
Metody používané ve farmaceutické technologii ke zvyšování biologické dostupnosti špatně rozpustných léčiv po perorálním podání
Methods used in pharmaceutical technology to increase bioavailability of poorly soluble drugs after oral administration
Bioavailability increasing of poorly soluble drugs has become one of the main topics of modern pharmaceutical technology. Many methods based on the chemical modification, physical modification or new technological processes have been already used to improve bioavailability. Some of these methods (e.g. micronization, preparation of solid dispersions, formulation of an inclusion complex, etc.) have been for many years successfully used by pharmaceutical companies. On the other hand, methods such as liquisolid system and self-emulsifying drug delivery systems are still in the early stages of their development. It is expected that this novel methods could play a significant role in the preparation of modern dosage forms. The aim of this paper is to provide the summary of methods improving bioavailability of poorly soluble drugs used in the field of pharmaceutical technology.
Key words:
bioavailability • poorly soluble drugs • micronization • solid dispersions • self-emulsifying drug delivery systems
Autoři:
Barbora Vraníková; Jan Gajdziok
Vyšlo v časopise:
Čes. slov. Farm., 2015; 64, 159-172
Kategorie:
Přehledy a odborná sdělení
Souhrn
Zvyšování biologické dostupností léčiv špatně rozpustných ve vodě se stalo jedním z trendů moderní farmaceutické technologie. Doposud byla popsána řada metod fungujících na principu chemické modifikace léčivé látky, její fyzikální úpravě či novém technologickém postupu. Řada metod, jako je např. mikronizace, příprava pevných disperzí, tvorba inkluzních komplexů atd., je již mnoho let s úspěchem využívána farmaceutickými firmami. Naopak některé z metod (systémy kapalina v pevné fázi, samoemulgující systémy aj.) jsou teprve v počátečních fázích svého vývoje a očekává se, že by mohly hrát významnou roli při přípravě moderních lékových forem. Cílem tohoto článku je přinést přehled metod používaných ve farmaceutické technologii ve snaze zvýšit biologickou dostupnost ve vodě špatně rozpustných léčivých látek.
Klíčová slova:
biologická dostupnost • špatně rozpustná léčiva • mikronizace • pevné disperze • samoemulgující systémy
Úvod
Biofarmaceutický klasifikační systém (BCS) rozděluje léčiva do čtyř základních skupin na základě jejich rozpustnosti ve vodě a gastrointestinální prostupnosti (permeabilitě)1, 2). V současné farmakoterapii narůstá množství léčivých látek, které jsou ve vodném prostředí gastrointestinálního traktu prakticky nerozpustné a je možné je zařadit do třídy II (nízká rozpustnost, vysoká prostupnost) nebo IV (nízká rozpustnost, nízká prostupnost). Tato léčiva způsobují řadu problémů při formulaci pevných p.o. lékových forem určených pro systémovou absorpci účinné látky, z důvodu jejich nízké biologické dostupnosti, která je úzce spjata právě se špatnou rozpustností ve vodném prostředí3).
Pro přípravu p.o. lékových forem byla popsána řada metod vedoucích ke zlepšení rozpustnosti léčiv, respektive jejich biologické dostupnosti. Tyto postupy se na základě svého charakteru zpravidla rozdělují na chemické a fyzikální modifikace léčivé látky a na technologické postupy (obr. 1). Chemické modifikace léčiva, mezi něž patří např. syntéza proléčiv, převedení účinné látky na její rozpustnější sůl, nebo vznik glykosylovaných derivátů, zahrnují chemickou přeměnu samotné látky, a spadají tedy do oboru farmaceutické chemie. Členění postupů na fyzikální modifikace a technologické postupy však není zcela přehledné, jelikož řada metod spadající mezi fyzikální úpravy léčivé látky (např. mikronizace, lyofilizace apod.) je běžně používaná i farmaceutickými technology v průběhu formulace finální lékové formy.

Zprostředkované rozpouštění
Pod pojmem zprostředkované rozpouštění (solubilizace) se rozumí zlepšení rozpustnosti léčiva přidáním vhodné pomocné látky tzv. solubilizátoru, který napomáhá jeho převedení do roztoku. Mezi nejčastěji používané metody solubilizace se řadí použití kosolventu, efekt hydrotropismu, zvýšení smáčivosti a micelární solubilizace4).
Přídavek kosolventu
Přídavek kosolventu je vysoce účinným a zároveň jednoduchým způsobem přípravy vodných roztoků řady málo rozpustných látek. Jako kosolventy jsou označovány s vodou mísitelné kapaliny, které mají nižší povrchové napětí než voda4). Pro přípravu vodných roztoků léčiv se jako kosolventy přidávají nejčastěji organická rozpouštědla přípustná pro daný způsob použití. V preklinických studiích (toxikologie, farmakologický účinek aj.) se obvykle používají jako kosolventy dimethylsulfoxid nebo dimethylacetamid mající vysokou solubilizační kapacitu a relativně nízkou toxicitu (letální dávka dimethylsulfoxidu pro potkana je 14,5 g/kg a dimethyacetamidu 4,9 g/kg5)). Ve farmaceutické technologii jsou oblíbenými kosolventy glycerol, ethanol, propylenglykol a kapalné makrogoly6). Účinným kosolventem je rovněž 2-pyrrolidon, který v nižších koncentracích působí i jako komplexační činidlo4). Kosolventy zvyšují rozpustnost jen částečně, proto se často kombinují s tenzidy nebo s látkami upravujícími pH6). Solubilizací vhodnými kosolventy a jejich kombinací lze zvýšit rozpustnost léčiv až 1000násobně4, 7).
Příkladem zvýšení rozpustnosti léčiva za použití kosolventů může být studie publikovaná Seedherem a Kanojiou8), kde se testovala rozpustnost sedmi antidiabetik (gliklazidu, glyburidu, glipizidu, glimepiridu, repaglinidu, pioglitazonu a roziglitazonu) ve vodě s přídavkem některého z kosolventů (makrogol 400, makrogol 8000, propylenglykol, glycerol, ethanol a ethanol v kombinaci s propylenglykolem). Z výsledků je patrné, že přídavek kosolventu zlepšuje rozpustnost všech použitých antidiabetik ve vodě až 430krát8).
Efekt hydrotropismu
Efekt hydrotropismu byl poprvé popsán Neubergem v roce 19169). Jako hydrotropní látky (hydrotropy) se označuje různorodá skupina látek, které při dostatečných koncentracích (tzv. minimální hydrotropní koncentrace) selektivně zvyšují (100–200krát) rozpustnost nepolárních sloučenin ve vodě částečným narušením její asociované struktury. Od klasických povrchově aktivních látek se odlišují přítomností krátké hydrofobní oblasti7, 10). Mezi běžně používané hydrotropní látky je možné zařadit např. močovinu, nikotinamid, lysin, tryptofan, kyselinu citronovou, benzoan sodný, salicylan sodný, aromatické sulfonové kyseliny a jejich sodné soli a řadu dalších9, 11, 12). Mezi výhody této metody patří její jednoduchost, nízké náklady a toxicita. Nevýhodou je pak potřeba vysokých koncentrací hydrotropní látky a možnost vzniku interakcí mezi léčivou látkou a použitým hydrotropem9).
Nalezením nejvhodnějšího hydrotropu pro cytostatikum paklitaxel se zabývala studie Leea et al.13). Zkoumalo se 60 potenciálních hydrotropních látek, přičemž jako nejvhodnější byl vybrán N,N-diethylnikotinamid.
Zvyšování smáčivosti
Jako smáčedla jsou označovány tenzidy, které zvyšují smáčivost tuhých částic tím, že snižují mezipovrchové napětí mezi tuhou a kapalnou fází. Tyto látky se adsorbují na povrchu částic a vytvářejí film, který brání aglomeraci a umožnují lepší průnik rozpouštědla k povrchu tuhé částice4, 7). Za smáčedla se obvykle označují tenzidy s hodnotou hydrofilně-lipofilní rovnováhy (HLB) mezi 7 a 9, zatímco povrchově aktivní látky s HLB 10 a více se řadí mezi micelární solubilizátory (tab. 1)4). Mezi nejpoužívanější smáčedla ve farmaceutické technologii patří: natrium-lauryl-sulfát, benzalkonium-chlorid, makrogol-stearát, sorbitanové estery (Spany), polyethylované sorbitanové estery (Polysorbáty), oxyethylovaný ricinový olej (Cremophor® EL) apod.5, 7)

Micelární solubilizace
Micelární solubilizace je proces, během kterého dochází k rozpuštění léčiva díky vratným interakcím s micelami povrchově aktivní látky (tenzidu) za vzniku termodynamicky stabilního izotropního roztoku. Určité množství tenzidu umožňuje solubilizaci pouze určitého množství nerozpustné látky, které bývá označováno jako maximální aditivní koncentrace. K vytvoření micel dochází rozpouštěním tenzidů ve vodě v koncentraci vyšší, než je kritická micelární koncentrace (CMC)7, 14). Pro většinu povrchově aktivních látek se hodnota CMC pohybuje v rozmezí 0,05–0,10 % a závisí na chemických vlastnostech rozpouštěné fáze4, 15). Kromě zlepšené rozpustnosti zvyšuje inkorporace léčiva do struktury micel také míru jeho transportu přes střevní epitel, a umožňuje tak zlepšit jeho biologickou dostupnost16).
Micely mohou mít sférický, cylindrický nebo planární tvar a dosahovat velikosti mezi 5 až řádově 100 nm. Tvar a velikost micel je možné ovlivnit změnou chemické struktury tenzidu nebo podmínek přípravy, jako je teplota roztoku, koncentrace a zastoupení jednotlivých tenzidů, iontová síla a pH. Na těchto parametrech závisí také rozpouštěcí kapacita tenzidu, přičemž bylo zjištěno, že neiontové povrchově aktivní látky jsou obvykle lepší solubilizační činidla pro hydrofobní léčiva než tenzidy iontové, a to vzhledem k jejich nižší hodnotě CMC14).
Léčivo je možné do micely začlenit na základě jeho povahy několika způsoby, jak ukazuje obrázek 2. Hydrofilní léčivo je zpravidla adsorbováno na povrch micely (1.), zatímco léčivo ve vodě částečně rozpustné (amfifilní látku) je možné začlenit do micely mezi hydrofilní „hlavy“ tenzidů (např. polyethylenoxidové) (2.) nebo do palisádové vrstvy mezi hydrofilními skupinami a několika atomy uhlíku hydrofobních skupin, které tvoří vnější část jádra (3.). Lipofilní látky jsou pak inkorporovány do vnitřního jádra micely mezi hydrofobní „řetězce“ tenzidu (4.)14).

Mezi povrchově aktivní látky využívané k zlepšování rozpustnosti léčiv za pomoci micelární solubilizace patří natrium-lauryl-sulfát17), poloxamery (Pluronic® P-85)18), makrogoly (Carbowax® 19), Carbowax Sentry® 19)), Polysorbát 2019), polyoxyethylované ethery (Brij® 30)19) apod.
Micelární solubilizace se využila například ke zvýšení rozpustnosti antiflogistika ibuprofenu. Jako povrchově aktivní látky tvořící micely se použily natrium-lauryl-sulfát, dodecyltrimethylamonium bromid a n-dodecylocta(ethylenoxid) v koncentraci 0–85 mM. Z výsledků studie vyplývá, že rozpustnost ibuprofenu lineárně narůstá s rostoucí koncentrací jednotlivých tenzidů. Nejvyšší solubilizační kapacitu (množství rozpuštěné látky v molech, které je rozpuštěno jedním molem micel tenzidů) pak vykazoval dodecyltrimethylamonium bromid (až 16krát)17).
Změna pH
Rozpustnost a míra absorpce celé řady léčivých látek jsou závislé na pH prostředí. Jeho hodnota se však výrazně mění napříč gastrointestinálním traktem, proto dochází u léčiv, která mají charakter slabých kyselin, slabých zásad nebo jejích solí, k nestejnoměrnému uvolňování a rozpouštění20).
U léčivých látek, jejichž uvolňování je závislé na hodnotě pH, je problematické zajištění dostatečné biologické dostupnosti s ohledem na kratší časový úsek, ve kterém se musí léčiva látka rozpustit. Začleněním pufrovacích pomocných látek (pH modifikátorů) do formulace lze vytvořit mikrooblasti s pH zajišťujícím rovnoměrnou rozpustnost léčivé látky bez ohledu na pH okolního prostředí. Jinými slovy přidání pH modifikátoru do formulace se může tento časový horizont pro rozpouštění účinné látky prodloužit díky vzniku mikroprostředí se změněným pH v závislosti na použitém pH modifikátoru4).
