, Iglewski BH (1985) The role of exoenzyme S in infections with Pseudomonas aeruginosa. J Infect Dis 152 : 716–721. 2995500
57. Tang H, Kays M, Prince A (1995) Role of Pseudomonas aeruginosa pili in acute pulmonary infection. Infect Immun 63 : 1278–1285. 7890385
58. Tang HB, DiMango E, Bryan R, Gambello M, Iglewski BH, et al. (1996) Contribution of specific Pseudomonas aeruginosa virulence factors to pathogenesis of pneumonia in a neonatal mouse model of infection. Infect Immun 64 : 37–43. 8557368
59. Amidon GL, Lee HJ (1994) Absorption of peptide and peptidomimetic drugs. Annu Rev Pharmacol Toxicol 34 : 321–341. 8042854
60. Lee YH, Sinko PJ (2000) Oral delivery of salmon calcitonin. Adv Drug Deliv Rev 42 : 225–238.
61. Bachmanov AA, Reed DR, Beauchamp GK, Tordoff MG (2002) Food intake, water intake, and drinking spout side preference of 28 mouse strains. Behav Genet 32 : 435–443. 12467341
62. Padmanabhan P, Grosse J, Asad AB, Radda GK, Golay X (2013) Gastrointestinal transit measurements in mice with 99mTc-DTPA-labeled activated charcoal using NanoSPECT-CT. EJNMMI Res 3 : 60. doi: 10.1186/2191-219X-3-60 23915679
63. Andrews SC, Robinson AK, Rodriguez-Quinones F (2003) Bacterial iron homeostasis. FEMS Microbiol Rev 27 : 215–237. 12829269
64. Hunter RC, Asfour F, Dingemans J, Osuna BL, Samad T, et al. (2013) Ferrous iron is a significant component of bioavailable iron in cystic fibrosis airways. MBio 4.
65. Konings AF, Martin LW, Sharples KJ, Roddam LF, Latham R, et al. (2013) Pseudomonas aeruginosa uses multiple pathways to acquire iron during chronic infection in cystic fibrosis lungs. Infect Immun 81 : 2697–2704. doi: 10.1128/IAI.00418-13 23690396
66. Evans DF, Pye G, Bramley R, Clark AG, Dyson TJ, et al. (1988) Measurement of gastrointestinal pH profiles in normal ambulant human subjects. Gut 29 : 1035–1041. 3410329
67. Moura E, Verheul AF, Marx JJ (1998) A functional defect in hereditary haemochromatosis monocytes and monocyte-derived macrophages. Eur J Clin Invest 28 : 164–173. 9541131
68. van Asbeck BS, Marx JJ, Struyvenberg A, Verhoef J (1984) Functional defects in phagocytic cells from patients with iron overload. J Infect 8 : 232–240. 6736664
69. Patel RM, Myers LS, Kurundkar AR, Maheshwari A, Nusrat A, et al. (2012) Probiotic bacteria induce maturation of intestinal claudin 3 expression and barrier function. Am J Pathol 180 : 626–635. doi: 10.1016/j.ajpath.2011.10.025 22155109
70. Grillot R, Portmann-Coffin V, Ambroise-Thomas P (1994) Growth inhibition of pathogenic yeasts by Pseudomonas aeruginosa in vitro: clinical implications in blood cultures. Mycoses 37 : 343–347. 7746293
71. Kerr JR (1994) Suppression of fungal growth exhibited by Pseudomonas aeruginosa. J Clin Microbiol 32 : 525–527. 8150966
72. Kerr JR, Taylor GW, Rutman A, Hoiby N, Cole PJ, et al. (1999) Pseudomonas aeruginosa pyocyanin and 1-hydroxyphenazine inhibit fungal growth. J Clin Pathol 52 : 385–387. 10560362
73. Ray TL, Payne CD (1988) Scanning electron microscopy of epidermal adherence and cavitation in murine candidiasis: a role for Candida acid proteinase. Infect Immun 56 : 1942–1949. 3294180
74. Scherwitz C (1982) Ultrastructure of human cutaneous candidosis. J Invest Dermatol 78 : 200–205. 7035576
75. Purschke FG, Hiller E, Trick I, Rupp S (2012) Flexible survival strategies of Pseudomonas aeruginosa in biofilms result in increased fitness compared with Candida albicans. Mol Cell Proteomics 11 : 1652–1669. doi: 10.1074/mcp.M112.017673 22942357
76. Peleg AY, Tampakakis E, Fuchs BB, Eliopoulos GM, Moellering RC Jr., et al. (2008) Prokaryote-eukaryote interactions identified by using Caenorhabditis elegans. Proc Natl Acad Sci U S A 105 : 14585–14590. doi: 10.1073/pnas.0805048105 18794525
77. Akagawa G, Abe S, Yamaguchi H (1995) Mortality of Candida albicans-infected mice is facilitated by superinfection of Escherichia coli or administration of its lipopolysaccharide. J Infect Dis 171 : 1539–1544. 7769289
78. Burd RS, Raymond CS, Dunn DL (1992) Endotoxin promotes synergistic lethality during concurrent Escherichia coli and Candida albicans infection. J Surg Res 52 : 537–542. 1528027
