
Association between Common Variation at the Locus and Changes in Body Mass Index from Infancy to Late Childhood: The Complex Nature of Genetic Association through Growth and Development
An age-dependent association between variation at the FTO locus and BMI in children has been suggested. We meta-analyzed associations between the FTO locus (rs9939609) and BMI in samples, aged from early infancy to 13 years, from 8 cohorts of European ancestry. We found a positive association between additional minor (A) alleles and BMI from 5.5 years onwards, but an inverse association below age 2.5 years. Modelling median BMI curves for each genotype using the LMS method, we found that carriers of minor alleles showed lower BMI in infancy, earlier adiposity rebound (AR), and higher BMI later in childhood. Differences by allele were consistent with two independent processes: earlier AR equivalent to accelerating developmental age by 2.37% (95% CI 1.87, 2.87, p = 10−20) per A allele and a positive age by genotype interaction such that BMI increased faster with age (p = 10−23). We also fitted a linear mixed effects model to relate genotype to the BMI curve inflection points adiposity peak (AP) in infancy and AR. Carriage of two minor alleles at rs9939609 was associated with lower BMI at AP (−0.40% (95% CI: −0.74, −0.06), p = 0.02), higher BMI at AR (0.93% (95% CI: 0.22, 1.64), p = 0.01), and earlier AR (−4.72% (−5.81, −3.63), p = 10−17), supporting cross-sectional results. Overall, we confirm the expected association between variation at rs9939609 and BMI in childhood, but only after an inverse association between the same variant and BMI in infancy. Patterns are consistent with a shift on the developmental scale, which is reflected in association with the timing of AR rather than just a global increase in BMI. Results provide important information about longitudinal gene effects and about the role of FTO in adiposity. The associated shifts in developmental timing have clinical importance with respect to known relationships between AR and both later-life BMI and metabolic disease risk.
Published in the journal:
. PLoS Genet 7(2): e32767. doi:10.1371/journal.pgen.1001307
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001307
Summary
An age-dependent association between variation at the FTO locus and BMI in children has been suggested. We meta-analyzed associations between the FTO locus (rs9939609) and BMI in samples, aged from early infancy to 13 years, from 8 cohorts of European ancestry. We found a positive association between additional minor (A) alleles and BMI from 5.5 years onwards, but an inverse association below age 2.5 years. Modelling median BMI curves for each genotype using the LMS method, we found that carriers of minor alleles showed lower BMI in infancy, earlier adiposity rebound (AR), and higher BMI later in childhood. Differences by allele were consistent with two independent processes: earlier AR equivalent to accelerating developmental age by 2.37% (95% CI 1.87, 2.87, p = 10−20) per A allele and a positive age by genotype interaction such that BMI increased faster with age (p = 10−23). We also fitted a linear mixed effects model to relate genotype to the BMI curve inflection points adiposity peak (AP) in infancy and AR. Carriage of two minor alleles at rs9939609 was associated with lower BMI at AP (−0.40% (95% CI: −0.74, −0.06), p = 0.02), higher BMI at AR (0.93% (95% CI: 0.22, 1.64), p = 0.01), and earlier AR (−4.72% (−5.81, −3.63), p = 10−17), supporting cross-sectional results. Overall, we confirm the expected association between variation at rs9939609 and BMI in childhood, but only after an inverse association between the same variant and BMI in infancy. Patterns are consistent with a shift on the developmental scale, which is reflected in association with the timing of AR rather than just a global increase in BMI. Results provide important information about longitudinal gene effects and about the role of FTO in adiposity. The associated shifts in developmental timing have clinical importance with respect to known relationships between AR and both later-life BMI and metabolic disease risk.