Optimalizují se tak podmínky pro uvolnění léčiva, jeho absorpci i biologickou dostupnost21).
Působení modifikátorů pH je závislé na jejich vlastnostech, jako je rozpustnost, rychlost uvolňování, ionizační konstanta apod. Efektivita modifikátoru se zvyšuje s jeho rostoucí silou, která je dána disociační konstantou pKa, a klesá s jeho rostoucí rozpustností v disolučním prostředí21).
Zásadité modifikátory pH jsou přidávány do formulací s obsahem slabých kyselin a jejich solí (kyselina acetylsalicylová, metotrexát, furosemid aj.) pro zvýšení jejich rozpustnosti v kyselém prostředí žaludku (pH 1,5–2,9) a proximální části tenkého střeva (pH 6,0–6,8)4, 21–23). Naopak kyselé modifikátory pH se přidávají k léčivům povahy slabých zásad a jejich solí (atropin, kodein, tolbutamid apod.), které jsou lépe rozpustné v kyselém prostředí. Kyselé modifikátory pak úpravou lokálního pH zlepšují rozpustnost léčiv nejčastěji v oblasti tenkého střeva (pH 6,8–7,4)21, 23, 24). Přehled nejčastěji používaných pH modifikátorů je uveden v tabulce 2.

Příkladem využití pufrovacích pomocných látek je studie Amarala et al.25), ve které se k modifikaci pH pro uvolňování antiflogistika naproxenu použily uhličitan sodný, uhličitan vápenatý a citronan sodný. Jako nejvhodnější pH modifikátor se jevil uhličitan sodný, který díky úpravě pH mikroprostředí navýšil množství uvolněného léčiva z matrice ze 40 % na 90 %25).
Úprava velikosti částic
Míra a především rychlost uvolňování léčiva z lékové formy narůstá se zvyšujícím se povrchem, respektive se snižující se velikostí jednotlivých částic, jak popisuje Noyes-Whitneyho rovnice [1]. Z tohoto důvodu je jednou z nejstarších a také nejčastěji používaných technik ke zvyšování biologické dostupnosti léčiv špatně rozpustných ve vodě úprava velikosti jejich částic obvykle mechanickým mletím26). Cílem je obvykle dosáhnout až velikosti částic menší než 10 μm, kdy se uplatňují aktivní transportní mechanismy, nebo nanometrových částic, které prostupují střevními endocyty3).

kde dn – množství látky, které se rozpustí v časovém intervalu (dt), D – difuzní koeficient rozpouštěné látky v použitém rozpouštědle, S – celková plocha fázového rozhraní mezi rozpouštěnou látkou a roztokem, δ – tloušťka difuzní vrstvy, cs – koncentrace nasyceného roztoku rozpouštěné látky na fázovém rozhraní, c – koncentrace rozpouštěné látky v celkovém objemu roztoku v uvažovaném čase.
Mechanické rozdrobňování
Nejčastěji používanou metodou vedoucí k mikronizaci částic je mechanické rozmělňování/rozdrobňování (např. drcení, mletí) větších částic, které může probíhat suchou nebo mokrou cestou, popřípadě mletím zmražené látky (kryomletí). Během mletí v suchém stavu se obvykle používají méně účinné kladivové, oscilační a kulové mlýny (částice v desítkách μm) nebo účinnější tryskové mlýny (částice o velikosti 100–10–1 μm). Tento postup však i přes své široké uplatnění není ideální, jelikož výstupní vlastnosti (velikost, tvar, morfologie, povrchové vlastnosti atd.) léčivé látky je možné kontrolovat jen omezeně. Navíc částice během suchého mletí získávají silný elektrostatický náboj vedoucí k tvorbě aglomerátů, které ve styku s kapalinou zabraňují průniku rozpouštědla k jednotlivým částicím3, 27–30). V případech, kdy není možné použít suché mletí, se uplatňuje mletí vlhké za použití klasických kulových, koloidních nebo moderních perlových mlýnů. Jako disperzní prostředí se používá nejčastěji voda s příměsí povrchově aktivních látek nebo vhodného oleje3). Tyto metody však nejsou vhodné pro látky s nízkou teplotou tání, vysokou elasticitou nebo naopak tvrdostí, u kterých je častěji využíváno kryogenní mletí. V tomto případě je látka podchlazena ve zkapalněném plynu (např. oxidu uhličitém, dusíku, vodíku nebo freonech). Takto zmražená látka je vysoce křehká a snadno se v průběhu mletí drtí3).
Jino et al. ve své studii porovnávali vliv použité metody mikronizace (mletí v kladivovém mlýnu, v tryskovém mlýnu a sprejovým sušením tzv. metoda NanoCrystal®). Ve všech třech případech došlo ke zlepšení disolučního profilu antitrombotika cilostazolu. U suspenzí připravených mletím v kladivovém a tryskovém mlýnu byl však pozorován vliv potravy na rychlost uvolňování léčiva, zatímco rozpouštění léčiva ze suspenze připravené metodou NanoCrystal® bylo na přísunu potravy nezávislé26).
Příkladem využití vlhkého mletí může být příprava nanosuspenze protinádorových léčiv (piposulfan, etoposid, kamptothecin a paklitaxel). Jako povrchové aktivní látky stabilizující suspenzi v průběhu mletí se použily Pluronic® a Tetronic®. Výsledky studie ukázaly, že vlhké mletí je vhodnou metodou přípravy vodných nanosuspenzí těchto léčiv31).
Metoda kryogenního mletí se použila pro zjištění rozpustnosti antihypertenziva nifedipinu a antiflogistika indomethacinu v kopolymeru polyvinylpyrolidinu a polyacetátu. Směs polymeru a léčiva zde byla mleta za snížené teploty a následně se za pomocí diferenciální skenovací kalorimetrie hodnotila míra rozpustnosti účinných látek v daném polymeru32).
Řízená krystalizace
Novější metodou úpravy velikosti částic v porovnání s mechanickým rozdrobňováním je např. řízená krystalizace, která umožňuje získat krystaly požadované velikosti již během jejich přípravy bez nutnosti dalšího zmenšování částic či použití speciálních zařízení. Nejčastěji probíhá řízená krystalizace metodou výměny rozpouštědla nebo změnou pH. Tvar krystalů je možné ovlivnit použitými přísadami (stabilizátory), které se přednostně adsorbují na některé krystalové plochy, a tím zpomalují jejich nárůst. Plocha krystalů je totiž tvořená rozdílně orientovanými molekulami a stabilizátor se specificky váže na plochy podle této orientace3, 27). Jako stabilizátor se nejčastěji uplatňuje hypromelosa33–36), avšak je možné použít také další deriváty celulosy (např. sodnou sůl karmelosy, hyetelosu, methylcelulosu apod.), dále pak hydroxyethylškrob, agar, želatinu, natrium-alginát, pektin, polyvinylalkohol, povidon, makrogoly a řadu dalších látek27, 37).
Řízená krystalizace jako technika zmenšování částic se využila k redukci velikosti částic ibuprofenu, itrakonazolu a ketokonazolu. Výsledkem byly disperze s homogenní velikosti částic okolo 2 a méně μm. Výběr stabilizátoru měl pak vliv na velikost vzniklých krystalů, přičemž nejmenší krystaly vznikly při použití hypromelosy 400037).
Mikronizace za přítomnosti superkritických médií
Pro úpravu velikosti částic je dále možné použít mikronizaci za přítomnosti superkritických médií, která umožňuje získat částice o velikosti několika nanometrů. Nejčastěji používaným superkritickým médiem je oxid uhličitý38–40), méně často se pak uplatňují také např. oxid uhličitý v kombinaci s ethanolem, oxid dusný, fluoroform, dimethylether apod.39, 41, 42). V případě, že je léčivo rozpuštěno v superkritickém médiu a následně rychle vstřikováno skrze trysku do okolního vzduchu, hovoříme o tzv. mikronizaci rychlou expanzí superkritického roztoku (rapid expansion of supercritical solution). Vysoký stupeň saturace doprovázený rychlým snížením tlaku vede k homogenní nukleaci a vzniku dobře dispergovaných částic43). Naopak pokud má léčiva látka nízkou rozpustnost v superkritickém médiu, je vhodné použít tzv. mikronizaci superkritickým anti-rozpouštědlem (supercritical anti-solvent micronization), kdy je roztok léčivé látky v organickém rozpouštědle za pomoci vhodné trysky vstřikován do komory s obsahem superkritického média, které zde funguje jako anti-rozpouštědlo. Velikost a morfologii výsledných částic (krystalů) je možné ovlivnit typem a vnitřním průměrem trysky, výběrem organického rozpouštědla a provozními podmínkami (tlak, teplota a rychlost průtoku vstřikovaného roztoku)44). Mezi výhody metod využívajících superkritická média patří možnost získání částic o velmi malém průměru (méně než 500 nm) a upravovat velikost částic látek citlivých na teplo díky použití nízkých provozních teplot43, 44).
Mikronizace rychlou expanzí superkritického roztoku se použila pro zmenšení částic např. griseofulvinu43), β-sitosterolu43), ibuprofenu45), cyklosporinu A46) a dalších, zatímco metoda mikronizace superkritickým anti-rozpouštědlem se s úspěchem využila u felodipinu47), atorvastatinu48) apod.
Sprejové sušení
Kromě výše zmíněných postupů se k redukci velikosti částic používá také metoda sprejového sušení, kterou je možné získat mikronizované a zároveň sférické částice. Léčivá látka se v tomto případě rozpustí, popřípadě suspenduje ve vhodné kapalině nebo směsi kapalin. Vzniklý roztok nebo suspenze se pak obvykle za zvýšené teploty nastřikuje do expanzní nádoby a suší v proudu vzduchu či inertního plynu. Změnou podmínek celého procesu je možné získat částice požadovaného tvaru, velikosti a hustoty3, 49).
Metodou sprejového sušení se připravily například pevné samoemulgující systémy s obsahem špatně rozpustného antihypertenziva nimodipinu50), suchá emulze (sprejově vysušená emulze) 5-fenyl-1,2-dithiol-3-thionu51) nebo bezvodá amorfní forma hypolipidemika atorvastatinu52).
Příkladem léčivých přípravků a potravinových doplňků dostupných na českém trhu v mikronizované podobě může být Novofem (17β-estradiol), Lipanthyl 267 M (fenofibrát), Detralex (diosminol a hesperidin) nebo Mobivenal Micro (diosminol a hisperidon)53).
Lyofilizace
Lyofilizace neboli mrazová sublimace (sušení) je proces, v jehož průběhu dochází k šetrnému vysoušení látek nebo jejich roztoků, popřípadě suspenzí, ve zmraženém stavu, kdy se rozpouštědlo odstraní nejdříve jeho sublimací (primární sušení) a následně desorpcí (sekundární sušení) ve vakuu. Zbylá pevná část si zachovává specifickou pórovitou strukturu, která je tvořena kanálky vzniklými po sublimovaném rozpouštědle. Tato struktura po styku s vodou umožňuje rychlý průnik disolučního média do lyofilizátu, jeho energeticky málo náročné rozrušení a následné zrychlené rozpouštění3, 54, 55).
Tradiční proces lyofilizace se skládá ze tří základních kroků: zmrazení, primární a sekundární sušení. V průběhu fáze mrazení je roztok, popřípadě suspenze, ochlazována (obvykle na teplotu –40 až –50 °C), dokud se nezačnou tvořit krystaly ledu, které postupně narůstají. Následuje primární sušení neboli sublimace ledu za sníženého tlaku. Po dokončení primárního sušení obsahuje vzniklý produkt zpravidla ještě 15–20 % vody, která se desorbuje v průběhu sekundárního sušení. Sekundární sušení probíhá při vyšší teplotě a nízkém tlaku a dovoluje dosáhnout požadované nízké vlhkosti produktu56, 57). Celý proces je vysoce energeticky, finančně a časově náročný, což tvoří jeho hlavní nevýhody56–58).
Ekonomicky a časově méně náročnou metodou je pak sprejová lyofilizace, která spojuje techniku sprejového sušení s lyofilizací, a umožňuje tak vznik vysoce porézních sférických částic o požadované velikosti. Sprejová lyofilizace se stejně jako klasická mrazová sublimace skládá ze tří základních kroků. Prvním z nich je rozptýlení roztoku do podoby malých kapek následované jejich zmražením a sublimací použitého rozpouštědla59).
