
A Population Genetic Approach to Mapping Neurological Disorder Genes Using Deep Resequencing
Deep resequencing of functional regions in human genomes is key to identifying potentially causal rare variants for complex disorders. Here, we present the results from a large-sample resequencing (n = 285 patients) study of candidate genes coupled with population genetics and statistical methods to identify rare variants associated with Autism Spectrum Disorder and Schizophrenia. Three genes, MAP1A, GRIN2B, and CACNA1F, were consistently identified by different methods as having significant excess of rare missense mutations in either one or both disease cohorts. In a broader context, we also found that the overall site frequency spectrum of variation in these cases is best explained by population models of both selection and complex demography rather than neutral models or models accounting for complex demography alone. Mutations in the three disease-associated genes explained much of the difference in the overall site frequency spectrum among the cases versus controls. This study demonstrates that genes associated with complex disorders can be mapped using resequencing and analytical methods with sample sizes far smaller than those required by genome-wide association studies. Additionally, our findings support the hypothesis that rare mutations account for a proportion of the phenotypic variance of these complex disorders.
Published in the journal:
. PLoS Genet 7(2): e32767. doi:10.1371/journal.pgen.1001318
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001318
Summary
Deep resequencing of functional regions in human genomes is key to identifying potentially causal rare variants for complex disorders. Here, we present the results from a large-sample resequencing (n = 285 patients) study of candidate genes coupled with population genetics and statistical methods to identify rare variants associated with Autism Spectrum Disorder and Schizophrenia. Three genes, MAP1A, GRIN2B, and CACNA1F, were consistently identified by different methods as having significant excess of rare missense mutations in either one or both disease cohorts. In a broader context, we also found that the overall site frequency spectrum of variation in these cases is best explained by population models of both selection and complex demography rather than neutral models or models accounting for complex demography alone. Mutations in the three disease-associated genes explained much of the difference in the overall site frequency spectrum among the cases versus controls. This study demonstrates that genes associated with complex disorders can be mapped using resequencing and analytical methods with sample sizes far smaller than those required by genome-wide association studies. Additionally, our findings support the hypothesis that rare mutations account for a proportion of the phenotypic variance of these complex disorders.
Introduction
Genome-wide interrogation approaches for mapping genes often are designed to detect the common variants associated with common phenotypes or disease, generally leaving rare variants undetected or untested [1]–[5]. The Rare Allele–Major Effects (RAME) model postulates that rare (minor allele frequency <0.01 to 0.05) penetrant variants are key to the genetic etiology of common disease. Functional mutations that lead to an altered amino acid are often deleterious and potentially disease causing. Such mutations are subjected to natural selection, which either removes these alleles from the population or maintains them at low frequencies relative to the neutral expectations [3], [4], [6]–[9]. Partial genome or candidate gene resequencing of a large number of individuals holds the promise of finding both common and rare variants associated with clinically relevant phenotypes [3], [4]. While a number of studies have mapped common mutations or structural variants associated with neurological disorders like Autism Spectrum Disorder (ASD) (e.g. [10]) and Schizophrenia (e.g. [11]), the RAME model is better suited for modelling diseases that occur sporadically in families, and for testing whether rare variants contribute to these disorders.
ASD is a neurodevelopmental disorder characterized by stereotyped and repetitive behaviours and impairments in social interactions. Schizophrenia is a chronic psychiatric syndrome characterized by a profound disruption in cognition, behaviour and emotion, which begins in adolescence or early adulthood. The incidence of both ASD and schizophrenia is higher in males than in females [12], [13], which points to an important role of X-chromosome genes in the two diseases. There is significant clinical variability among ASD and schizophrenia patients, suggesting that they are etiologically and genetically heterogeneous. For ASD, genetics clearly plays an important role in the etiology, as revealed by twin and familial studies [14]–[16]. Some susceptibility regions have been identified through whole genome linkage analyses [17], [18], although they rarely coincide among the different studies [19]. Additionally, de novo mutations have been observed [20], [21] and are candidate variants in sporadic cases of either ASD or schizophrenia, but they explain a small percentage of the phenotypic variation. Together, these observations suggest that disruption of numerous genes by rare yet penetrant mutations could represent a major cause of ASD. Schizophrenia has an estimated heritability of 80% [22], and it has been recently associated with common variants at the MHC locus [11], [23], [24]. There are also several observations suggesting a causal link between rare mutations and schizophrenia, such as the fact that patients mostly have no affected close relatives, or the associations of both paternal age and decreased fertility with the disease [2]. Recent studies also reported de novo copy number variants (CNVs) in schizophrenia, providing further support for the rare variant hypothesis in non-familial cases [25]–[28], as well as an excess of rare inherited CNVs in familial cases [29].
We hypothesized that capturing genetic variation at low frequencies (minor allele frequency ≥0.005) in a large set of genes expressed in the brain will significantly contribute to our understanding of the genetic basis of ASD and schizophrenia. If the RAME model is relevant to these two diseases, the expectation is an enrichment of rare deleterious mutations among individuals diagnosed with ASD and schizophrenia. Here, we use population genetic and other statistical methods to analyze a resequencing dataset to map genes associated to these two disorders. Using this approach, we identified candidate genes by testing for genes harboring an excess of rare missense variants among individuals affected with ASD and schizophrenia, testing for selection at the gene level in each disease cohort, and assessing the impact of the variants found in candidate genes on the study-wide distribution of missense allele frequencies for each disease cohort.
Results
SNPs Discovered from Deep Resequencing
We resequenced 408 selected brain-expressed genes (Table S1) in 142 ASD and 143 schizophrenia-affected individuals [21]. The ASD and schizophrenia cohorts had global ethnicity representation yet were predominantly European with a large French Canadian sub-group. It is crucial to exclude ethnic and genetic outliers when analyzing rare variants because such samples contain private alleles from other populations. While the results from the software structure [30] revealed no population structure, we identified and removed potential ethnic outliers from the analysis using self reported ethnicity and a principle component analysis using eignesoft [31] (see Materials and Methods, Figure S1). The result was a sample of individuals of European ancestry: ASD (n = 102) and schizophrenia (n = 138). A total of 285 samples from the Québec Newborn Twin Study (QNTS) [32] were screened for self reported European ancestry (n = 240) and were used as controls. Thirty-eight (19 autosomal, 19 X-linked) of the 408 brain expressed genes were sequenced in the QNTS controls, including any gene with de novo mutations previously described in one of the disease cohorts [21] or with potential protein disrupting mutations.