Introduction
Genome-wide association studies on body mass index (BMI) and adiposity have reliably identified its association with variation at the fat mass and obesity related locus (FTO) in adult and child populations [1]–[5]. In meta-analyses, the addition of each minor (A) allele at the single nucleotide polymorphism (SNP) rs9939609 within the first intron of FTO has been shown to be associated with a higher BMI of up to 0.33 kg/m2 or approximately 0.1 standard deviations [2], [6]. The biological mechanisms behind this association are yet to be fully determined, however evidence from both population based analyses and functional investigations have suggested that this locus is likely involved in the hypothalamic regulation of appetite or energy expenditure and metabolic rate [7]–[13]. Indeed, following a series of investigations noting the correlation between differential Fto expression, fat mass, food consumption [14], [15] and of raised FTO mRNA levels in the subcutaneous adipose tissue of obese individuals [16]–[18], ubiquitous overexpression of Fto has been shown recently to lead to a dose-dependent increase in body and fat mass irrespective of diet type [19]. Coincident observation of increases in dietary consumption, reduced leptin levels and further studies showing high expression levels in cerebellum, hippocampus and hypothalamus [11], [20], [21], point towards an important role for Fto/FTO in the regulation of dietary intake.
Until recently, most replication efforts concentrating on variation in FTO have employed singlepoint analyses of cross-sectional data. These have been conducted from ages as low as 2 weeks to old age (>70 years) and have demonstrated, with differing degrees of reliability, associations between variation at FTO, BMI and related traits [22], [23]. Although limited, available evidence suggests that the cross-sectional FTO/BMI association varies by age [24]–[26]. Specifically, at early ages up to and around 7 years, the association between common variation at FTO and BMI appears to be reduced in magnitude, with smaller studies being unable to detect association [24]–[26]. This pattern then changes in early adulthood with peak effect sizes (approximately 0.3kg/m2/minor allele) occurring by age 20 [24], [26]. Following this peak, this association appears to diminish absolutely (not relatively) in a manner one would expect for coincident reduction in adiposity levels with later age [23].
Haworth et al simultaneously examined the FTO/BMI association and the heritability of BMI in a longitudinal twin collection with data at ages 4, 7, 10 and 11 years [25]. They found that the association between BMI and variation at FTO was age dependent, that the heritability for BMI increased with age and that the proportion of variance in BMI explained by shared environment diminished over the same period. Consistent with the idea that BMI and adiposity related traits may be determined by a complex interplay between genetic and environmental features, these findings suggest that with age and dietary autonomy, loci such as FTO may be able to exert a greater effect on BMI. A Finnish twin study has since suggested an increase in the heritability of BMI throughout adolescent years [27].
Individuals vary considerably in their rate of growth and differ in their rate of development so that some children mature faster and reach milestones such as puberty earlier [28]. These two processes can be distinguished from each other. For example most inter-individual variability in pubertal height velocity can be explained in terms of developmental age/timing and adjusting for this allows their growth velocity curves to be superimposed [29]. So if developmental age explains variability in pubertal height, might it also explain BMI variability in childhood?
The timing of BMI adiposity rebound (AR) is inversely related to the risk of later obesity [30]–[34], and it is also positively correlated with the timing of puberty [35]. In terms of adiposity, the age of AR can be viewed as a developmental marker and an early AR implies an advanced developmental age [36]. This suggests that variation in the pattern of BMI development may arise from differences on the developmental age scale that are both independent of and in addition to differences on the BMI scale. In fact, there is good evidence to suggest that different processes are involved in the pattern of high BMI throughout life as opposed to high adult BMI preceded by average or low BMI in infancy followed by early AR [36].
Despite the published work on longitudinal differences in the associations between the variation at FTO and BMI or related traits, lack of dense lifecourse data and inadequate statistical power has made interpretation of findings difficult. In this investigation, we aimed to assess the relationship between variation at rs9939609 and changes in BMI from after birth until 13 years of age, by meta-analysing data from eight cohorts. We also aimed to explore the possibility of differences in developmental age between FTO genotypes by scaling the ages for each genotype appropriately, so as to minimise the differences in the pattern of BMI development through childhood. Lastly, we aimed to fit individual growth curves in order to explore directly the relationship of this variant to critical change points in BMI; all features known to be related to health in later life [36]–[40].