Ve farmaceutickém průmyslu se lyofilizace používá hlavně u hromadně vyráběných injekčních přípravků s léčivými látkami nestabilními ve vodných roztocích a pro zlepšení stability při dlouhodobém skladování nestabilních léčiv, především pak terapeutických proteinů54, 57). Proces mrazové sublimace je dále předmětem několika patentovaných technologických postupů (Zydis®, Lyoc® a Quisksolv®) pro přípravu orálně dispergovatelných tablet (ODT). Novinkou jsou pak lyofilizované filmy vhodné pro přívod léčiva přes bukální sliznici, nebo určené pro aplikaci na rány60).
Příkladem na českém trhu dostupných lyofilizátů jsou Neoclaritin (desloratadin), Maxalt (rizatriptan), Grazax 75 000 SQ-T (extrakt z trávního pylu) nebo Imunor (vepřový přenosový faktor)53).
Komplexy s cyklodextriny
Tvorba komplexů léčiva s cyklodextriny (tzv. inkluzní komplexy) je ve farmaceutickém průmyslu jednou z nejoblíbenějších a nejčastěji využívaných metod pro zvyšování rozpustnosti a zároveň také permeability (prostupnosti) léčiv přes membrány v gastrointestinálním traktu61, 62). Cyklodextriny (CD) byly poprvé izolovány v roce 1891 jako cyklické oligosacharidy vznikající degradací škrobu glukosyltransferasami bakteriálního původu (Bacilus macerans). Jejich schopnost tvořit komplexy s léčivy však byla objevena až roku 1948. Kromě běžně se vyskytujících přírodních cyklodextrinů (α, β a γ známý také pod označením τ) (tab. 3), existuje řada hydrofilních, hydrofobních a iontových derivátů s rozdílnými fyzikálně-chemickými vlastnostmi, lepší stabilitou v porovnání s běžně se vyskytujícími přírodními cyklodextriny, nižší parenterální toxicitou a vyšší inkluzní kapacitou. Nejpoužívanějším CD pro perorální lékové formy je β-CD63, 64) a jeho deriváty (např. hydroxypropyl-β-cyklodextrin65, 66), dimethyl-β-cyklodextrin67) apod.) díky snadné dostupnosti a nízké ceně, avšak vysoká nefrotoxicita a relativně nízká rozpustnost ve vodě (1,85 g/100 ml) omezují jeho častější použití především v parenterálních přípravcích62, 68, 69).

Uspořádání glukosových jednotek dává CD specifický prstencovitý tvar dutého komolého kuželu, kde vzniklá dutina je nepolární (lipofilní) povahy, zatímco vnější povrch molekuly je díky přítomnosti volných hydroxylových skupin povahy hydrofilní. Lipofilní dutina vytváří mikroprostředí, do kterého může být začleněna vhodně velká nepolární (lipofilní) skupina či celé léčivo, které je těžce rozpustné ve vodě, za vzniku inkluzního komplexu s dobrou rozpustností (obr. 3)69). Léčivo zde není vázáno pevně ani trvale a dochází jen k nekovalentním interakcím mezi léčivou látkou a nosným CD (van der Waalsovy, disperzní a vodíkové vazby), jejichž síla závisí na tom, jak zapracovávané léčivo „pasuje“ do dutiny CD a na specifických interakcích mezi povrchovými atomy4, 69).
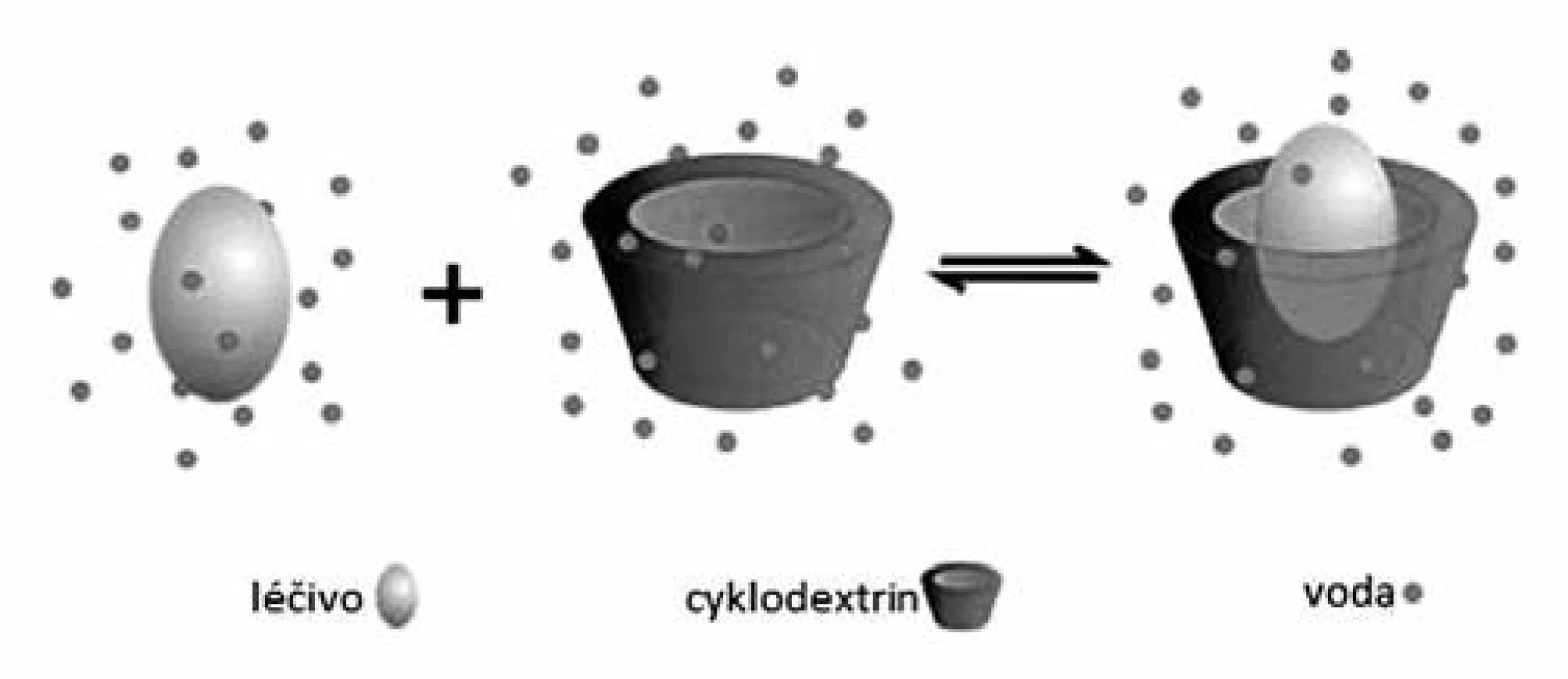
Příprava inkluzních komplexů může probíhat několika způsoby. První z nich je prosté smísení požadovaných molárních množství léčiva a CD. Mísení probíhá intenzivním třením po dobu několika hodin71). Další technikou přípravy je tzv. „metoda hnětením“, kdy jsou CD navlhčeny malým množstvím rozpouštědla (voda, vodný roztok ethanolu či methanolu apod.). K takto vzniklé hmotě je následně přidáno léčivo a celá směs se potřebnou dobu hněte. Získaná směs se vysuší, a pokud je to nezbytné, také přesítuje72). Inkluzní komplexy je možné získat také metodou spolusrážení (koprecipitace), kdy je roztok léčiva po kapkách přidáván k roztoku CD a následně několik hodin míchán. Výsledný produkt se opět vysuší. Obdobou je metoda odpařování rozpouštědla, kde je k roztoku léčiva v organickém rozpouštědle po kapkách za stálého míchání přidáváno požadované množství roztoku CD v horké vodě. Po dostatečně dlouhém promíchávání se vzniklé komplexy odfiltrují a vysuší71). Novější metodou je pak ozáření směsi léčiva, cyklodextrinu a minimálního množství rozpouštědla (obvykle směs vody a organického rozpouštědla) mikrovlnami. Směs je ozařována po krátkou dobu (obvykle 1–2 minuty) při 60 °C v mikrovlnné troubě. Po dokončení reakce je k reakční směsi přidáno dostatečné množství použitého rozpouštědla, aby došlo k odstranění přebytečného léčiva a volného cyklodextrinu. Získaná sraženina je pak odfiltrována a vysušena72, 73). Výhodou této metody je její vysoká výtěžnost a krátký reakční čas73).
Kromě zvyšování rozpustnosti, a tím i biologické dostupnosti léčiv je možné komplexy s cyklodextriny použít pro zvýšení stability, zlepšení smáčivosti, k zapracování kapalných léčiv do podoby mikrokrystalického prášku, dále pak k omezení interakcí mezi léčivy nebo léčivem a pomocnými látkami, ke snížení podráždění gastrointestinálního traktu a maskování nepříjemné chuti či zápachu léčiva61, 72, 74). Další výhodou je jejich přírodní charakter, biodegradabilita a fakt, že jsou vyráběny z obnovitelných zdrojů (škrob)75). K jejich nevýhodám se pak řadí toxicita především méně hydrofilních typů a specifické požadavky na velikost a strukturu začleňovaného léčiva72).
Na českém trhu je řada dostupných inkluzních komplexů, mezi něž je možné zařadit např. Flamexin (piroxikam), Voltaren Ophtha CD (diclofenak sodná sůl), Silymarin Duo (extrakt z ostropestřce mariánského) nebo Prostavasin (alprostadil)53).
Pevné disperze
Pevné disperze mohou být obecně definovány jako molekulární směsi ve vodě špatně rozpustného léčiva a hydrofilního nosiče, jehož vlastnosti ovlivňují rychlost rozpouštění a uvolňování účinné látky76), neboť disperze léčiva v prostředí pomocné látky je rozpustnější než samotná krystalická látka4). První zmínka o pevných disperzích se objevila již v roce 196177), avšak od té doby prošly tyto systémy rozsáhlým vývojem, na jehož základě je lze rozdělit do čtyř generací (obr. 4).
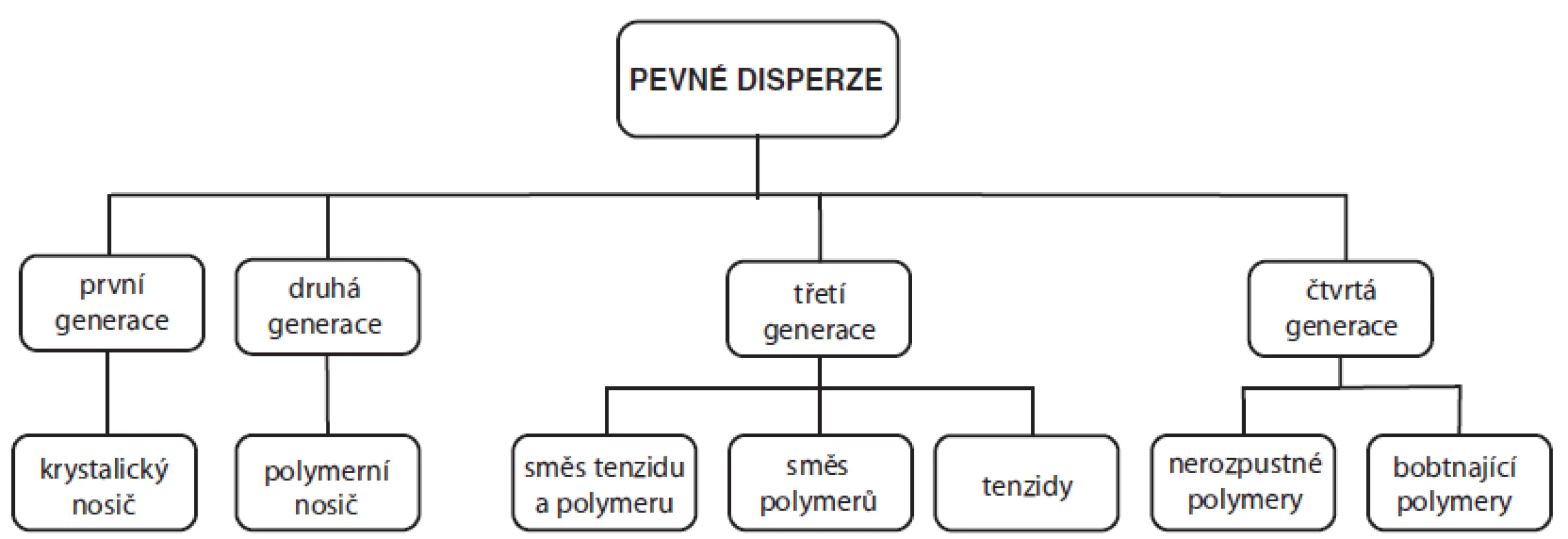
První generace pevných disperzí používala jako vhodné hydrofilní nosiče krystalické látky, jako je např. močovina78) nebo cukry79). Jejich nevýhodou však bylo formování krystalických pevných disperzí, které byly termodynamicky stálé, a proto neuvolňovaly léčivo tak rychle jako disperze amorfní76). Z tohoto důvodu vznikla druhá generace pevných disperzí obsahující amorfní nosiče, kterými jsou nejčastěji polymery. Tyto nosné polymery je pak možné rozdělit na základě jejich původu na syntetické, kam se řadí např. povidon80), krospovidon81), pevné makrogoly82) a polymethakryláty83), a na polosyntetické, které zahrnují deriváty celulosy (např. hypromelosa84) a hyprolosa85)) a deriváty škrobu (cyklodextriny86)). Tyto amorfní pevné disperze je pak možné rozdělit na základě molekulárních interakcí mezi léčivem a nosičem na homogenní disperze (tzv. tuhé roztoky, popř. pevné roztoky), heterogenní disperze (tzv. tuhé suspenze, popř. pevné suspenze) a jejich směsi4, 76). V případě homogenních disperzí vytvoří léčivo s nosičem molekulární, popřípadě iontovou disperzi, zatímco v heterogenní disperzi zůstane léčivá látka oddělena v tuhém stavu4).