We identified a total of 5,396 segregating sites in the disease cohorts, including 1,111 missense and 11 nonsense variants (Table 1 and Table 2), from lymphoblastoid cell DNA. As expected, there was a reduction in nucleotide diversity (π) on the X chromosome relative to autosomes by a ratio of 0.76 (Table 1), consistent with neutral expectations for the reduced effective population size of the X chromosome [33]. The ratio of the male to female population mutation rate [34] was estimated to be 6.37, slightly higher but similar to previous estimates of the male mutation rate being four times the female mutation rate [35]. The population mutation rate (θW per base pair) for all variants (including nonsense mutations) was 3.471×10−4 and 3.47×10−4 for Schizophrenia and ASD, respectively (Table 1).


Rare Missense Variants Show Increased Predicted Detrimental Effects
The fact that both ASD and Schizophrenia are highly heritability and that the common variant associations have been difficult to replicate support rare variants as an important component of the genetic etiology of these diseases. If rare variants are contributing to these diseases, we expect the site frequency spectrum (SFS) of missense variants to show an excess of deleterious, low frequency variants in our disease cohorts relative to either neutral expectations or controls. We analyzed the proposed detrimental effects of missense variants by estimating the potential functional effect of the missense variants observed using the software mapp [36]. Mapp was used to predict the severity of a missense mutation based on conservation in a multispecies protein alignment and the physiochemical properties of the amino acids. Severity scores indicated that 19% of the missense variants, in 47% of the genes with one or more missense variants, were likely to adversely affect protein function, considering a threshold for mapp scores of ten (approximately P = 0.01). Mapp scores are significantly higher for rare versus common variants (average 7.59 vs. 5.91, Mann-Whitney test P = 1.45×10−4, Figure S2), and the proportion of variants with high mapp scores (>10) is also significantly higher in the rare variants (21%) than in the common variants (11%) (χ2 = 8.3, P = 0.0039). These results are consistent with the presence of deleterious alleles being maintained at low frequency by selection. However, we did not observe an excess of high mapp scores in the cases relative to those in the QNTS controls indicating that rare mutations are likely subject to selection across all populations and not just cognitive related genes screened among our ASD and SCZ patients.
Individual Genes with Excess of Missense and Rare Missense Variants
We applied three different methods to identify genes harboring an excess of rare missense variants in the disease cohorts. First, we identified genes with an excess of missense variants relative to the number of silent variants observed. For each gene, we used Fisher's Exact Test to test the ratio of missense to silent variants observed within the gene relative to the ratio of missense to silent variants found in the remaining pooled genes within each cohort, and separately for X-linked and autosomal loci. The ratio of missense to silent variants allows us to control for demographic effects on variation in each cohort and detect genes subject to natural selection. In both cohorts, the autosomal gene MAP1A exhibited a significant excess of missense compared to silent variants (Figure S3, Table S2) (ASD P = 0.04, schizophrenia P = 0.03 after Bonferroni correction) and 23 of the 29 missense variants described in this gene are rare (minor allele frequency <0.03). MAP1A also exhibits an excess of missense variants when compared to the total counts of missense and silent variants in the control cohort (ASD-Control P = 0.009, Schizophrenia-Control P = 0.008). To further test the total predicted effect of the missense variants; for each gene, we summed the mapp scores for all missense variants within a gene, calculated the ratio of summed mapp scores to the number of silent variants, and compared these relative the same ratio in all other genes combined, separately for autosomal and X-linked loci. While the ratio for MAP1A was not significant after multiple testing correction, GRIN3A remained significant, relative to all other genes (P = 0.011) for the ASD cohort.
Second, we tested for an excess of individuals bearing rare missense variants, using Li and Leal's collapsing method [37]. We 1) contrasted the ASD and schizophrenia cohorts to each other (n = 277 genes with one or more missense variant in at least one cohort), 2) compared ASD to the QNTS controls (n = 26 genes with one or more missense variant), and 3) compared the schizophrenia cohort to the QNTS controls (n = 26 genes with one or more missense variant), and corrected for multiple testing. For every gene, the number of individuals carrying at least one rare missense variant, and the number of individuals without rare missense variants, is compared between two cohorts (see Materials and Methods). Although the ASD–Schizophrenia comparison revealed no significant results for any gene, we observed an excess of individuals with rare missense variants for two genes in the ASD cohort relative to the controls (GRIN2B and CACNA1F, Bonferroni adjusted P = 0.026, and P = 0.031 respectively, Table S3). The schizophrenia cohort had a significant excess of individuals hosting rare missense variants at GRIN2B (P = 0.041). We repeated this analysis considering only missense variants predicted to have detrimental effects on protein function (mapp scores >10). We found GRIN3A to exhibit an excess of individuals hosting rare detrimental missense variants in the ASD cohort relative to QNTS controls (P = 0.034).
Finally, we used an extension of the McDonald Krietman test, implemented in mkprf [38], to obtain estimates of γ per gene, based on our observed polymorphisms within humans and substitutions between humans and an outgroup (Pan troglodytes, see Materials and Methods). Estimates of the population selection parameter (γmkprf = 2Nes, where Ne is the effective population size and s is the selection coefficient) will be negative when amino acid replacements are deleterious [39]. Here, we compared the per gene selection coefficients between cohorts to detect genes enriched for missense variants in one cohort compared to another. The overall distribution of γmkprf values among genes was similar between ASD and schizophrenia (Figure 1). When we contrasted ASD and schizophrenia γmkprf estimates to those estimated from a Western-European population [39], we found both the ASD and schizophrenia cohorts have significantly more negative γmkprf distributions (Wilcoxon paired test, ASD P = 0.0026, schizophrenia P = 0.0005), while the QNTS controls do not show significant differences relative to the Western-European dataset (P = 0.18). We also found individual genes that differed significantly among our cohorts. For example, we observed a significant difference in γmkprf between the QNTS and ASD cohorts for CACNA1F (Figure 1B), with the disease cohort having the lower γmkprf estimate.