Results
Cross-sectional results
For the eight studies in the cross-sectional meta-analysis of the association between rs9939609 and BMI, the average sample size per age stratum was 9143. Table 1 gives the stratum and study-specific subject characteristics. Genotypic frequencies at rs9939609 were broadly consistent across studies and in accordance with expectations for population samples of European origin. All genotypic sampling adhered to Hardy Weinberg equilibrium (Table S1).

In meta-analyses above the age of 5.5 years (childhood) the minor allele (A) was associated additively with a higher BMI, though this was not detectable in the age stratum 11 to 13 years where the sample was small and where age associated increase in variance compromises analytical power. Expressed as a percentage change, the additive effect of each minor allele (A) was 0.7% (95% CI: 0.3, 1.1), 1.0% (95% CI: 0.6, 1.3), and 1.3% (95% CI: 0.6, 2.0) at 5.5–7, 7–9 and 9–11 years respectively. Maximum heterogeneity was high with I2 = 69.6% (95% CI: 22, 88). In contrast to this, each minor allele was associated with a lower BMI before the age of 2.5 years. The additive effect of each minor allele was −0.4% (95% CI: −0.6, −0.1), −0.3% (95% C%I: −0.6, −0.1) and −0.3% (95% CI: −0.5, 0.0) at age 0–0.5, 0.5–1.5 and 1.5–2.5 years respectively. Between 2.5 and 5.5 years there was no association between rs9939609 genotype and BMI. For these periods, maximum heterogeneity was high with I2 = 44.1% (95% CI: 0, 81). Figure 1 shows meta-analysis results representing major observations throughout the age range. Similar results (not shown) were found for weight/heightp.

To clarify the age trends in BMI for each genotype, median curves were estimated using the LMS method to adjust for age-specific heteroscedasticity and skewness. Figure 2 shows the median curves by genotype, where comparison shows three distinct features: (i) BMI is higher for A carriers later in childhood, but lower early in childhood; (ii) AR is earlier for A carriers; (iii) the A allele effects are additive in that the TA group is consistently midway between AA and TT. Curves for weight and height (Figure 3) show genotype differences for weight that emerge only after age 4, and height differences that are small at all ages. The differences in age at AR (Figure 2) can be removed by estimating a developmental age scaling effect per A allele of 2.79% (95% CI: 2.35, 3.23), such that the age scale in the AA group is shrunk, and in the TT group stretched, by 2.79% relative to TA. However there remains a rising BMI trend in AA relative to TT which is confirmed by fitting a log age by genotype interaction (coefficient 0.039 kg/m2 (0.031, 0.046) per log age unit per A allele, p = 10−23). Fitting the interaction reduces the optimal age scaling slightly to 2.37% (95% CI: 1.87, 2.87, p = 10−20), but provides evidence suggesting that the two processes together, developmental differences on the age scale and on the BMI scale, explain the complex age-related genotype effects on BMI in childhood. Figure 4 shows the estimation of this optimal scaling (top) and the curves of Figure 2 scaled by this amount (bottom), where now AR occurs at the same developmental ‘age’ for all groups. For comparison the optimal age scale to adjust for genotype differences in height (Figure 3) is 0.3% (0.2, 0.4), a value attaining statistical confidence, but far smaller than for BMI. Thus FTO appears to affect BMI developmental age much more than it does height developmental age.