Nedávno bylo prokázáno, že uvolňování léčiva z lékové formy může být urychleno, pokud je nosič povrchově aktivní nebo má samoemulgující vlastnosti. Proto se pro přípravu pevných disperzí třetí generace používají povrchově aktivní látky (např. deriváty inulinu (Inutec SP1)87), glycerol-dibehenát (Compritol 888 ATO)88), polyoxylglyceridy (Gelucire 44/14)89), poloxamery (Poloxamer 407)90), Polysorbát 8091) apod.) nebo jejich směsi85, 92), popřípadě směs tenzidů a amorfního polymeru93, 94). Tyto přípravky pak vykazují nejvyšší zlepšení biologické dostupnosti špatně rozpustných léčiv a vysokou stabilitu výsledné disperze bez projevů rekrystalizace léčiva76). Novější literatura pak uvádí i čtvrtou generaci, která zahrnuje pevné disperze s řízeným uvolňováním léčiv. V těchto přípravcích je zvýšená rozpustnost těchto léčiv kombinována s prodloužením jejich uvolňování z finální matrice nejčastěji za použití nerozpustných (ethylcelulosa95), amonioalkylmethakrylátové kopolymery (Eudragit® RL a RS)96), polyethylenoxid97)) nebo bobtnajících polymerů (hyprolosa85), hypromelosa97) a karbomer98))99).
Pevné disperze je možné získat za pomocí metody tavení nebo odpařování rozpouštědla100). Příprava tavením je první volbou v průběhu formulace pevných disperzí101). Léčiva látka je zde nejdříve zahřívaná spolu s nosičem a následně se vzniklá směs ochladí. Chlazení vede k přesycení, avšak vzhledem k souběžně probíhajícímu tuhnutí se léčivo uzavírá do matrice nosiče. Vznik molekulové disperze závisí na stupni přesycení, mísitelnosti nosiče a léčiva v nataveném stavu a rychlosti chlazení. Mletím vzniklé směsi se pak získají částice potřebné velikosti. Důležitým předpokladem pro použití této metody je stabilita účinné látky a nosiče za zvýšené teploty76, 101). Limitujícím krokem může být také nedostatečná mísitelnost natavených látek díky vyšší viskozitě nosiče. Aby se předešlo těmto omezením, začalo se používat několik modifikací metody tavením, mezi něž je možné zařadit extruzi tavenin, aglomeraci tavenin, metodu MeltrexTM (speciální patentovaná technologie využívající dvoušnekového extrudéru a dvou nezávislých násypek, ve kterých se teplota může pohybovat v širokém rozsahu teplot) a řada dalších76, 99, 101).
Při přípravě pevných disperzí metodou odpařování rozpouštědla dojde nejdříve k rozpuštění léčiva společně s nosičem v těkavém, nejčastěji organickém rozpouštědle (chloroform, ethanol, methanol, aceton, směs ethanolu a dichlormethanu aj.). V některých případech může být polymer použit také ve formě suspenze namísto roztoku. Rozpouštědlo je pak odstraněno (ve vakuové odparce, sprejovým sušením, lyofilizací) za vzniku pevného roztoku, který je následně upraven mletím na požadovanou velikost částic. Vzhledem k toxicitě řady organických rozpouštědel je důležité odstranit rozpouštědlo z finálního přípravku76, 99, 101). Dalšími nevýhodami této metody jsou vysoké náklady a fakt, že i malá změna podmínek odpařování rozpouštědla může vést k výrazným změnám ve vlastnostech finálního produktu76).
Hlavní výhodou pevných disperzí je zvýšená rozpustnost a biologická dostupnost léčiv špatně rozpustných ve vodě redukcí jejich velikosti částic, zvýšením pórovitosti, zlepšením smáčivosti a polymorfními změnami léčiva. Interakce mezi nosičem a léčivem navíc zabraňují aglomerací částic léčivé látky, která je z lékové formy uvolňována v přesyceném stavu, což je výhodné pro její rychlou absorpci. Pevné disperze jsou také jednoduše připravitelné a navíc mohou být formulovány do podoby běžných pevných lékových forem, které jsou oblíbené u pacientů. Kromě nevýhod spojených s metodou přípravy, které jsou popsány výše, vykazují tyto přípravky také zhoršenou stabilitu díky rekrystalizaci léčiva z jeho amorfní podoby v průběhu skladování, což může vést ke snížení jeho biologické dostupnosti. Dále může docházet k precipitaci léčiva v disolučním médiu, avšak vzniklé částice obvykle nepřekračují velikost 1 μm99).
V současné době je na českém trhu dostupných několik přípravků formulovaných metodou pevných disperzí. Příkladem mohou být tablety Certican (everolimus), Intelence (etravirin) nebo Crestor (rosuvastatin)53, 99).
Interaktivní práškové směsi
Pojem interaktivní prášková směs popisuje soustavu skládající se ze dvou monodisperzních složek: mikronizované léčivé látky a volně se sypajícího plniva (plniva s dobrými až výbornými tokovými vlastnostmi) ve vzájemné interakci. Jejich hlavním využitím ve farmaceutické technologii je zlepšení přesnosti dávkování malých množství léčivých látek především v práškových inhalátorech a u přípravy tablet přímým lisováním4, 102). Od roku 1980 jsou pak interaktivní směsi testovány také z hlediska zvýšení míry rozpustnosti léčiva inkorporovaného do podoby interaktivní práškové směsi103). Dodáním energie interaktivním mícháním se mění krystalický povrch léčiva na amorfní charakterizovaný vyšší rozpustností ve vodě. Tato změna je ovšem dočasná, jelikož amorfní podíl postupně uvolňuje nabytou energii a vrací se do původního uspořádaného stavu102). Navíc navázáním mikronizovaného léčiva na povrch hydrofilního nosiče dochází ke zvětšení styčné plochy léčivé látky s kapalinou, čímž dojde ke zrychlení rozpouštění a případně zvýšení rozpustnosti léčiva4).
Principem této metody je dlouhodobé mísení směsi malého množství léčiva s pomocnými látkami řádově většího rozměru částic (nosič)4). Nejčastěji používaným nosičem pro jejich přípravu je monohydrát α-laktosy, avšak je možné použít také např. manitol, trehalosu, sorbitol, chlorid sodný aj.102, 104). Vznik interaktivních směsí je závislý na velikosti kohezní síly mezi částicemi léčiva a adhezní síly mezi léčivou látkou a nosičem. V případě, že je adhezivní síla malá dochází ke vzniku směsi se zbytky aglomerovaného léčiva, naopak pokud je příliš velká hrozí zejména u přípravků určených k inhalaci, že nedojde k uvolnění mikronizovaného léčiva a celá směs ulpí v horních cestách dýchacích104).
Ve studii Thiela a Sberna105) se připravila interaktivní prášková směs z mikronizované kyseliny salicylové (velikost částic 2–5 μm) a nosiče, kterým byla v tomto případě sprejově sušená laktosa. Díky přípravě interaktivní práškové směsi se dosáhlo homogenního rozložení kyseliny salicylové, která zároveň vykazovala vysokou míru adheze na použitý nosič (90–95 %).
Samoemulgující systémy
Samoemulgující systémy se řadí mezi tzv. lipofilní formulace a jsou definovány jako izotropní směsi léčiva, olejů, povrchově aktivních látek a v některých případech také hydrofilních kosolventů nebo dalších emulgátorů. Tyto systémy se vyznačují především schopností po podání do gastrointestinálního traktu, respektive po mírném promísení s trávicími tekutinami (vodnou fází), samovolně tvořit jemné emulze (popřípadě mikroemulze, či nanoemulze) typu olej ve vodě (o/v)106–109). Velikost částic vzniklé emulze souvisí s typem samoemulgujícího systému, který vznikne. Pokud vzniká samoemulgující systém vzniká „jemná emulze“ s velikosti vzniklých kapek 0,25–5 μm, u samomikroemulgujích systémů vzniká „mikroemulze“ s velikostí kapek mezi 100 a 250 nm a u samonanoemulgujících systémů „nanoemulze“ s velikostí kapek pod 100 nm. Tato terminologie používaná v odborné literatuře mezi autory není zcela přesná (už u samoemulgujících systémů vzniká emulze s kapkami vnitřní fáze o velikosti μm, tedy mikroemulze, zatímco u samomikroemulgujících je velikost kapek řádově v nanometrech, a je tedy možné hovořit o nanoemulzi) ani jednotná.
Tato emulze vzniklá v trávicím traktu je termodynamicky stálá vzhledem k relativně malému objemu dispergované olejové fáze, úzkému rozmezí distribuce velikosti vzniklých kapek a jejich polaritě110). Malé olejové kapky (< 5 μm) poskytují velký povrch dostupný pro pankreatickou lipázu, která hydrolyzuje triglyceridy, a tím napomáhá k rychlejšímu uvolnění léčiva, a/nebo pro tvorbu micel žlučových solí a léčiva111). Tyto micely po průchodu přes enterocyty (transcelulární přestup) tvoří chylomikrony, které uvolňují léčivo do lymfatického oběhu namísto do oběhu krevního, čímž je omezena metabolizace léčiva v játrech (tzv. „first-pass efekt“) a následně dochází ke zvýšení jeho biologické dostupnosti po perorálním podání112).
Samoemulgující formulace je možné na základě vlastností vzniklé emulze rozdělit na:
- samoemulgující systémy (self-emulsifying drug delivery systems, SEDDS), což jsou izotropní směsi léčiva, oleje, emulgátoru akoemulgátoru, které po mírném promísení s vodnou fází tvoří emulzi o/v s velikosti kapek v rozmezí 0,25–5 μm113,114).
- samomikroemulgující systémy (self-microemulsifying drug delivery systems, SMEDDS), které se od samoemulgujících systémů liší velikosti vzniklých kapek, která se v případě SMEDDS pohybuje mezi 100 a250 nm113).
- samonanoemulgující systémy (self-nanoemulssifying drug delivery systems, SNEDDS), jejichž velikost částic je menší než 100 nm. Velmi malá velikost kapek vnitřní fáze zaručuje těmto systémům vysoce účinnou absorpci olejové fáze s léčivem113, 115).
- samodvojemulgující systémy (self-double-emulsifying drug delivery systems, SDEDDS) jsou nové formulace objevené Qi et al.116), které mohou samovolně po smísení s vodnou fází tvořit dvojité emulze typu voda v oleji ve vodě (v/o/v). Samodvojemulgující systémy jsou vhodné především pro hydrofilní léčiva, která jsou uzavřena ve vnitřní vodné fázi vzniklé dvojité emulze, aproto je u nich vysoký předpoklad navýšení biologické dostupnosti léčiv s vysokou rozpustností a špatnou prostupností (III. třída BCS)116).
- samoemulgující fosfolipidové suspenze (self-emulsifying phospholipid suspensions, SEPS) jsou samoemulgující formulace obsahující vysoké množství fosfolipidů a relativně malá množství emulgátorů/koemulgátorů117).