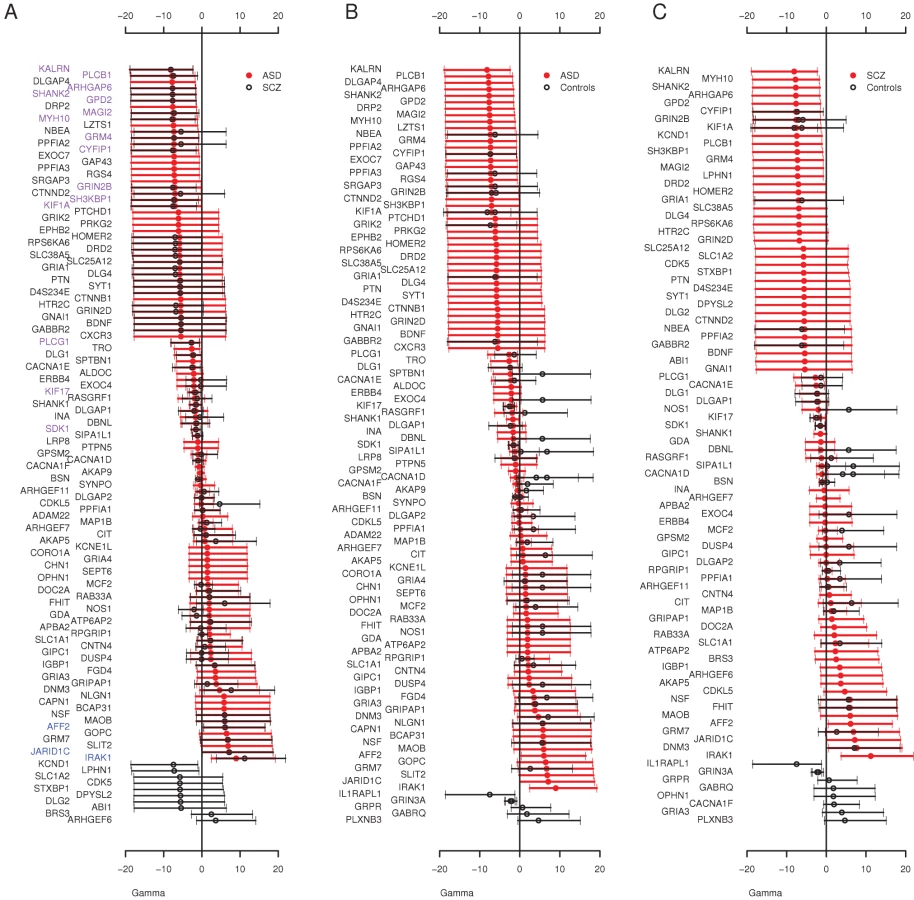
The Overall Frequency Spectrum Reveals an Excess of Rare Deleterious Variants at Autosomal Loci among ASD and Schizophrenia Individuals
Having identified a number of individual genes with an excess of deleterious rare alleles, we examined the contribution of these individual genes to estimates of the population selection parameters, γprfreq, across all variants within each cohort. Estimates of γprfreq will be negative when an excess of low frequency variants are observed, suggesting an accumulation of deleterious variants [39] through negative selection. We used the Poisson Random Fields method implemented in prfreq [40] to infer the demographic and selection parameters from cohort specific site frequency spectrum (SFS) and ask 1) whether the SFS of the missense variants showed evidence of selection relative to the SFS of silent and intronic variants; 2) if these selection estimates significantly differed between the disease cohorts and the control cohort; and 3) how removing the disease associated genes mapped with other approaches (see above) contribute to the SFS γprfreq estimates.
First, we estimated the demographic parameters for the different cohorts using the silent and intronic variants discovered among all autosomal genes. Only genes having at least one or more variant (265 for disease cohorts, 19 for controls) were included. Next, while fixing the estimated demographic parameters, the population selection parameters (γprfreq) were then estimated from the missense variants SFS (198 genes with one or more missense variant in the disease cohorts, 15 such genes for the controls; Table S4 and Figure S4). In each case, we estimated the likelihood for that model including the likelihood for a Wright-Fisher neutral model. The demographic-only model was a significant improvement relative to the neutral model (two times the difference in log likelihood, P<0.001), and the demographic-with-selection model was a significant improvement relative to the demographic-only model (P<0.001)(Table S4). The estimates of γprfreq were more negative in the two disease cohorts (γprfreqASD = −920, and γprfreqSCZ = −1,100, Table S4 and Figure S4) than for the control samples (γprfreqcontrol = −740), Specifically, the γprfreqSCZ estimate was significantly more negative than the γprfreqcontrol estimate (P = 0.01), although this pattern was not observed for ASD (P = 0.12) (see Materials and Methods). These observations indicate an excess of low frequency missense variants in our disease cohorts.
To assess each gene's contribution to the overall SFS, we developed an empirical distribution of γprfreq estimates by removing each gene from the SFS and re-estimating γprfreq. For ASD, when excluding MAP1A, GRIN3B and RPGRIP1, either individually or combined, the γprfreq values became more positive (implying less deleterious effects; γprfreqASD = −880, P[γ≥−880| empirical distribution] = 0.015 and −800 respectively) than when estimated for all variants (γprfreqASD = −920) (Figure 2). These values are also closer to the γprfreq value estimated in controls (γprfreqcontrol = −740, see above) and are at the most positive end of the empirical distribution of γprfreq values (Figure 2A). In the schizophrenia cohort, we estimated γprfreq values when removing MAP1A and GRIN3B were γprfreqSCZ = −1,020 (P[γ≥−1020 | empirical distribution] = 0.01) and γprfreqSCZ = −980 (P[γ≥−920 | empirical distribution] = 0.005), respectively (Figure 2B). When variants in both the MAP1A and GRIN3B loci were excluded from the SFS, the estimated γprfreqSCZ increased to −880, again a value similar to that observed in controls. These results indicate that the presence of a few genes, enriched with rare missense variants in the disease cohorts, is enough to alter the global SFS among all of our candidate genes. In our case, the overall excess of deleterious missense variants and the more negative γprfreq estimate in the two disease cohorts as compared to a neutral cohort is mainly caused by a very few candidate genes.