Longitudinal results
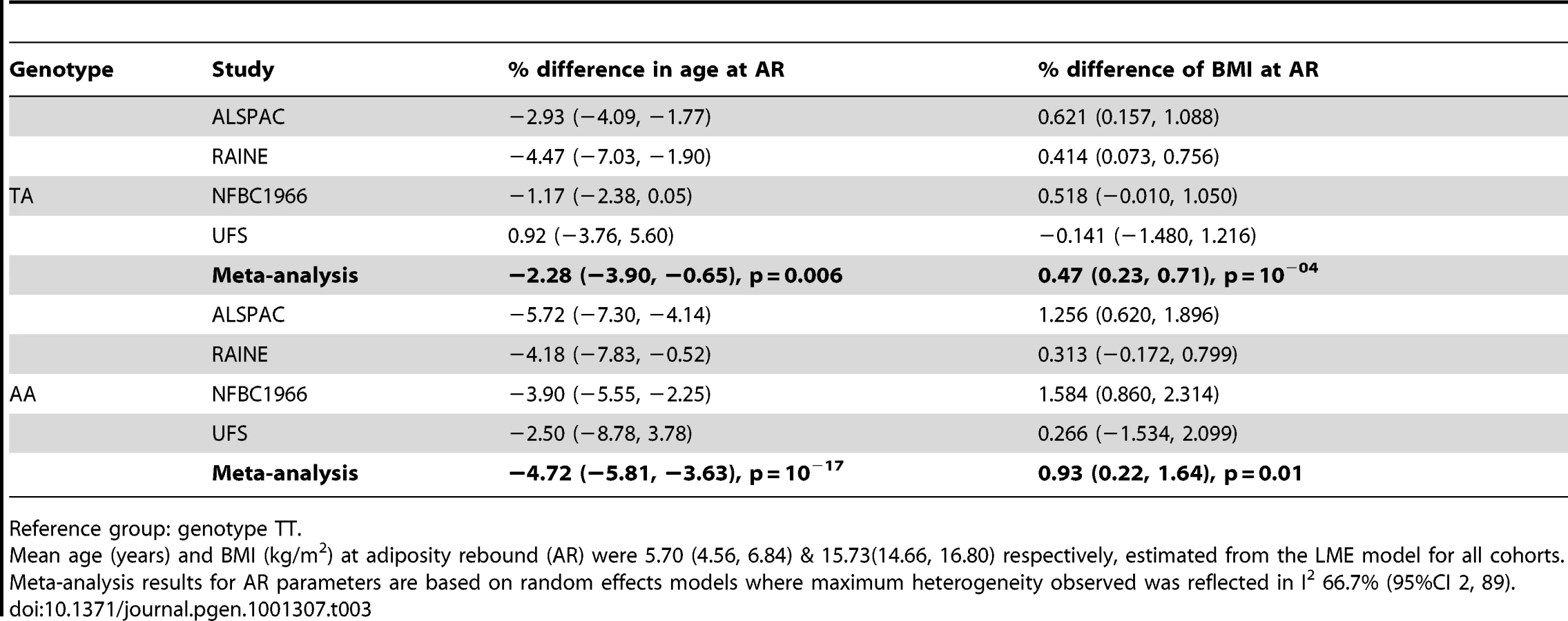
We analysed the richest data sets longitudinally for age and BMI at adiposity peak (AP) and adiposity rebound (AR). Mean ages at AP and AR were 0.75 (0.73, 0.78) and 5.70 years (4.56, 6.84) and mean BMIs 17.87 (17.11, 18.62) and 15.73 kg/m2 (14.66, 16.80) respectively. There was weak evidence for a lower BMI at AP only in the carriers of two minor alleles (AA) compared to the reference group (TT): −0.40% (95%CI: −0.74, −0.06), p = 0.02 (Table 2). In contrast, there was evidence a per minor allele difference in BMI of 0.47% (95%CI: 0.17, 0.77) at AR. This was realised as carriers of two minor alleles (AA) having a 0.93% (95%CI: 0.22, 1.64), p = 0.01 higher BMI at AR than those in the reference group (TT) (Table 3). There was no evidence for genotypic association with age at AP, but there was evidence for an additive relationship between carriage of minor alleles (A) at rs9939609 and earlier AR. Per minor allele, there was a −2.31% (95%CI: −3.05, −1.57) difference in age at AR. Indeed, carriers of one minor allele showed −2.28% earlier AR (95%CI: −3.90, −0.65), p = 0.006 and carriers of two minor alleles −4.72% earlier AR (−5.81, −3.63), p = 10−17 versus baseline genotype TT). There was evidence of heterogeneity between cohorts, lower for AP (maximum I2 30% (0, 74)) than for AR (I2 67% (2, 89)). The estimated effect on age at AR, and hence the scaling of developmental age, was very similar with the cross-sectional and longitudinal data; 2.4% and 2.3% age shrinkage per allele respectively.