Výše uvedená terminologie samoemulgujících formulací, především pak samomikroemulgujících systémů a samonanoemulgujících systémů, však není vždy jednotná. Označení „samonanoemulgující systémy“ je používáno řadou zahraničních autorů pro prekoncentráty, které vedou ke vzniku jak mikroemulzí, tak i nanoemulzí. Zatímco druhá skupina používá termín SNEEDS pouze v případě, že získaná disperze je obecně nanoemulze118).
Samoemulgující formulace jsou obvykle připravovány jako kapaliny nebo polotuhé přípravky, kde je léčivá látka rozpuštěna ve směsi pomocných látek (oleje, emulgátoru apod.). Získaná kapalina (popř. polotuhý přípravek) je následně plněna do měkkých, či speciálně uzavřených tvrdých tobolek. Tato podoba SEDDS má řadu nevýhod, jako jsou např. vyšší výrobní náklady, nízká stabilita, možné inkompatibility mezi složkami systémů a pomocnými látkami obalu, možnost nevratného vysrážení léčiva nebo pomocných látek a omezené možnosti podoby finální lékové formy50, 107, 114). Některé z těchto nevýhod mohou být odstraněny převedením SEDDS do pevné podoby za vzniku tzv. pevných samoemulgujících systémů. K formulaci pevných SEDDS je používaná celá řada metod, mezi něž patří např. sprejové sušení50, 119), extruze/sferonizace120, 121) nebo příprava systémů kapalina v pevné fázi122).
Jednu z nejdůležitějších skupin pomocných látek pro přípravu SEDDS představují oleje, a to nejen proto, že se v nich rozpouští léčivo nebo usnadňují samoemulgaci, ale především proto, že navyšují míru absorpce léčiva z gastrointestinálního traktu106, 113, 123). Zvýšená absorpce díky přítomnosti olejové složky může být vysvětlena několika fyziologickými mechanismy, jako je např. změna motility žaludku a střev, zvýšená produkce žluči, která díky přítomnosti žlučových solí usnadňuje rozpouštění léčiva, zvýšení propustnosti sliznice a zvýšená lymfatická absorpce124). Mezi používaná olejová rozpouštědla se řadí rostlinné oleje (bavlníkový, kukuřičný, ricinový, slunečnicový, arašídový, sezamový, sójový)125–127), střední nasycené triacylglyceroly (Labrafac®)128), makrogol-6-glyceridy (Labrafil®)119) a řada dalších.
Další skupinou pomocných látek jsou emulgátory, jejichž optimální hodnota hydrofilně-lipofilní rovnováhy pro formulaci SEDDS by se měla pohybovat okolo 10129) a jejich obvyklá koncentrace v přípravku je v rozmezí 30–60 %109, 123). Hlavním kritériem pro výběr vhodného emulgátoru je však jeho bezpečnost. Emulgátory přírodního původu jsou obecně považovány za bezpečnější než syntetické, avšak jejich samoemulgující kapacita je jen omezená. Neiontové emulgátory jsou méně toxické než iontové, avšak jejich použití může vést k nevratným změnám v permeabilitě střevní stěny106, 109). Stejně jako oleje i emulgátory se podílejí na zvyšování biologické dostupnosti několika mechanismy včetně zlepšení rozpustnosti léčiva, zvýšeni propustnosti střevní membrány a zvýšení permeability těsných spojů (tight junctions)111, 119). K nejběžněji používaným emulgátorům patří Polysorbáty130), makrogol-8-glyceridy (Labrasol®)131, 132), Labrafac® 111), Cremophor® 133), diethylenglykolmonoethylether (Transcutol®)132) a Gelucire® 127) atd.
K navýšení rozpustnosti léčiva mohou být do samoemulgujících systému přidány také kosolventy ze skupiny organických rozpouštědel vhodných pro perorální podání (ethanol126, 127), propylenglykol127, 133), kapalné makrogoly127, 133) apod.). Alkoholy a těkavá rozpouštědla se používají pro přípravu SEDDS jen výjimečně z důvodu jejich odpařování z formulace a následnému vysrážení léčiva v přípravku106, 109, 113).
Hlavní výhodou samoemulgujících systémů je zvýšení biologické dostupnosti lipofilních léčivých látek, což umožňuje snížení podávané dávky léčiva. K dalším výhodám pak patří snížení vlivu potravy na rychlost a množství uvolněné účinné látky, ochrana léčiva před agresivním prostředím žaludku, konstantní rychlost vstřebávání a možnost formulace samoemulgujících lékových forem s řízeným uvolňováním123, 124, 130, 134–136). Nevýhodou je problematické zapracování léčiv, která jsou špatně rozpustná jak v hydrofilních, tak i lipofilních rozpouštědlech. Tyto účinné látky pak vyžadují vyšší množství emulgátorů, což představuje zvýšené riziko vysrážení léčiva nebo jeho rekrystalizace během skladování a možné podráždění trávicího traktu117).
Příkladem samoemulgujících systémů dostupných na českém trhu mohou být přípravky Sandimmun (cyklosporin A), Vesanoid (tretionin), Aptivus (tipranavir) a samomikroemulgující systém Sandimmun Neoral (cyklosporin A)53, 137).
Další metody vedoucí ke zvýšení biologické dostupnosti
Systémy kapalina v pevné fázi
Systémy kapalina v pevné fázi, často také označovány na základě jejich anglického názvu jako liquisolid systémy (LSS), představují moderní formulace schopné zvyšovat biologickou dostupnost špatně rozpustných léčiv. Principem jejich přípravy je nasorbování (prosté smísení, nástřik) léčiva v kapalné fázi na porézní nosič, který je následně obalen materiálem s velkým měrným povrchem částic, za vzniku suchého nepřilnavého prášku138). Modernost liquisolid systémů spočívá především v jejich schopnosti zlepšovat biologickou dostupnost léčivých látek zapracovaných v kapalné podobě. Léčivo je tak v tabletě/prášku/granulátu přítomno v již rozpuštěné podobě a ihned dostupné pro absorpci v gastrointestinálním traktu. Liquisolid systémy tak spojují výhody kapalných a pevných lékových forem, jako je např. rychlá dostupnost léčiva a dobrá stabilita přípravku. Podrobněji je tato perspektivní metoda vedoucí ke zvýšení biologické dostupnosti špatně rozpustných léčivých látek zpracována v předchozích publikacích137, 139, 140).
Volba fyzikální formy
Rozpustnost (rychlost rozpouštění) účinné látky lze modifikovat i volbou její fyzikální formy. V případě, že má léčivo schopnost krystalovat ve více krystalových strukturách (polymorfie), potom se jeho polymorfy budou lišit svými vlastnostmi a tedy i mírou rozpustnosti, respektive biologické dostupnosti. Rozpustnost je také ovlivněna krystalickým nebo amorfním stavem léčivé látky, přičemž obecně platí, že amorfy jsou lépe rozpustné než krystalické formy3, 141).
Vyšší rozpustnost amorfní podoby potvrzuje studie provedená Hancockem a Parksem142). Jako modelová léčiva se použily antiflogistikum indomethacin, antidiabetikum glibenklamid, antimykotikum griseofulvin a diuretika hydrochlorothiazid a polythiazid. U všech léčivých látek bylo pozorováno, že amorfní forma je více rozpustná než forma krystalická.
Nanosuspenze
Příprava nanosuspenzí se uplatňuje především u léčivých látek s nízkou rozpustností ve vodě i v olejích. Jedná se o dvoufázové systémy obsahující léčivo v nanonizované podobě stabilizováno povrchově aktivními látkami a/nebo polymery. Velikost jednotlivých částic je menší než 1 μm s průměrnou hodnotou v rozmezí 200–600 nm143). Jejich hlavní výhody spočívají v dosažení rychlého nástupu účinku, snížení vlivu potravy na rychlost vstřebávání, zlepšení rozpustnosti a biologické dostupnosti144).
Nanosuspenze mohou být připraveny metodou srážení145), kdy je k roztoku léčiva přidáno anti-rozpouštědlo za vysrážení nanokrystalů léčivé látky. Výhodou této metody je její jednoduchost a nízká cena, nevýhodou je požadavek na dobrou rozpustnost léčiva alespoň v jednom rozpouštědle, které je mísitelné s anti-rozpouštědlem143). Další metodou přípravy je vlhké mletí v perlových mlýnech146), kdy je léčivo při velmi vysokých otáčkách mleto spolu s perlami (skleněné, zirkonium oxidové), vodou a stabilizátorem po dobu několika dní (2–7 dní). Třetí metodou přípravy je homogenizace za zvýšeného tlaku147), v průběhu které je suspenze léčiva a povrchově aktivní látky hnána za zvýšeného tlaku přes ventil homogenizátoru s otvorem o velikosti v řádech nanometrů143).
Příkladem navýšení rozpustnosti, respektive biologické dostupnosti mohou být nanosuspenze spasmolytika tarazepidu148), antiflogistika ibuprofenu149) nebo naproxenu144).
Mikrogranulace
Mikrogranulací léčiva dochází k navýšení styčného povrchu mezi léčivou látkou a tekutinami gastrointestinálního traktu, čímž dochází ke zvýšení rozpustnosti a rychlosti uvolňování léčiva z lékové formy. Mikrogranuláty jsou tvořeny směsí pomocných a účinných látek, které jsou speciálními postupy upraveny do formy mikrogranulí, jejichž velikost je zpravidla méně než 200 μm4, 150).
Mikrogranuláty mohou být připraveny několika postupy, mezi něž je možné zařadit vysokoobrátkovou agregační granulaci, fluidní rotogranulaci, termoplastickou granulaci, fluidní nástřik na mikročástice nebo novější techniku impregnace porézní směsi pomocných látek. Další alternativou je pak úprava velikosti částic u granulátu, který byl připraven jednou z klasických metod granulace4).
Pevné lipidické nanočástice
Další metodou vedoucí ke zvýšení rozpustnosti, respektive biologické dostupnosti léčivých látek špatně rozpustných ve vodě je příprava pevných lipidických nanočástic (SLN – solid lipid nanoparticles). Jedná se o částice o velikosti 50–1000 nm, které jsou složeny z fyziologicky dobře tolerovaných lipidů. SLN spojují výhody klasických nanočástic, olejových emulzí a liposomů – jsou netoxické a mohou být použity jak pro zlepšení biologické dostupnosti, tak i pro řízené nebo místně specifické uvolňování léčiv151).
Müller et al. porovnávali biologickou dostupnost cyklosporinu A podaného v podobě pevných lipidických nanočástic a nanokrystalů. Výsledky studie ukázaly, že cyklosporin v podobě SLN vykazuje nižší variabilitu biologické dostupnosti v porovnání s nanokrystaly a komerčně dostupnými přípravky (Sandimmun)152).
Liposomy
Liposomy jsou měchýřkovité útvary tvořené fosfolipidy o velikosti 0,6–2,0 μm s tloušťkou membrány 5 nm (obr. 5). Nejčastěji jsou liposomy využívány jako nosičové systémy s řízeným uvolňováním hydrofilních léčiv, avšak díky jejich bifázické struktuře mohou být použity ke zvýšení rozpustnosti, respektive biologické dostupnosti léčiv lipofilní povahy. Inkorporace špatně rozpustných účinných látek do struktury liposomu je však závislá nejen na fyzikálně-chemických vlastnostech léčiva, ale také na složení lipidové membrány samotného liposomu153).

Příkladem jejich použití pro zlepšení biologické dostupnosti špatně rozpustných léčivých látek mohou být studie s antiflogistikem ibuprofenem153) nebo hypolipidemikem felodipinem154).
Enhancery absorpce
Kromě výše uvedených metod, které se zaměřují především na navýšení rozpustnosti dané léčivé látky, spadá pod postupy vedoucí ke zlepšení biologické dostupnosti také použití enhancerů střevní absorpce. Jejich přidání do finální lékové formy je výhodné především u léčivých látek s nízkou prostupností přes membrány gastrointestinálního traktu (III. a IV. třída dle Biofarmaceutického klasifikačního systému), mezi něž patří mimo jiné amoxicilin, atenolol, kodein, metformin, pravastatin apod.155, 156). Mezi enhancery střevní absorpce je možné zařadit např. alkylmaltosidy157), chitosan a jeho deriváty158), polyoxyethylen cetylether v kombinaci s kyselinou olejovou159) aj.