Discussion
Using approaches that specifically analyze both the function and accumulation of rare variants in disease cohorts, we have found a number of candidate genes associated with ASD and schizophrenia in two cohorts with sample sizes substantially smaller than those required for GWA studies. Population genetic approaches are designed to deal with data where sample sizes are limited compared to the sizes required in GWA studies. In both the ASD and schizophrenia cohorts, GRIN2B and MAP1A contain a statistically significant excess of rare missense variants, while the excess of rare missense variants in CACNAF1 was restricted to the ASD cohort. The involvement of a particular gene (e.g. GRIN2B or MAP1A) in the etiology of both disorders may reflect a critical role for neurodevelopment processes and suggests a pleiotropic effect [41]. Population genetic models incorporating demographic and selection processes, implemented in prfreq, corroborated this result for MAP1A and CACNA1F, and identified three new candidate genes: GRIN3B, NOS1, RPGRIP1 (Table 3).

Most of our mapped genes (Table 3, Table S5) have been previously implicated in neurological disorders or in neurodevelopment. MAP1A, a member of the microtubule-associated MAP1 proteins family, is predominantly expressed in adult neurons and is involved in axon and dendrite development. Among other interactions, MAP1A participates in the linking of DISC1 to microtubules [42]. DISC1 is a protein that was described as related to the pathogenesis of schizophrenia by linkage analysis of a Scottish family [43], [44] and confirmed among Finnish cohorts [45], [46]. The association between schizophrenia and eleven genes interacting with DISC1 (including MAP1A) was explored in Finish families, with significant results in three of the candidates but not for MAP1A [47]. Among calcium channel classes of genes, such as CACNA1F, we found a significant excess of rare variants and two segregating inframe indels at CDS position 807 falling in a glutamic acid-rich coiled domain. CACNA1F has been previously associated to schizophrenia [48] and mutations have also been described for other neurological disorders [49]. Finally, two independent meta-analyses corroborate our findings for a role of GRIN2B in the etiology of schizophrenia [50], [51]. GRIN2B codes a subunit of the glutamate and N-methyl-D-aspartate (NMDA) receptor. There exist several NMDA receptors that are constructed with one or more isoforms of the NR1 subunit (GRIN1) in different combinations with NR2 (GRIN2A, GRIN2B, GRIN2C and GRIN2D genes) and NR3 subunits (GRIN3A and GRIN3B) [52]. Some studies have suggested a relationship of decreased expression and abnormalities in NMDA receptors with schizophrenia [53], [54]. Additionally, we observed key results for other NMDA related genes. GRIN3B had an excess of detrimental variants, two different analyses revealed significantly higher mapp scores in GRIN3A in the ASD cohort, while we observed a nonsense mutation in both GRIN2C and GRIN3A in our cohorts. In the case of GRIN2B, we also observed a coding indel which results in an amino acid insertion at cds position 1,353. This collection of rare and functional changes in the GRIN gene family points to an important role of NMDA receptors in these neurological disorders.
For some cases in our study, different methods identified different loci associated with ASD and Schizophrenia. Observing signficant disease associations for the same genes using all the methods employed would be ideal, however it is not expected as each statistical test evaluates a different component of variation within the data. Furthermore, the ability to use each test varies by chromosome (X vs. autosomal) and cohort. The Li and Leal collapsing method [37] evaluates the accumulation of rare missense variants in genes in cases relative to controls, and is independent of silent variants. The ratio of missense to silent mutations is cohort specific and is used to identify genes that have an excess of missense mutations conditional on the number of silent mutations, relative to the cohort-wide average. As an example, a gene can have a minimum of two rare missense variants and generate a significant result with the collapsing method, but might not have sufficient power to test the ratio of rare missense to silent variants.
In contrast, we do expect genes with high missense to silent variant ratios, to have negative γmkprf estimates, and to contribute to more negative γprfreq estimates overall. We observed these patterns for the GRIN2B gene (Table 3) in both diseases which was supported by a significant collapsing method test result. Like GRIN2B, CACNA1F also shows concordance among methods within the ASD cohort; including a nominally significant missense to silent variant ratio, a significant collapsing method result, and a negative shift in the γmkprf estimate relative to the control cohort. Finally, MAP1A has a significant ratio of missense to silent variants relative to the rest of the genes in both cohorts, and appears to have a signficant impact on the overall γprfreq estimates for both the ASD and SCZ cohorts. Due to limited resequencing data in controls, we were unable to apply the collapsing method to MAP1A. In conclusion, for GRIN2B, MAP1A, and CACNA1F, we see concordant results among multiple methods when data availability allows testing by the different methods.
Previous studies demonstrate that genes associated with Mendelian disease or cancer have more negative population selection parameters compared to genes implicated in complex diseases [55]. This pattern may be explained by a late-onset effect, or by a potential enrichment of positively selected genes, among the loci involved in complex disease [55], [56]. Another explanation is that genes which accumulate either rare inherited or de novo mutations are also more likely to accumulate rare mutations which individually have either a high impact, as in the case of Mendelian disorders, or have an intermediate impact in aggregate.
In this paper we hypothesized that rare variants in neurologically expressed genes are enriched in ASD and schizophrenia cohorts. Our findings support a rare allele-major effect model as we have uncovered significant excess of rare variants in our disease cohorts. It remains an open question if ASD and schizophrenia are caused by variants found in a reduced set of genes such as DISC1 or NMDA receptor related genes, or in a larger number of genes associated to the same functional class or pathway/network.