79. Neely AN, Law EJ, Holder IA (1986) Increased susceptibility to lethal Candida infections in burned mice preinfected with Pseudomonas aeruginosa or pretreated with proteolytic enzymes. Infect Immun 52 : 200–204. 2420722
80. Peters BM, Noverr MC (2013) Candida albicans-Staphylococcus aureus polymicrobial peritonitis modulates host innate immunity. Infect Immun 81 : 2178–2189. doi: 10.1128/IAI.00265-13 23545303
81. Bodey GP (2001) Pseudomonas aeruginosa infections in cancer patients: have they gone away? Curr Opin Infect Dis 14 : 403–407. 11964856
82. Angebault C, Djossou F, Abelanet S, Permal E, Ben Soltana M, et al. (2013) Candida albicans is not always the preferential yeast colonizing humans: a study in Wayampi Amerindians. J Infect Dis 208 : 1705–1716. doi: 10.1093/infdis/jit389 23904289
83. Reichart PA, Khongkhunthian P, Samaranayake LP, Yau J, Patanaporn V, et al. (2005) Oral Candida species and betel quid-associated oral lesions in Padaung women of Northern Thailand. Mycoses 48 : 132–136. 15743432
84. Xu J, Mitchell TG (2003) Geographical differences in human oral yeast flora. Clin Infect Dis 36 : 221–224. 12522756
85. Bougnoux ME, Diogo D, Francois N, Sendid B, Veirmeire S, et al. (2006) Multilocus sequence typing reveals intrafamilial transmission and microevolutions of Candida albicans isolates from the human digestive tract. J Clin Microbiol 44 : 1810–1820. 16672411
86. Kam AP, Xu J (2002) Diversity of commensal yeasts within and among healthy hosts. Diagn Microbiol Infect Dis 43 : 19–28. 12052625
87. Xu J, Boyd CM, Livingston E, Meyer W, Madden JF, et al. (1999) Species and genotypic diversities and similarities of pathogenic yeasts colonizing women. J Clin Microbiol 37 : 3835–3843. 10565893
88. Almeida RS, Brunke S, Albrecht A, Thewes S, Laue M, et al. (2008) the hyphal-associated adhesin and invasin Als3 of Candida albicans mediates iron acquisition from host ferritin. PLoS Pathog 4: e1000217. doi: 10.1371/journal.ppat.1000217 19023418
89. Cornelis P, Bodilis J (2009) A survey of TonB-dependent receptors in fluorescent pseudomonads. Environ Microbiol Rep 1 : 256–262. doi: 10.1111/j.1758-2229.2009.00041.x 23765855
90. Cuiv PO, Keogh D, Clarke P, O'Connell M (2007) FoxB of Pseudomonas aeruginosa functions in the utilization of the xenosiderophores ferrichrome, ferrioxamine B, and schizokinen: evidence for transport redundancy at the inner membrane. J Bacteriol 189 : 284–287. 17056746
91. Llamas MA, Mooij MJ, Sparrius M, Vandenbroucke-Grauls CM, Ratledge C, et al. (2008) Characterization of five novel Pseudomonas aeruginosa cell-surface signalling systems. Mol Microbiol 67 : 458–472. 18086184
92. Ochsner UA, Johnson Z, Vasil ML (2000) Genetics and regulation of two distinct haem-uptake systems, phu and has, in Pseudomonas aeruginosa. Microbiology 146 (Pt 1): 185–198. 10658665
93. Cartron ML, Maddocks S, Gillingham P, Craven CJ, Andrews SC (2006) Feo—transport of ferrous iron into bacteria. Biometals 19 : 143–157. 16718600
94. Wang Y, Newman DK (2008) Redox reactions of phenazine antibiotics with ferric (hydr)oxides and molecular oxygen. Environ Sci Technol 42 : 2380–2386. 18504969
95. Chatzinikolaou I, Abi-Said D, Bodey GP, Rolston KV, Tarrand JJ, et al. (2000) Recent experience with Pseudomonas aeruginosa bacteremia in patients with cancer: Retrospective analysis of 245 episodes. Arch Intern Med 160 : 501–509. 10695690
96. Micol JB, de Botton S, Guieze R, Coiteux V, Darre S, et al. (2006) An 18-case outbreak of drug-resistant Pseudomonas aeruginosa bacteriemia in hematology patients. Haematologica 91 : 1134–1138. 16885056
97. Brandl K, Plitas G, Mihu CN, Ubeda C, Jia T, et al. (2008) Vancomycin-resistant enterococci exploit antibiotic-induced innate immune deficits. Nature 455 : 804–807. doi: 10.1038/nature07250 18724361
98. Brandtzaeg P (2010) Food allergy: separating the science from the mythology. Nat Rev Gastroenterol Hepatol 7 : 380–400. doi: 10.1038/nrgastro.2010.80 20606633
99. Beach RC, Menzies IS, Clayden GS, Scopes JW (1982) Gastrointestinal permeability changes in the preterm neonate. Arch Dis Child 57 : 141–145. 7065710
100. Rouwet EV, Heineman E, Buurman WA, ter Riet G, Ramsay G, et al. (2002) Intestinal permeability and carrier-mediated monosaccharide absorption in preterm neonates during the early postnatal period. Pediatr Res 51 : 64–70. 11756641