Závěr
Biologická dostupnost léčiva je závislá na řadě faktorů, mezi něž je možné zařadit fyzikálně-chemické vlastnosti léčivé látky, fyziologické a patologické aspekty organismu, konzumaci potravy a nápojů, intra - a interindividuální variabilitu mezi jedinci a v neposlední řadě také typ lékové formy. Odhaduje se, že až 40 % běžně používaných léčivých látek vykazuje nízkou rozpustnost ve vodě, a s tím související nedostatečnou biologickou dostupnost po perorálním podání. Z tohoto důvodu se postupy vedoucí k přípravě lékových forem zajišťujících zvýšenou rozpustnost, respektive biologickou dostupnost účinné látky staly jedněmi z hlavních směrů moderní farmaceutické technologie.
Střet zájmů: žádný.
Došlo 2. září 2015
Přijato 12. října 2015
doc. PharmDr. Jan Gajdziok, Ph.D., B. Vraníková
Ústav technologie léků FaF VFU
Palackého třída 1–3, 612 42 Brno
e-mail: gajdziokj@vfu.cz
Zdroje
1. Yazdanian M., Briggs K., Jankovsky C., Hawi A. The „high solubility“ definition of the current FDA guidance on biopharmaceutical classification system may be too strict for acidic drugs. Pharm. Res. 2004; 21, 293–299.
2. Yu L. X., Amidon G. L., Polli J. E., Zhao H., Mehta M. U., Conner D. P., Shah V. P., Lesko L. J., Chen M., Lee V. H. L., Hussain A. S. Biopharmaceutics Classification System: The Scientific Basis for Biowaiver Extensions. Pharm. Res. 2002; 19, 921–925.
3. Okáčová L., Vetchý D., Franc A., Rabišková M., Kratochvíl B. Zvýšení biodostupnosti těžce rozpustných léčivých látek jejich modifikací. Chem. Listy 2010; 104, 21–26.
4. Okáčová L., Vetchý D., Franc A, Rabišková M. Zvýšení biodostupnosti těžce rozpustných léčivých látek technologickými postupy usnadňujícími jejich rozpouštění. Chem. Listy 2011; 105, 34–40.
5. Rowe R. C., Sheskey P. J., Owen S. C. Handbook of Pharmaceutical Excipients. 5. vyd. London: Pharmaceutical Press 2006.
6. Kawakami K., Oda N., Miyoshi K., Funaki T., Ida Y. Solubilization behavior of a poorly soluble drug under combined use of surfactants and cosolvents. Eur. J. Pharm. Sci. 2006; 28, 7–14.
7. Komárek P., Rabišková M. Technologie léků. 3. přepracované a doplněné vydání. Praha: Galén 2006.
8. Seedher N., Kanojia M. Co-solvent solubilization of some poorly-soluble antidiabetic drugs. Pharm. Dev. Technol. 2009; 14, 185–192.
9. Sonali J., Kamaldeep Y., Bhumika S., Sanjay J., Kumar M. R. Hydrotropy: A novel approach in estimation of poorly aqueous soluble drugs by TLC. International Journal of Pharmacy and Pharmaceutical Sciences 2013; 5, 176–178.
10. Srinivas V., Rodley G. A., Ravikumar K., Robinson W. T., Turnbull M. M., Balasubramanian D. Molecular organization in hydrotrope assemblies. Langmuir 1997; 13, 3235–3239.
11. Janakiraman B., Sharma M. M. Enhancing rates of multiphase reactions through hydrotropy. Chem. Eng. Sci. 1985; 40, 2156–2158.
12. Patil A. E., Devtalu S. V., Bari M. M., Barhate S. D. A review on: Novel solubility enhancement technique hydrotropy. Indo American Journal of Pharmaceutical Research. 2013; 3, 4670–4679.
13. Lee J., Lee S. C., Acharya G., Chang C. J., Park K. Hydrotropic solubilization of paclitaxel: analysis of chemical structures for hydrotropic property. Pharm. Res. 2003; 20, 1022–1030.
14. Rangel-Yagui C. O., Pessoa A. Jr., Travares L. C. Micellar solubilization of drugs. J. Pharm. Pharm. Sci. 2005; 8, 147–165.
15. Sikarra D., Shukla V., Kharia A. A., Chatterjee D. P. Enhancement of poorly soluble drugs: An overview. Journal of Medical Pharmaceutical and Allied Sciences. 2012; 1, 1–22.
16. Atanacković M., Posa M., Heinle H., Gojković-Bukarica L., Cvejić J. Solubilization of resveratrol in micellar solutions of different bile acids. Colloids Surf. B. Biointerfaces 2009; 72, 148–154.
17. Rangel-Yagui C. O., Hsu H. W. L., Pessoa A. Jr., Travares L. C. Micellar solubilization of ibuprofen – influence of surfactant head groups on the extent of solubilization. Rev. Bras. Cienc. Farm. 2005; 41, 237–246.
18. Hagan S. A., Coombes A. G. A., Garnett M. C., Dunn S. E., Davies M. C., Illum L., Davis S. S. Polylactide-poly(ethylene glycol) copolymers as drug delivery systems. 1. Characterization of water dispersible micelle-forming systems. Langmuir 1996; 12, 2153–2161.
19. Blondino F. E., Byron P. R. Surfactant dissolution and water solubilization in chlorine-free liquifield gas propellants. Drug Dev. Ind. Pharm. 1998; 24, 935–945.
20. Vraníková B., Gakdziok J. Biologická dostupnost léčiva a možnosti jejího ovlivňování. Ces. slov. Farm. 2015; 64, 7–13.
21. Dvořáčková K. Principy uvolňování léčiv z perorálních matricových tablet obsahujících hypromelosu. Chem. Listy 2009; 103, 66–72.
22. Lindahl A., Ungell A. L., Knutson L., Lennernäs H. Characterization of fluids from the stomach and proximal jejunum in men and women. Pharm. Res. 1997; 14, 497–502.
23. Zhou D., Qui Y. Oral absorption and the biopharmaceutics classification systém. Journal of Validation Technology 2009; 15, 62–72.
24. Fallingborg J. Intraluminal pH of the human gastrointestinal tract. Dan. Med. Bull. 1999; 46, 183–196.
25. Amaral M. H., Lobo J. M. S., Ferreira D. C. Effect of hydroxypropyl methylcellulose and hydrogenated castor oil on naproxen release from sustained-release tablets. AAPS PharmSciTech. 2001; 2, 14–21.
26. Jinno J., Kamada N., Miyake M., Yamada K., Mikai T., Odomi M., Toguchi H., Liversidge G. G., Higaki K., Kimura T. Effect of particle size reduction on dissolution and oral absorption of poorly water-soluble drug, cilostazol, in beagle dogs. J. Control. Release 2006; 111, 56–64.
27. Vandana K. R., Raju Y. P., Chowdary V. H., Sushma M., Kumar V. N. An overview on in situ micronization technique – An emerging novel concept in advanced drug delivery. Saudi. Pharm. J. 2014; 22, 283–289.
28. Serrano D. R., Gallagher K. H., Healy A. M. Emerging nanonisation technologies: tailoring crystalline versus amorphous nanomaterials. Cur. Top. Med. Chem. 2015; 15, 2327–2340.
29. Rasenack N., Müller B. W. Micron-size drug particles: common and novel micronization techniques. Pharm. Dev. Technol. 2004; 9, 1–13.
30. Han X., Ghoroi C., To D., Chen Y., Davé R. Simultaneous micronization and surface modification for improvement of flow and dissolution of drug particles. Int. J. Pharm. 2011; 415, 185–195.
31. Merisko-Liversidge E., Sarpotdar P., Bruno J., Hajj S., Wei L., Peltier N., Rake J., Shaw J. M., Pugh S., Polin L., Jones J., Corbett T., Cooper E., Liversifge G. G. Formultion and antitumor activity evaluation of nanocrystalline suspensions of poorly soluble anticancer drugs. Pharm. Res. 1996; 13, 272–278.
32. Tao J., Sun Y., Zhang G. G., Yu L. Solubility of small-molecule crystals in polymers: D-mannitol in PVP, indomethacin in PVP/VA, and nifedipine in PVP/VA. Pharm. Res. 2009; 26, 855–864.
33. Steckel H., Rasenack N., Müller B. W. In-situ-micronization of disodium cromoglycate for pulmonary delivery. Eur. J. Pharm. Biopharm. 2003; 55, 173–180.
34. Rasenack N., Steckel H., Müller B. W. Micronization of anti.inflamatory drugs for pulmonary delivery by a controlled crystallization process. J. Pharm. Sci. 2003; 92, 35–44.
35. Steckel H., Rasenack N., Villax P., Müller B. W. In vitro characterization of jet-milled and in-situ-micronized fluticasone-17 - -propionate. Int. J. Pharm. 2003; 258, 65–75.
36. Bajerová M., Gajdziok J., Dvořáčková K., Masteiková R., Kollár P. Polosyntetické deriváty celulosy jako základ hydrofilních gelových systémů. Čes. slov. Farm. 2008; 57, 63–69.
37. Rasenack N., Müller B. W. Dissolution rate enhancement by in situ micronization of poorly water-soluble drugs. Pharm. Res. 2002; 19, 1894–1900.
38. Kim Y. H., Shing K. S. Supercritical fluid-micronized ipratropium bromide for pulmonary drug delivery. Powder Technol. 2008; 182, 25–32.
39. Yildiz N., Tuna ŞŞ., Döker O, Çalimli A. Micronization of salicylic acid and taxol (paclitaxel) by rapid expansion of supercritical fluids (RESS). The Journal of Supercritical Fluids. 2007; 41, 440–451.
40. Zhiyi L., Jingzhi J., Xuewu L., Huihua T., Wei W. Experimental investigation on the micronization of aqueous cefadroxil by supercritical fluid technology. The Journal of Supercritical Fluids 2009; 48, 247–252.
41. Perrut M., Jung J., Leboeuf F. Enhancement of dissolution rate of poorly-soluble active ingredients by supercritical fluid processes. Part I: Micronization of neat particles. Int. J. Pharm. 2005; 288, 3–10.
42. Jung J., Perrut M. Particle design using supercritical fluids: Literature and patent survey. The Journal of Supercritical Fluids 2001; 20, 179–219.
43. Türk M., Hils P., Helfgen B., Schaber K., Martin H. J., Wahl M. A. Micronization of pharmaceutical substances by the Rapid Expansion of Supercritical Solutions (RESS): a promising method to improve bioavailability of poorly soluble pharmaceutical agents. The Journal of Supercritical Fluids. 2002; 22, 75–84.
44. Boonnoun P., Nerome H., Machmudah S., Goto M., Shotipruk A. Supercritical anti-solvent micronization of marigold-derived lutein dissolved in dichloromethane and ethanol. The Journal of Supercritical Fluids. 2013; 77, 103–109.
45. Charoenchaitrakool M., Dehghani F., Foster N. R. Micronization by rapid expansion of supercritical solutions to enhance the dissolution rates of poorly water-soluble pharmaceuticals. Ind. Eng. Chem. Res. 2000; 39, 4794–4802.
46. Young T. J., Mawson S., Johnston K. P., Henriksen I. B., Pace G. W., Mishra A. K. Rapid expansion from supercritical to aqueous solution to produce submicron suspensions of water-insoluble drugs. Biotechnol. Prog. 2000; 16, 402–407.
47. Won D. H., Kim M. S., Lee S., Park J. S., Hwang S. J. Improved physicochemical characteristics of felodipine solid dispersion particles by supercritical anti-solvent precipitation process. Int. J. Pharm. 2005; 301, 199–208.
48. Zhang H. X., Wang J. X., Zhang Z. B., Le Y., Shen Z. G., Chen J. F. Micronization of atorvastatin calcium by antisolvent precipitation process. Int. J. Pharm. 2009; 374, 106–113.
49. Steckel H., Brandes H. G. A novel spray-drying technique to produce low density particles for pulmonary delivery. Int. J. Pharm. 2004; 278, 187–195.
50. Yi T., Wan J., Xu H., Yang X. A new solid self-microemulsifying formulation prepared by spray-drying to improve the oral bioavailability of poorly water soluble drugs. Eur. J. Pharm. Biopharm. 2008; 70, 439–444.
51. Dollo G., Le Corre P., Guérin A., Chevanne F., Burgot J. L., Leverge R. Spray-dried redispersible oil-in-water emulsion to improve oral bioavailability of poorly soluble drugs. Eur. J. Pharm. Sci. 2003; 19, 273–280.
52. Kim J. S., Kim M. S., Park H. J., Jin S. J., Lee S., Hwang S. J. Physicochemical properties and oral bioavailability of amorphous atorvastatin hemi-calcium using spray-drying and SAS process. Int. J. Pharm. 2008; 359, 211–219.