Materials and Methods
Candidate Gene Selection
In total, 408 genes were selected for sequencing: 122 in the X chromosome and 286 autosomal genes [21] (Table S1) from a comprehensive list of potential synaptic genes (n = 5,000) based on published studies and databases [57]–[62]. X-chromosome synaptic genes were chosen due to the excess of affected males as compared to females in individuals affected with schizophrenia [12] and ASD [13], and since many genes affecting neurodevelopmental brain diseases happen to be on the X-chromosome [63]. Autosomal genes implicated in synapse function, including those encoding glutamate receptors and their interactors were also chosen, because glutamate signalling is strongly implicated in synapse function [64]. A total of 38 genes (19 autosomal and 19 X-linked) with de novo mutations [21] or potentially disrupting protein mutations in the disease cohorts were chosen for sequencing in the controls.
Samples
Autism Spectrum Disorder (ASD) subjects
Subjects diagnosed with autism spectrum disorders and both of their parents were recruited in clinics specializing in the diagnosis of Pervasive Developmental Disorders (PDD), rehabilitation centers, and specialized schools in the Montreal and Quebec regions, Canada [65]. Subjects with ASD were diagnosed by child psychiatrists and psychologists specialized in the evaluation of ASD. All subjects were diagnosed using the Diagnostic and Statistical Manual (DSM) of Mental Disorders criteria from patients in the Montreal and surrounding area, and depending on the recruitment site, either the Autism Diagnostic Interview-Revised or the Autism Diagnostic Observation Schedule was used. In addition, the Autism Screening Questionnaire (ASQ) was also completed for all of our subjects. All samples were French-Canadian. Furthermore, all proband medical charts were reviewed by a child psychiatrist expert in PDD to confirm diagnoses. Exclusion criteria were: (1) an estimated mental age <18 months, (2) a diagnosis of Rett Syndrome or Childhood Disintegrative Disorder and (3) evidence of any other psychiatric and neurological conditions, specifically: birth anoxia, rubella during pregnancy, fragile-X disorder, encephalitis, phenylketonuria, tuberous sclerosis, Tourette and West syndromes. Subjects with these conditions were excluded based on parental interview and chart review. However, participants with a co-occurring diagnosis of semantic-pragmatic disorder (due to its large overlap with PDD), attention deficit hyperactivity disorder (seen in a large number of patients with AD during development) and idiopathic epilepsy (which is related to the core syndrome of AD) were eligible for the study.
Schizophrenia subjects
The schizophrenia subjects were selected from among several large schizophrenia clinical genetic research centers worldwide. These include: (A) The L.E. Delisi cohort collected in the USA and Europe [66]. Dr. DeLisi and her collaborators had identified and collected over 500 families with schizophrenia or schizoaffective disorder in at least two siblings over the last two decades. Diagnoses were done using the DSM-III-R criteria on the basis of structured interviews, review of medical records from all hospitalizations or other relevant treatments, and structured information obtained from at least one reliable family member about each individual. Two independent diagnoses (one made by Dr. DeLisi) were made for each individual in the study. In cases of disagreement between the diagnosing clinicians, a third diagnostician was consulted, and final diagnoses were made by consensus after discussion. In cases where there was a sibling diagnosed with schizophrenia, the case with earlier age of onset and more definite schizophrenia diagnosis was selected for the initial screening. (B) The R. Joober cohort: Dr. Joober has collected over 300 schizophrenia families in Montreal in the past 10 years [67]. The same clinical assessment procedures have been followed as in (A). In addition, extensive pharmacological data have been collected in this cohort. (C) The J. Rapoport cohort (USA): collection includes Childhood Onset Schizophrenia cases [68]. Individuals in this cohort known to carry the VCFS deletion on chromosome 22q11 were excluded. All patients met DSM-IIIR/DSM-IV criteria for schizophrenia or psychosis not otherwise specified, had premorbid full-scale IQ scores of 70 or above and onset of psychotic symptoms by age 12 years. (D) Marie-Odile Krebs cohort (4 cases) was collected in France: All subjects were examined according to the standardized Diagnostic Interview for Genetic Studies (DIGS 3.0) [69]. Family histories of psychiatric disorders were also collected using the Family Interview for Genetic Studies (FIGS). All DIGS and FIGS have been reviewed by two or more psychiatrists for a final consensus diagnosis based on DSM-IIIR or DSM-IV at each centre. Exclusion criteria for all subjects included neurologic hard signs (referring to any symptoms or neurological conditions that can come with psychosis (and not related to schizophrenia such as Parkinson, Alzheimers, etc.), a history of head trauma and substance abuse or dependence. Institutional ethical approval for the study and informed consent was obtained for all study participants.
From over 500 ASD and 1,000 schizophrenia families, 122 males and 20 females were selected for the ASD cohort, and 95 males and 48 females were selected for the schizophrenia cohort [21]. The random population cohort (150 males; 135 females) [21] consist of unrelated individuals collected for the Quebec Newborn Twin Study (QNTS) [32] where DNA samples were available for both parents and both twins (either monozygotic or dizygotic), however only one sibling was chosen randomly for sequencing.
DNA Preparation, Sequencing, and SNP Calling
DNA samples were available for all affected and unaffected individuals and parents. For certain individuals where blood DNA was limited, we used DNA isolated from an Epstein-Barr Virus transformed lymphoblastoid cell line derived from the individual for the screen. The ASD cell lines samples have been frozen or regrown a maximum of two times. Genomic DNA was extracted from peripheral blood lymphocytes for each individual using Puregene extraction kits (Gentra System, USA). In all cases, rare variants were confirmed by sequencing both parents and using blood-derived DNA to rule out variations having arisen during production or growth of the lymphoblastoid cell line. Parentage was tested using 17 microsatellite markers. Primers were designed using the Exon Primer program from the UCSC genome browser. PCR products were sequenced at the Genome Quebec Innovation Centre in Montreal, Canada (www.genomequebecplatforms.com/mcgill/) on a 3730XL DNA Analyzer System. PolyPhred (v6.0) and Mutation Surveyor (v3.10, Soft Genetics Inc.) were used for mutation detection analysis. Initial screens were done on cell-line DNA from samples to conserve blood sample DNA. PolyPhred (v6.0) scores of 40 or higher were used as the threshold cut-off for all sequencing reads. When reads did not meet this criterion they were resequenced. Chromatograms for all rare variants (singletons or homozygous doubletons) were manually checked. The sensitivity of singleton detection was previously assessed by contrasting the heterozygote calls from the sequencing to heterozygote calls made from Affy SNPChip genotyping, obtaining an estimated false negative rate of heterozygote calls of ∼2% [21]. Variants passing all validation steps were retained for analysis, resulting in a high confidence dataset.