101. Finck-Barbancon V, Goranson J, Zhu L, Sawa T, Wiener-Kronish JP, et al. (1997) ExoU expression by Pseudomonas aeruginosa correlates with acute cytotoxicity and epithelial injury. Mol Microbiol 25 : 547–557. 9302017
102. Kudoh I, Wiener-Kronish JP, Hashimoto S, Pittet JF, Frank D (1994) Exoproduct secretions of Pseudomonas aeruginosa strains influence severity of alveolar epithelial injury. Am J Physiol 267: L551–556. 7977765
103. Vallis AJ, Finck-Barbancon V, Yahr TL, Frank DW (1999) Biological effects of Pseudomonas aeruginosa type III-secreted proteins on CHO cells. Infect Immun 67 : 2040–2044. 10085057
104. Allewelt M, Coleman FT, Grout M, Priebe GP, Pier GB (2000) Acquisition of expression of the Pseudomonas aeruginosa ExoU cytotoxin leads to increased bacterial virulence in a murine model of acute pneumonia and systemic spread. Infect Immun 68 : 3998–4004. 10858214
105. Chang W, Small DA, Toghrol F, Bentley WE (2005) Microarray analysis of Pseudomonas aeruginosa reveals induction of pyocin genes in response to hydrogen peroxide. BMC Genomics 6 : 115. 16150148
106. Swanson BL, Colmer JA, Hamood AN (1999) The Pseudomonas aeruginosa exotoxin A regulatory gene, ptxS: evidence for negative autoregulation. J Bacteriol 181 : 4890–4895. 10438759
107. Swanson BL, Hamood AN (2000) Autoregulation of the Pseudomonas aeruginosa protein PtxS occurs through a specific operator site within the ptxS upstream region. J Bacteriol 182 : 4366–4371. 10894751
108. Goodman AL, Kallstrom G, Faith JJ, Reyes A, Moore A, et al. (2011) Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc Natl Acad Sci U S A 108 : 6252–6257. doi: 10.1073/pnas.1102938108 21436049
109. Lopez-Medina E, Neubauer MM, Pier GB, Koh AY (2011) RNA isolation of Pseudomonas aeruginosa colonizing the murine gastrointestinal tract. J Vis Exp.
110. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5 : 621–628. doi: 10.1038/nmeth.1226 18516045
111. Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11: R106. doi: 10.1186/gb-2010-11-10-r106 20979621
112. Samuel BS, Gordon JI (2006) A humanized gnotobiotic mouse model of host-archaeal-bacterial mutualism. Proc Natl Acad Sci U S A 103 : 10011–10016. 16782812
113. Savli H, Karadenizli A, Kolayli F, Gundes S, Ozbek U, et al. (2003) Expression stability of six housekeeping genes: A proposal for resistance gene quantification studies of Pseudomonas aeruginosa by real-time quantitative RT-PCR. J Med Microbiol 52 : 403–408. 12721316
114. Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP (1998) A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212 : 77–86. 9661666
115. Mettrick KA, Lamont IL (2009) Different roles for anti-sigma factors in siderophore signalling pathways of Pseudomonas aeruginosa. Mol Microbiol 74 : 1257–1271. doi: 10.1111/j.1365-2958.2009.06932.x 19889096
116. Morgan AF (1979) Transduction of Pseudomonas aeruginosa with a mutant of bacteriophage E79. J Bacteriol 139 : 137–140. 110777
117. Chuanchuen R, Narasaki CT, Schweizer HP (2002) Benchtop and microcentrifuge preparation of Pseudomonas aeruginosa competent cells. Biotechniques 33 : 760, 762–763. 12398182
118. Riemer J, Hoepken HH, Czerwinska H, Robinson SR, Dringen R (2004) Colorimetric ferrozine-based assay for the quantitation of iron in cultured cells. Anal Biochem 331 : 370–375. 15265744
119. Stookey LL (1970) Ferrozine—a new spectrophotometric reagent for iron. Anal Chem 42 : 779–781.
120. Carmi R, Carmeli S, Levy E, Gough FJ (1994) (+)-(S)-dihydroaeruginoic acid, an inhibitor of Septoria tritici and other phytopathogenic fungi and bacteria, produced by Pseudomonas fluorescens. J Nat Prod 57 : 1200–1205. 7798954
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 8
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Human Non-neutralizing HIV-1 Envelope Monoclonal Antibodies Limit the Number of Founder Viruses during SHIV Mucosal Infection in Rhesus Macaques
- Type VI Secretion System Toxins Horizontally Shared between Marine Bacteria
- Illuminating Targets of Bacterial Secretion
- Are Human Intestinal Eukaryotes Beneficial or Commensals?