53. SÚKL – Státní ústav pro kontrolu léčiv. www.sukl.cz (13. 7. 2015)
54. Rabišková M., Vetchý D. Orálně dispergovatelné tablety. Praktické lékárenství 2007; 4, 181–183.
55. Nireesha G. R., Divya L., Sowmya C., Venkateshan N., Babu M. N., Lavakumar V. Lyophilization/Freeze drying – An review. International Journal of Novel Trends in Pharmaceutical Sciences 2013; 3, 87–98.
56. Tang X., Pikal M. J. Design of freeze-drying processes for pharmaceuticals: Practical advice. Pharm. Res. 2004; 21, 191–200.
57. Kasper J. C., Friess W. The freezing step in lyophilization: physico-chemical fundamentals, freezing methods and consequences on process performance and quality attributes of biopharmaceuticals. Eur. J. Pharm. Biopharm. 2011; 78, 248–263.
58. Wang W., Chen M., Chen G. Issues in freeze drying of aquesous solutions. Chinese J. Chem. Eng. 2010; 20, 551–559.
59. Wanning S., Süverkrüp R., Lamprecht A. Pharmaceutical spray freeze drying. Int. J. Pharm. 2015; 488, 136–153.
60. Kasper J. C., Winter G., Friess W. Recent advances and futher challenges in lyophilization. Eur. J. Pharm. Biopharm. 2013; 85, 162–169.
61. Carrier R. L., Miller L. A., Ahmed I. The utility of cyclodextrins for enhancing oral bioavailability. J. Control. Release 2007; 123, 78–99.
62. Challa R., Ahuja A., Ali J., Khar R. K. Cyclodextrins in drug delivery: An updated review. AAPS PharmSciTech.l 2005; 6, 329–357.
63. Uekama K., Otagiri M., Uemura Y., Fujinaga T., Arimori K., Matsuo N., Tasaki K., Sigii A. Improvement of oral bioavailability of prednisolone by beta-cyclodextrin complexation in humans. J. Phamacobiodyn. 1983; 6, 124–127.
64. Ghorab M. K., Adeyeye M. C. Enhanced bioavailability of process-induced fast-dissolving ibuprofen congranulated with beta-cyclodextrin. J. Pharm. Sci. 2003; 92, 1690–1697.
65. Barone J. A., Moskovitz B. L., Guarnieri J., Hassell A. E., Colaizzi J. L., Bierman R. H., Jessen L. Enhanced Bioavailability of itraconazole in hydroxypropyl-β-cyclodextrin solution versus capsules in healthy volunteers. Antimicrob. Agents. Chemother. 1998; 42, 1862–1865.
66. Freedman K. A., Klein J. W., Crosson C. E. Beta-cyclodextrins enhance bioavailability of pilocarpine. Curr. Eye. Res. 1993; 12, 641–647.
67. Miyake K., Arima H., Irie T., Hirayama F., Uekama K. Enhanced absorption of cyclosporine A by complexation with dimethyl-beta-cyclodextrin in bile duct-cannulated and noncannulated rats. Biol. Pharm. Bull. 1999; 22, 66–72.
68. Uekama K. Design and evaluation of yclodextrine-based drug formulation. Chem. Pharm. Bull. 2004; 52, 900–915.
69. Del Valle E. M. M. Cyclodextrins and their uses: a review. Process Biochem. 2004; 39, 1033–1046.
70. Baden-Württemberg. http://www.ua-bw.de/pub/beitrag.asp?subid =0&Thema_ID=3&ID=1242&Pdf=No. (22. 7. 2015).
71. Ghosh A., Biswas S., Ghosh T. Preparation and evaluation of silymarin β-cyclodextrin molecular inclusion complexes. J. Young Pharm. 2011; 3, 205–210.
72. Gowardhane A. P., Kadam N. V., Dutta S. Review on enhancement of solubilization process. Journal of Pharmacy and Phytotherapeutics 2013; 2, 28–38.
73. Wen X., Tan F., Jing Z., Liu Z. Preparation and study the 1 : 2 inclusion complex of carvediol with beta-cyclodextrin. J. Pharm. Biomed. Anal. 2004; 34, 517–523.
74. Loftsson T., Brewster M. E. Pharmaceutical applications of cyclodextrins. 1. Drug solubilization and stabilization. J. Pharm. Sci. 1996; 85, 1017–1025.
75. Kraus T. Cyclodextriny. http://www.uochb.cas.cz/Zpravy/Post Grad2004/8_Kraus.pdf (22. 7. 2015).
76. Vasconcelos T., Sarmento B., Costa P. Solid dispersions as strategy to improve oral bioavailability of poorly water soluble drugs. Drug. Discov. Today 2007; 12, 1068–1075.
77. Sekiguchi K., Obi N. Studies on absorption of eutectic mixture. I. A comparison of the behavior of eutectic mixture of sulfathiazole and that of ordinary sulfathiazole in man. Chem. Pharm. Bull. 1961; 9, 866–872.
78. Ford J. L., Rubinstein M. H. Preparation, properties and ageing of tablets prepared from the chlorpropamide-urea solid dispersion. Int. J. Pharm. 1981; 8, 311–322.
79. Allen L. V., Levinson R. S., Martono D. D. Dissolution rates of hydrocortisone and prednisone utilizing sugar solid dispersion systems in tablet form. J. Pharm. Sci. 1978; 67, 979–981.
80. Patel M. M., Patel D. M. Fast dissolving Valdecoxib tablets containing solid dispersion of Valdecoxib. Ind. J. Pharm. Sci. 2006; 68, 222–226.
81. Dahima R., Pachori A., Netam S. Formulation and evaluation of mouth dissolving tablet containing amlodipine besylate solid dispersion. International Journal of ChemTech Research 2010; 2, 706–715.
82. Law S. L., Lo W. Y., Lin F. M., Chaing C. H. Dissolution and absorption of nifedipine in polyethylene glycol solid dispersion containing phosphatidylcholine. Int. J. Pharm. 1992; 84, 161–166.
83. Nazzal S., Guven N., Reddy I. K., Khan M. A. Preparation and characterization of coenzyme Q10-Eudragit solid dispersion. Drug Dev. Ind. Pharm. 2002; 28, 49–57.
84. Verreck G., Six K., Van den Mooter G., Baert L., Peeters J., Brewster M. E. Characterization of solid dispersions of itroconazole and hydroxypropylmethylcellulose prepared by melt extrusion – Part I. Int. J. Pharm. 2003; 251, 165–174.
85. Kohda Y., Kobayashi H., Baba Y., Yuasa H., Ozeki T., Kanaya Y., Sagara E. Controlled release of lidocaine hydrochloride from buccal mucosa-adhesive films with solid dispersion. Int. J. Pharm. 1997; 158, 147–155.
86. Nagarsenker M. S., Meshram R. N., Ramprakash G. Solid dispersion of hydroxypropyl beta-cyclodextrin and ketorolac: enhancement of in-vitro dissolution rates, improvement in anti-inflammatory activity and reduction in ulcerogenicity in rats. J. Pharm. Pharmacol. 2000; 52, 949–956.
87. van den Mooter G., Weuts I., De Ridder T., Blaton N. Evaluation of Inutec SP1 as a new carrier in the formulation of solid dispersions for poorly soluble drugs. Int. J. Pharm. 2006; 316, 1–6.
88. Jagdale S., Patil S., Kuchekar B., Chabukswar A. Preparation and characterization of metformin hydrochloride – Compritol 888 ATO Solid Dispersion. J. Young. Pharm. 2011; 3, 197–204.
89. Damian F., Blaton N., Naesens L., Balzarini J., Kinget R., Augustijns P., Van den Mooter G. Physicochemical characterization of solid dispersions of the antiviral agent UC-781 with polyethylene glycol 6000 and Gelucire 44/14. Eur. J. Pharm. Sci. 2000; 10, 311–322.
90. Vyas V., Sancheti P., Karekar P., Shah M., Pore Y. Physicochemical characterization of solid dispersion systems of tadalafil with poloxamer 407. Acta Pharm. 2009; 59, 453–461.
91. Dannenfelser R. M., He H., Joshi Y., Bateman S., Serajuddin A. T. Development of clinical dosage forms for a poorly water soluble drug I: Application of polyethylene glycol-polysorbate 80 solid dispersion carrier system. J. Pharm. Sci. 2004; 93, 1165–1175.
92. Yüksel N., Karataş A., Ozkan Y., Savaşer A., Ozkan S. A., Baykara T. Enhanced bioavailability of piroxicam using Gelucire 44/14 and labrasol: in vitro and in vivo evaluation. Eur. J. Pharm. Biopharm. 2003; 53, 453–459.
93. Janssens S., Humbeeck J. V., Van den Mooter G. Evaluation of the formulation of solid dispersions by co-spray drying itraconazole with Inutec SP1, a polymeric surfactant, in combination with PVPPA 64. Eur. J. Pharm. Biopharm. 2008; 70, 500–505.
94. Srinarong P., Hämäläinen S., Visser M. R., Hinrichs W. L., Ketolainen J., Frijlink H. W. Surface-active derivative of inulin (Inutec® SP1) is a superior carrier for solid dispersions with a high drug load. J. Pharm. Sci. 2011; 100, 2333–2342.
95. Ohara T., Kitamura S., Kitagawa T., Terada K. Dissolution mechanism of poorly water-soluble drug from extended release solid dispersion system with ethylcellulose and hydroxypropylmethylcellulose. Int. J. Pharm. 2005; 302, 95–102.
96. Otsuka M., Onoe M., Matsuda Y. Hygroscopic stability and dissolution properties of spray-dried solid dispersions of furosemide with Eudragit. J. Pharm. Sci. 1993; 82, 32–38.
97. Ozeki T., Yuasa H., Kanaya Y. Application of the solid dispersion method to the controlled release of medicine. IX. Difference in the release of flurbiprofen from solid dispersions with poly(ethylene oxide) and hydroxypropylcellulose and the interaction between medicine and polymers. Int. J. Pharm. 1997; 155, 209–217.
98. Ozeki T., Yuasa H, Kanaya Y. Controlled release from solid dispersion composed of poly(ethylene oxide) – Carbopol interpolymer complex with various cross-linking degrees of Carbopol. J. Control. Release. 2000; 63, 287–295.
99. Vo C. L., Park C., Lee B. J. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur. J. Pharm. Biopharm. 2013; 85, 799–813.
100. Serajuddin A. T. Solid dispersion of poorly water-soluble drugs: early promises, subsequent problems, and recent breakthroughs. J. Pharm. Sci. 1999; 88, 1058–1066.
101. Leuner C., Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 2000; 50, 47–60.
102. Zatloukal Z. Interaktivní práškové směsi. Čes. slov. Farm. 2004; 53, 165–171.
103. Allahham A., Stewart P. J. Enhancement of the dissolution of indomethacin in interactive mixtures using added fine lactose. Eur. J. Pharm. Biopharm. 2007; 67, 732–742.
104. Lohrmann M., Kappl M., Butt H. J., Urbanetz N. A., Lippold B. C. Adhesion forces in interactive mixtures for dry powder inhalers – Evaluation of a new measuring method. Eur. J. Pharm. Biopharm. 2007; 67, 579–586.
105. Thiel W. J., Sberna F. J. Fluidized bed film coating of an interactive powder mixture to produce microencapsulated 2–5 μm particles. J. Pharm. Pharmacol. 1986; 38, 166–171.
106. Gursoy R. N., Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed. Pharmacother. 2004; 58, 173–182.
107. Tang B., Cheng G., Gu J. C., Xu C. H. Development of solid self-emulsifying drug delivery systems: preparation techniques and dosage forms. Drug. Discov. Today. 2008; 13, 606–612.
108. Hong J. Y., Kim J. K., Song Y. K., Park J. S., Kim C. K. A new self-emulsifying formulation of itraconazole with improved dissolution and oral absorption. J. Control. Release 2006; 110, 332–338.
109. Gershanik T., Benita S. Self-dispersing lipid formulations for improving oral absorption of lipophilic drugs. Eur. J. Pharm. Biopharm. 2000; 50, 179–188.
110. Matuszewska B., Hettrick L., Bondi J. V., Storey D. E. Comparative bioavailability of L-683,453, a 5-alpha-reductase inhibitor, from a self-emulsifying drug delivery in Beagle dogs. Int. J. Pharm. 1996; 136, 147–154.