Population Structure
Structure [30] was used to analyze the degree of population structure. Analysis of all samples showed no significant population structure as a large proportion of samples were of the same ethnicity. Principal Component Analysis (PCA) is more susceptible to samples with excess of private alleles and revealed genetic variability between individuals. We removed samples of non-European ancestry (self-reported ethnicity, ethnic outliers) and used eigensoft [31] to identify and remove remaining genetic outliers (defined below). All autosomal variants excluding those with calls in less than 20 per cohort were used for PCA analysis (4,645 SNPs). We used the linkage disequilibrium correction and calculated the top 10 principal components (PCs) and removed individuals with PC projections greater than two standard deviations or more from the mean, for all significant principal components, using 10 iterations. Individuals exhibiting excess of rare variants genome-wide are likely to be genetic outliers and readily identifiable with PCA and removed, while individuals with an excess of rare variants in specific genes are retained. To ensure the PCA outliers were true outliers, we used a one-sided t-test to assess if the proportion of missense singletons in each PCA outlier was higher than in PCA retained samples across all autosomal genes (see Text S1). Structure (admixture model) was used to reassess levels of population structure within the final sample set (Figure S1C).
Computational Inferences of Mutation Severity
Mapp was used to predict severity and assign scores to each missense variant. Orthologous protein sequences were obtained using the Galaxy Browser (http://main.g2.bx.psu.edu/) and the UCSC Human Genome Browser (hg18, http://genome.ucsc.edu/cgi-bin/hgGateway) to generate columns of aligned orthologous amino acids. Mapp scores and P values were calculated as shown in Stone and Sidow [36]. Mapp assesses variation observed at each amino acid position with respect to six physicochemical properties (hydropathy, polarity, charge, side-chain volume, free energy in alpha-helical conformation, and free energy in beta-sheet conformation) after weighting each protein sequence to mitigate the influence of phylogeny.
Statistical Analysis
Estimating population mutation rates and selection coefficients
Hclust [70] was used to calculate θW, and π for each gene in each cohort. Demographic parameters were inferred from the folded site frequency spectrum (SFS) of silent and intronic variants using prfreq [40]. Conditioning on the demographic parameters from the silent and intronic SFS, the likelihood of the missense SFS was estimated for three models: a Wright-Fisher neutral population, the demographic model inferred form the silent and intronic variants, and a demographic with a selection parameter (γprfreq) model. P values of model improvement were estimated by comparing the likelihoods, assuming a χ2 distribution of two times the differences in the likelihoods between the models. To compare the γprfreq values estimated in the ASD and schizophrenia cohort to the γprfreq values estimated in the control cohort, we computed the likelihood of the control parameters estimates in the schizophrenia and ASD cohorts, and compared these likelihoods to the likelihoods of the ASD and schizophrenia parameters estimates. We analyzed the effect of each gene with at least one missense variant (n = 198 autosomal genes) on the SFS by estimating γ in all the SFSs resulting from excluding the missense variants of each individual gene, in each cohort.
Estimating individual gene selection coefficients
The gene specific selection coefficient, γmkprf, was calculated with the mkprf program [38] for each cohort (ASD, Schizophrenia, QNTS Controls). The number of synonymous and nonsynonymous changes between humans and chimpanzees was obtained from Bustamante, et al. [39] for 244 genes. Additionally, the European ancestry samples also reported in Bustamante, et al. [39] were used as a secondary control data set for this analysis.
Within-Gene Excess of (Rare) Missense Variants
Fisher's exact test was used to detect deviations in the missense to silent variant ratio within genes. For each gene, the ratio of missense (or rare missense) to silent variants was contrasted to the same ratio in all remaining genes. This analysis was conducted in the ASD and schizophrenia cohorts testing autosomal and X-linked genes separately. We used the Bonferroni correction for multiple tests (n = 277). Excess of predicted deleterious load within genes was evaluated by summing the mapp scores for all missense variants within a gene and testing the ratio of summed mapp scores to silent variants within the gene relative to all other genes. This was done separately for autosomes and X-linked genes, and separately for each disease cohort.
Genes with Excess of Individuals Harboring Rare Missense Variants
To identify genes with an excess of individuals bearing rare missense variants, we used Li and Leal's collapsing method [37]. For ASD vs schizophrenia, ASD vs QNTS, and schizophrenia vs QNTS controls, the number of individuals with at least one rare missense mutation and the number of individuals with no rare missense variants was determined for each cohort, and these counts made up the cells of the two by two table. We assessed statistical significance using Fisher's Exact test and used Bonferroni's correction for multiple tests (number of genes nASD-SCZ = 277, nASD – QNTS Controls = 26, nSCZ - QNTS Controls = 26). This analysis was repeated, considering only missense variants with a mapp score >10.