Discussion
We present an investigation into the association of rs9939609 with BMI in infancy and childhood. Results here not only confirm the increasing magnitude of associations between this variant and adiposity from the end of infancy through to childhood, but also suggest an inverse association at early ages—an event intimately linked with the timing of AR. With resolution afforded by a large sample size and dense anthropometry data, we have been able to demonstrate that rather than a null association between the rs9939609 adult adiposity associated variant and BMI before the age of 2.5 years, each extra minor (A) allele at this locus is associated with lower BMI. After the age of 5.5 years a positive association between rs9939609 and BMI emerges as seen in previous work [24]. However this study reveals further details of the association by examining the genotype-specific trends. Thus the two additive effects of the A allele are to accelerate developmental age by around 2.4% per allele, corresponding to an early AR, and at the same time to increase BMI accretion by about 0.1 kg/m2 per allele from 1 to 13 years. This is confirmed in longitudinal modelling of our richest data. High levels of adult BMI may be preceded by either raised BMI throughout childhood, or alternatively normal or low BMI during infancy with an early AR and steep climb to high adult BMI [36]. Results from this work suggest that, rather than predisposing to elevated adipose levels across the lifecourse, variation at FTO is associated with a shift in the timing of AR, and is entirely consistent with both the contrast between patterns of association between infancy and childhood and the later life BMI associations by which this locus was discovered [2].
The changing associations between rs9939609 and BMI over the course of infancy and childhood are consistent with what is known about the biology of this locus. It has been suggested previously that energy balance through early life may exert an influence on the timing of AR [36], [41], [42]. If FTO is operating through an influence on appetite and the amount of food consumed [7]–[9], [13], [14], [19], then it may be that as individuals are able to autonomously regulate dietary intake (and other relevant behavioural traits such as activity), realised differences in appetite will have an impact on the age at AR. Alternatively, before this period, it may be that differential metabolic activity according to genotype exerts influence on the patterns and timing of BMI change [10]. These possibilities reflect an anticipated interplay between genetic and environmental factors supported also by the only simultaneous study of the heritability and genetics of BMI to date [25]. Beyond this and without further functional understanding as to the action of the FTO locus, it is difficult to speculate as to the mechanism by which variation at this locus might lead to earlier AR.
Considering the implications of these findings, rapid early weight gain is a known risk factor for later obesity [43]–[45] and since weight gain is calculated as weight increment divided by time interval, shortening the developmental time interval has the same impact on gain as increasing the increment. More directly, earlier AR has been consistently associated with higher BMI and the risk of obesity in later life [30]–[32], [34]. Furthermore, this relationship has been shown to be both incremental [33] and predictive of downstream risk of diseases such as T2D and coronary heart disease [46], [47]. Overall, whilst the impact of AR being ∼5% earlier for AA carriers may be relatively small, the ultimate impact on BMI trajectory may have important lifecourse effects which go some way to explain the known associations between this genotype and the binary phenotype “obesity”. Secondary effects of such relationships may also be seen in features such as early puberty which is known to be associated with greater adiposity [48], [49] and corresponds to an advanced developmental age. This is in accordance with our second finding, that in addition to the earlier AR associated with minor allele carriage, BMI in this group is low early on, but subsequently rises faster. Thus we predict that suitably powered studies of BMI around puberty will show the minor allele at rs9939609 associated both with earlier puberty and greater adiposity.