111. Kommuru T. R., Gurley B., Khan M. A., Reddy I. K. Self-emulsifying drug delivery systems (SEDDS) of coenzyme Q10: formulation development and bioavailability assessment. Int. J. Pharm. 2001; 212, 233–246.
112. Jain S., Jain A. K. , Pohekar M., Thanki K. Novel self-emulsifying formulation of quercetin for improved in vivo antioxidant potential: implications on drug-induced cardiotoxicity and nephrotoxicicty. Free. Radic. Biol. Med. 2013; 65, 117–130.
113. Kohli K., Chopra S., Dhar D., Arora S., Khar R. K. Self-emulsifying drug delivery systems: an approach to enhance oral bioavailability. Drug. Discov. Today 2010; 15, 958–965.
114. Kallakunta V. R., Bandari S., Jukanti R., Veerareddy P. R. Oral self emulsifying powder of lercanidipine hydrochloride: Formulation and evaluation. Powder. Technol. 2012; 221, 375–382.
115. Rao S. V., Shao J. Self-nanoemulsifying drug delivery systems (SNEDDS) for oral delivery of protein drugs I. Formulation development. Int. J. Pharm. 2008; 362, 2–9.
116. Qi X., Wang L., Zhu J., Hu Z., Zhang J. Self-double-emulsifying drug delivery system (SDEDDS): A new way for oral delivery of drugs with high solubility and low permeability. Int. J. Pharm. 2011; 409, 245–251.
117. Shanmugam S., Park J. H., Kim K. S., Piao Z. Z., Yong C. S., Choi H. G., Woo J. S. Enhanced bioavailability and retinal accumulation of lutein from self-emulsifying phospholipid suspension (SEPS). Int. J. Pharm. 2011; 412, 99–105.
118. Niederquell A., Kuentz M. Proposal of stability categories for nano-dispersions obtained from pharmaceutical self-emulsifying formulations. Int. J. Pharm. 2013; 446, 70–80.
119. Balakrishnan P., Lee B. J., Oh D. H., Kim J. O., Hong M. J., Jee J. P., Kim J. A., Yoo B. K., Woo J. S., Yong C.S., Choi H. G. Enhanced oral bioavailability of dexibuprofen by a novel solid self-emulsifying drug delivery system (SEDDS). Eur. J. Pharm. Biopharm. 2009; 72, 539–545.
120. Abdalla A., Klein S., Mäder K. A new self-emulsifying drug delivery system (SEDDS) for poorly soluble drugs: Characterization, dissolution, in vitro digestion and incorporation into solid pellets. Eur. J. Pharm. Sci. 2008; 35, 457–464.
121. Wang Z., Sun J., Wang Y., Liu X., LiuY., Fu Q., Meng P., He Z. Solid self-emulsifying nitrendipine pellets: Preparation and in vitro/in vivo evaluation. Int. J. Pharm. 2010; 383, 1–6.
122. Zhao X., Zhou Y. Q., Potharaju S., Lou H., Sun H. M., Bruson E., Almoazen H., Johnson J. Development of a self micro-emulsifying tablet of cyclosporine A by the liquisolid compact technique. International Journal of Pharmaceutical Sciences and Research 2011; 2, 2299–2308.
123. Kumar A., Sharma S., Kamble R. Self emulsifying drug delivery system (SEDDS): future aspects. International Journal of Pharmacy and Pharmaceutical Sciences 2010; 2, 7–13.
124. Attama A. A., Nzekwe I. T., Nnamani P. O., Adikwu M. U., Onugu C. O. The use of solid self-emulsifying systems in the delivery of diclofenac. Int. J. Pharm. 2003; 262, 23–28.
125. Singh A. K., Chaurasiya A., Awasthi A., Mishra G., Asati D., Khar R. K., Mukherjee R. Oral bioavailability enhancement of exemestane from self-microemulsifying drug delivery system (SMEDDS). AAPS Pharmscitech 2009; 10, 906–916.
126. Khoo S. M., Humberstone A. J., Porter C. J. H., Edwards G. A., Charman W. N. Formulation design and bioavailability assessment of lipidic self-emulsifying formulations of halofantrine. Int. J. Pharm. 1998; 167, 155–164.
127. Cuiné J. F., McEvoy C. L., Charman W. N., Pouton C. W., Edwards G. A., Benameur H., Porter C. J. Evaluation of the mmpact of surfactant digestion on the bioavailability of danazol after oral administration of lipidic self-emulsifying formulations to dogs. J. Pharm. Sci. 2008; 97, 995–1012.
128. Atef E., Belmonte A. A. Formulation and in vitro and in vivo characterization of a phenytoin self-emulsifying drug delivery system (SEDDS). Eur. J. Pharm. Biopharm. 2008; 35, 257–263.
129. Perlman M. E., Murdande S. B., Gumkowski M. J., Shah T. S., Rodricks C. M., Thornton-Manning J., Freel D., Erhart L. C. Development of a self-emulsifying formulation that reduces the food effect for torcetrapib. Int. J. Pharm. 2008; 351, 15–22.
130. Patil P., Joshi P., Paradkar A. Effect of formulation variables on preparation and evaluation of gelled self-emulsifying drug delivery system (SEDDS) of ketoprofen. AAPS PharmSciTech. 2004; 5, 43–50.
131. Oh D. H., Kang J. H., Kim D. W., Lee B. J., Kim J. O., Yong C. S., Choi H. G. Comparison of solid self-microemulsifying drug delivery system (solid SMEDDS) prepared with hydrophilic and hydrophobic solid carrier. Int. J. Pharm. 2011; 420, 412–418.
132. Kale A. A., Patravale V. B. Design and evaluation of self-emulsifying drug delivery systems (SEDDS) of nimodipine. AAPS PharmSciTech. 2008; 9, 191–196.
133. Kang B. K., Lee J. S., Chon S. K., Jeong S. Y., Yuk S. H., Khang G., Lee H. B., Cho S. H. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int. J. Pharm. 2004; 274, 65–73.
134. Tiwari R., Tiwari G., Rai A. K. Self-emulsifying drug delivery system: An approach to enhance solubility. Systematic Reviews in Pharmacy 2010; 1, 133–140.
135. Craig D. Q. M., Barker S. A., Banning D., Booth S. W. An investigation into the mechanisms of self-emulsification using particle size analysis and low frequency dielectric spectroscopy. Int. J. Pharm. 1995; 114, 103–110.
136. Tran P.H., Tran T. T., Piao Z. Z., Vo T. V., Park J. B., Lim J., Oh K. T., Rhee Y. S., Lee B. J. Physical properties and in vivo bioavailability in human volunteers of isradipine using controlled release matrix tablet containing self-emulsifying solid dispersion. Int. J. Pharm. 2013; 450, 79–86.
137. Vraníková B., Franc A., Gajdziok J. Inovativní lékové formy pro těžce rozpustná léčiva. Remedi 2014; 24, 312–314.
138. Kavitha K., Lova Raju K. N. S., Ganesh N. S., Ramesh B. Effect of dissolution rate by liquisolid compacts approach: An Overview. Der Pharmacia Lettre 2011; 3, 71–83.
139. Gajdziok J., Vraníková B. Zvyšování biologické dostupnosti léčiv pomocí formulace liquisolid systémů. Čes. slov. Farm. 2015; 64, 55–66.
140. Vraníková B., Gajdziok J., Vetchý D., Kratochvíl B., Seilerová L. Systémy kapalina v pevné fázi jako moderní trend zvyšování biologické dostupnosti léčiva. Chem. Listy 2013; 107, 681–687.
141. Pudipeddi M., Serajuddin A. T. Trends in solubility of polymorphs. J. Pharm. Sci. 2005; 94, 929–939.
142. Hancock B. C., Parks M. What is the true solubility advantage for amorphous pharmaceuticals? Pharm. Res. 2000; 17, 397–404.
143. Savjani K. T., Gajjar A. K., Savjani J. K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 2012; 2012, 1–10.
144. Mishra B., Sahoo J., Dixit P. K. Formulation and process optimization of naproxen nanosuspensions stabilized by hydroxy propyl methyl cellulose. Carbohydr. Polym. 2015; 127, 300–308.
145. Xia D., Quan P., Piao H., Piao H., Sun S., Yin Y., Cui F. Preparation of stable nitrendipine nanosuspensions using the precipitation – ultrasonication method for enhancement of dissolution and oral bioavailability. Eur. J. Pharm. Sci. 2010; 40, 325–334.
146. van Eerdenbrugh B., Froyen L., Martens J. A., Blaton N., Augustijns P., Brewster M., van den Mooter G. Characterization of physico-chemical properties and pharmaceutical performance of sucrose co-freeze-dried solid nanoparticulate powders of the anti-HIV agent loviride prepared by media milling. Int. J. Pharm. 2007; 338, 198–206.
147. Xiong R., Lu W., Li J., Wang P., Xu R., Chen T. Preparation and characterization of intravenously injectable nimodipine nanosuspension. Int. J. Pharm. 2008; 350, 338–343.
148. Jacobs C., Kayser O., Müller R. H. Nanosuspensions as a new approach for the formulation for the poorly soluble drug tarazepide. Int. J. Pharm. 2000; 196, 161–164.
149. Kocbek P., Baumqartner S., Kristl J. Preparation and evaluation of nanosuspensions for enhancing the dissolution of poorly soluble drugs. Int. J. Pharm. 2006; 312, 179–186.
150. Yano Y., Fujimoto T., Hidaka T. Method for producing microgranulated particle. US 5547683 A.
151. Hu L., Tang X., Cui F. Solid lipid nanoparticles (SLNs) to improve oral bioavailability of poorly soluble drugs. J. Pharm. Pharmacol. 2004; 56, 1527–1535.
152. Müller R. H., Runge S., Ravelli V., Mehnert W., Thünemann A. F., Souto E. B. Oral bioavailability of cyclosporine: Solid lipid nanoparticles (SLN) versus drug nanocrystals. Int. J. Pharm. 2006; 317, 82–89.
153. Mohammed A. R., Weston N., Coombes A. G., Fitzgerald M., Perrie Y. Liposome formulation of poorly water soluble drugs: optimisation of drug loading and ESEM analysis of stability. Int. J. Pharm. 2004; 285, 23–34.
154. Chen Y., Lu Y., Chen J., Lai J., Sun J., HU F., Wu W. Enhanced bioavailability of the poorly water-soluble drug fenofibrate by using liposomes containing a bile salt. Int. J. Pharm. 2009; 376, 153–160.
155. Lindenberg M., Kopp S., Dressman J. B. Classification of orally administered drugs on the World Health Organization Model list of Essential Medicines according to the biopharmaceutics classification system. Eur. J. Pharm. Biopharm. 2004; 58, 265–278.
156. Wu C. Y., Benet L. Z. Predicting drug disposition via application of BCS: Transport/ bbsorption/ elimination interplay and development of biopharmaceutics drug disposition classification system. Pharm. Res. 2005; 22, 11–23.
157. Petersen S. B., Nolan G., Maher S., Rahbek U. L., Guldbrandt M., Brayden D. J. Evaluation of alkylmaltosides as intestinal permeation enhancers: Comparison between rat intestinal mucosal sheets and Caco-2 monolayers. Eur. J. Pharm. Sci. 2012; 47, 701–712.
158. Thanou M., Verhoef J. C., Junginger H. E. Chitosan and its derivatives as intestinal absorption enhancers. Adv. Drug. Deliv. Rev. 2001; 50, 91–101.
159. Neelam S., Puneet G., Arundhati B. Enhancement of intestinal absorption of poorly absorbed Ceftriaxone Sodium by using mixed micelles of Polyoxy Ethylene (20) Cetyl Ether & Oleic Acid as peroral absorption enhancers. Archives of Applied Science Research 2010; 2, 131–142.
Štítky
Farmacie FarmakologieČlánek vyšel v časopise
Česká a slovenská farmacie

2015 Číslo 5
- Aceklofenak v léčbě muskuloskeletálních onemocnění – srovnání s dalšími NSAIDs z hlediska účinnosti a bezpečnosti
- Využití diklofenaku ve formě gelu v terapii seboroické keratózy
- Budoucnost guaifenesinu jako OTC myorelaxans
Nejčtenější v tomto čísle
- Metody používané ve farmaceutické technologii ke zvyšování biologické dostupnosti špatně rozpustných léčiv po perorálním podání
- Formulační aspekty orodispergovatelných tablet
- Prof. RNDr. Jaroslav Květina, DrSc. dr.h.c. FCMA – 85letý
- Hodnocení sypných a konsolidačních vlastností prášků ve farmaceutické technologii