Supporting Information
Zdroje
1. MaDQ
CuccaroML
JaworskiJM
HaynesCS
StephanDA
2007 Dissecting the locus heterogeneity of autism: significant linkage to chromosome 12q14. Mol Psychiatry 12 376 384
2. McClellanJM
SusserE
KingMC
2007 Schizophrenia: a common disease caused by multiple rare alleles. Br J Psychiatry 190 194 199
3. PritchardJK
2001 Are rare variants responsible for susceptibility to complex diseases? Am J Hum Genet 69 124 137
4. PritchardJK
CoxNJ
2002 The allelic architecture of human disease genes: common disease-common variant…or not? Hum Mol Genet 11 2417 2423
5. SmithDJ
LusisAJ
2002 The allelic structure of common disease. Hum Mol Genet 11 2455 2461
6. CrowJF
2000 The origins, patterns and implications of human spontaneous mutation. Nat Rev Genet 1 40 47
7. Eyre-WalkerA
KeightleyPD
1999 High genomic deleterious mutation rates in hominids. Nature 397 344 347
8. GiannelliF
AnagnostopoulosT
GreenPM
1999 Mutation rates in humans. II. Sporadic mutation-specific rates and rate of detrimental human mutations inferred from hemophilia B. Am J Hum Genet 65 1580 1587
9. KryukovGV
PennacchioLA
SunyaevSR
2007 Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am J Hum Genet 80 727 739
10. WangK
ZhangH
MaD
BucanM
GlessnerJT
2009 Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 459 528 533
11. StefanssonH
OphoffRA
SteinbergS
AndreassenOA
CichonS
2009 Common variants conferring risk of schizophrenia. Nature 460 744 747
12. LewisDA
LevittP
2002 Schizophrenia as a disorder of neurodevelopment. Annu Rev Neurosci 25 409 432
13. SkuseDH
2000 Imprinting, the X-chromosome, and the male brain: explaining sex differences in the liability to autism. Pediatr Res 47 9 16
14. BaileyA
Le CouteurA
GottesmanI
BoltonP
SimonoffE
1995 Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med 25 63 77
15. SteffenburgS
GillbergC
HellgrenL
AnderssonL
GillbergIC
1989 A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. J Child Psychol Psychiatry 30 405 416
16. BoltonP
MacdonaldH
PicklesA
RiosP
GoodeS
1994 A case-control family history study of autism. J Child Psychol Psychiatry 35 877 900
17. GuptaAR
StateMW
2007 Recent advances in the genetics of autism. Biol Psychiatry 61 429 437
18. KlauckSM
2006 Genetics of autism spectrum disorder. Eur J Hum Genet 14 714 720
19. YangMS
GillM
2007 A review of gene linkage, association and expression studies in autism and an assessment of convergent evidence. Int J Dev Neurosci 25 69 85
20. SebatJ
LakshmiB
MalhotraD
TrogeJ
Lese-MartinC
2007 Strong association of de novo copy number mutations with autism. Science 316 445 449
21. AwadallaP
GauthierJ
MyersRA
CasalsF
HamdanFF
2010 Direct measure of the de novo mutation rate in autism and schizophrenia cohorts. Am J Hum Genet 87 316 324
22. TandonR
KeshavanMS
NasrallahHA
2008 Schizophrenia, “just the facts” what we know in 2008. 2. Epidemiology and etiology. Schizophr Res 102 1 18
23. PurcellSM
WrayNR
StoneJL
VisscherPM
O'DonovanMC
2009 Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460 748 752
24. ShiJ
LevinsonDF
DuanJ
SandersAR
ZhengY
2009 Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 460 753 757
25. WalshT
McClellanJM
McCarthySE
AddingtonAM
PierceSB
2008 Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320 539 543
26. XuB
RoosJL
LevyS
van RensburgEJ
GogosJA
2008 Strong association of de novo copy number mutations with sporadic schizophrenia. Nat Genet 40 880 885
27. StefanssonH
RujescuD
CichonS
PietilainenOP
IngasonA
2008 Large recurrent microdeletions associated with schizophrenia. Nature 455 232 236
28. The_International_Schizophrenia_Consortium 2008 Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455 237 241
29. XuB
WoodroffeA
Rodriguez-MurilloL
RoosJL
van RensburgEJ
2009 Elucidating the genetic architecture of familial schizophrenia using rare copy number variant and linkage scans. Proc Natl Acad Sci U S A 106 16746 16751
30. PritchardJK
StephensM
DonnellyP
2000 Inference of population structure using multilocus genotype data. Genetics 155 945 959
31. PriceAL
PattersonNJ
PlengeRM
WeinblattME
ShadickNA
2006 Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38 904 909
32. LemelinJP
BoivinM
Forget-DuboisN
DionneG
SeguinJR
2007 The genetic-environmental etiology of cognitive school readiness and later academic achievement in early childhood. Child Dev 78 1855 1869
33. HartlDL
ClarkAG
2007 Principles of population genetics Sunderland, Mass. Sinauer Associates xv, 652
34. MiyataT
HayashidaH
KumaK
MitsuyasuK
YasunagaT
1987 Male-driven molecular evolution: a model and nucleotide sequence analysis. Cold Spring Harb Symp Quant Biol 52 863 867
35. NachmanMW
CrowellSL
2000 Estimate of the mutation rate per nucleotide in humans. Genetics 156 297 304
36. StoneEA
SidowA
2005 Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res 15 978 986
37. LiB
LealSM
2008 Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet 83 311 321
38. BustamanteCD
NielsenR
SawyerSA
OlsenKM
PuruggananMD
2002 The cost of inbreeding in Arabidopsis. Nature 416 531 534
39. BustamanteCD
Fledel-AlonA
WilliamsonS
NielsenR
HubiszMT
2005 Natural selection on protein-coding genes in the human genome. Nature 437 1153 1157
40. BoykoAR
WilliamsonSH
IndapAR
DegenhardtJD
HernandezRD
2008 Assessing the evolutionary impact of amino acid mutations in the human genome. PLoS Genet 4 e1000083
41. CarrollLS
OwenMJ
2009 Genetic overlap between autism, schizophrenia and bipolar disorder. Genome Med 1 102
42. HalpainS
DehmeltL
2006 The MAP1 family of microtubule-associated proteins. Genome Biol 7 224
43. BlackwoodDH
FordyceA
WalkerMT
St ClairDM
PorteousDJ
2001 Schizophrenia and affective disorders–cosegregation with a translocation at chromosome 1q42 that directly disrupts brain-expressed genes: clinical and P300 findings in a family. Am J Hum Genet 69 428 433
44. St ClairD
BlackwoodD
MuirW
CarothersA
WalkerM
1990 Association within a family of a balanced autosomal translocation with major mental illness. Lancet 336 13 16
45. EkelundJ
HovattaI
ParkerA
PaunioT
VariloT
2001 Chromosome 1 loci in Finnish schizophrenia families. Hum Mol Genet 10 1611 1617
46. EkelundJ
LichtermannD
HovattaI
EllonenP
SuvisaariJ
2000 Genome-wide scan for schizophrenia in the Finnish population: evidence for a locus on chromosome 7q22. Hum Mol Genet 9 1049 1057
47. TomppoL
HennahW
LahermoP
LoukolaA
Tuulio-HenrikssonA
2009 Association between genes of Disrupted in schizophrenia 1 (DISC1) interactors and schizophrenia supports the role of the DISC1 pathway in the etiology of major mental illnesses. Biol Psychiatry 65 1055 1062
48. WeiJ
HemmingsGP
2006 A further study of a possible locus for schizophrenia on the X chromosome. Biochem Biophys Res Commun 344 1241 1245
49. HopeCI
SharpDM
Hemara-WahanuiA
SissinghJI
LundonP
2005 Clinical manifestations of a unique X-linked retinal disorder in a large New Zealand family with a novel mutation in CACNA1F, the gene responsible for CSNB2. Clin Experiment Ophthalmol 33 129 136
50. AllenNC
BagadeS
McQueenMB
IoannidisJP
KavvouraFK
2008 Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet 40 827 834
51. LiD
HeL
2007 Association study between the NMDA receptor 2B subunit gene (GRIN2B) and schizophrenia: a HuGE review and meta-analysis. Genet Med 9 4 8
52. Cull-CandyS
BrickleyS
FarrantM
2001 NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol 11 327 335
53. FunkAJ
RumbaughG
HarotunianV
McCullumsmithRE
Meador-WoodruffJH
2009 Decreased expression of NMDA receptor-associated proteins in frontal cortex of elderly patients with schizophrenia. Neuroreport 20 1019 1022
54. KristiansenLV
HuertaI
BeneytoM
Meador-WoodruffJH
2007 NMDA receptors and schizophrenia. Curr Opin Pharmacol 7 48 55
55. BlekhmanR
ManO
HerrmannL
BoykoAR
IndapA
2008 Natural selection on genes that underlie human disease susceptibility. Curr Biol 18 883 889
56. SabetiPC
SchaffnerSF
FryB
LohmuellerJ
VarillyP
2006 Positive natural selection in the human lineage. Science 312 1614 1620
57. BrachyaG
YanayC
LinialM
2006 Synaptic proteins as multi-sensor devices of neurotransmission. BMC Neurosci 7 Suppl 1 S4
58. CollinsMO
YuL
CobaMP
HusiH
CampuzanoI
2005 Proteomic analysis of in vivo phosphorylated synaptic proteins. J Biol Chem 280 5972 5982
59. CroningMD
MarshallMC
McLarenP
ArmstrongJD
GrantSG
2009 G2Cdb: the Genes to Cognition database. Nucleic Acids Res 37 D846 851
60. TakamoriS
HoltM
SteniusK
LemkeEA
GronborgM
2006 Molecular anatomy of a trafficking organelle. Cell 127 831 846
61. TrinidadJC
SpechtCG
ThalhammerA
SchoepferR
BurlingameAL
2006 Comprehensive identification of phosphorylation sites in postsynaptic density preparations. Mol Cell Proteomics 5 914 922
62. ZhangW
ZhangY
ZhengH
ZhangC
XiongW
2007 SynDB: a Synapse protein DataBase based on synapse ontology. Nucleic Acids Res 35 D737 741
63. LaumonnierF
CuthbertPC
GrantSG
2007 The role of neuronal complexes in human X-linked brain diseases. Am J Hum Genet 80 205 220
64. GrantSG
MarshallMC
PageKL
CumiskeyMA
ArmstrongJD
2005 Synapse proteomics of multiprotein complexes: en route from genes to nervous system diseases. Hum Mol Genet 14 Spec No. 2 R225 234
65. GauthierJ
BonnelA
St-OngeJ
KaremeraL
LaurentS
2005 NLGN3/NLGN4 gene mutations are not responsible for autism in the Quebec population. Am J Med Genet B Neuropsychiatr Genet 132B 74 75
66. DeLisiLE
ShawSH
CrowTJ
ShieldsG
SmithAB
2002 A genome-wide scan for linkage to chromosomal regions in 382 sibling pairs with schizophrenia or schizoaffective disorder. Am J Psychiatry 159 803 812
67. JooberR
RouleauGA
LalS
DixonM
O'DriscollG
2002 Neuropsychological impairments in neuroleptic-responder vs. -nonresponder schizophrenic patients and healthy volunteers. Schizophr Res 53 229 238
68. GochmanPA
GreensteinD
SpornA
GogtayN
NicolsonR
2004 Childhood onset schizophrenia: familial neurocognitive measures. Schizophr Res 71 43 47
69. GourionD
GoldbergerC
BourdelMC
BayleFJ
MilletB
2003 Neurological soft-signs and minor physical anomalies in schizophrenia: differential transmission within families. Schizophr Res 63 181 187
70. LucaD
RingquistS
KleiL
LeeAB
GiegerC
2008 On the use of general control samples for genome-wide association studies: genetic matching highlights causal variants. Am J Hum Genet 82 453 463
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 2
Nejčtenější v tomto čísle
- Meta-Analysis of Genome-Wide Association Studies in Celiac Disease and Rheumatoid Arthritis Identifies Fourteen Non-HLA Shared Loci
- MiRNA Control of Vegetative Phase Change in Trees
- Risk Alleles for Systemic Lupus Erythematosus in a Large Case-Control Collection and Associations with Clinical Subphenotypes
- The Cardiac Transcription Network Modulated by Gata4, Mef2a, Nkx2.5, Srf, Histone Modifications, and MicroRNAs