Although FTO impacts on developmental age it is important to stress that it applies to BMI and not to height. There are only minor differences in height growth between the three genotype groups (Figure 3), and they correspond to an age scaling per A allele of just 0.3%, which is only an eighth of the BMI age scaling effect. Thus it is not a generalised maturation effect but specific to adiposity accretion.
Despite the strengths of this study, it has limitations. Firstly, the definition of age windows in longitudinal analysis may not be optimal. There are still age groups for which we have limited data and this is reflected in the sampling error associated with these periods of growth and development. Furthermore, although the ability to examine the influence of this locus at different ages is aided by an analysis of many samples, the stratum specific estimates and their error terms are subject to the different measurement techniques and ages. The availability of further collections with broad age ranges and dense growth data and genotyping would increase the precision of findings documented here.
Secondly, a possible complication to the patterns of association seen between common variation at the FTO gene and BMI relates to the interplay between maternal and offspring genotypes. Whilst not within the bounds of this paper, the observation that mothers with greater BMI have, on average, offspring with greater birthweight who go on to be larger may be relevant in our interpretation of results [50]–[56]. Owing to the correlation between maternal and foetal genotypes, one may hypothesise that, on average, the elevation of adiposity in mothers carrying minor alleles at rs9939609 may translate to increased levels of birthweight or differential growth and development in early ages as shown observationally [57], [58]. This would theoretically counter the inverse association between minor (A) alleles at rs9939609 and BMI in offspring at very young ages that we have documented. Whilst this does not appear to be occurring, proper examination of this requires further large collections with available maternal genotypes (Table S2).
Lastly, the value of BMI as an assessment of adiposity at early ages has been questioned [40], [59], although BMI is still commonly used. In this investigation, we performed sensitivity analyses using the derived measure weight/heightp [60] to account for this limitation and found that results were largely consistent with weight/height2. For this reason and for consistency with later ages in childhood, we adopted the use of BMI throughout.
Overall, we conducted a large analysis of the association between common variation at FTO locus and BMI. We have noted that the effect of this locus appears to be age dependent. Importantly our results suggest an inversion of the known adult association between this locus and BMI at ages below 2.5 years, an observation which is consistent with relationships between variation in FTO and the timing of AR and which may help develop understanding of the biological mechanisms behind the association between common variation at this locus, adiposity related traits and disease risk. Further, specific, analyses will be required to confirm the age dependent associations and to investigate the clinical implications of associations between common genetic variation BMI and the timing of AR.
Materials and Methods
To examine the association between rs9939609 genotype and BMI from birth to 13 years of age we used the growth measurements from eight studies (Table 1). All subjects were unrelated children of white European ancestry, with multiple births excluded. When multiple siblings were present, only data from the oldest sibling were used. All studies have previously been described in detail, but brief descriptions are given below.
The Avon Longitudinal Study of Parents and Children (ALSPAC) is a prospective birth cohort in Bristol, UK, which recruited pregnant women with expected delivery dates in 1991–1992 (present analysis: 7,482 subjects). The Barry Caerphilly Growth Study (BCG) is a longitudinal study of infants born in the towns of Barry and Caerphilly in South Wales between 1972 and 1974 (569 subjects). The Christ's Hospital Cohort (CH) is a retrospective follow-up study comprised of former male students between the ages of 10 and 18 years of Christ's Hospital School born between 1927 and 1956 (812 subjects). As part of the Energy Balance Study (EBS), data were collected in 2002 and 2003 on pre-pubertal schoolchildren, ages 4 through 10 years, from north-eastern Scotland (2,604 subjects). The Generation R Study (GENR) is a prospective birth cohort from early foetal life onwards based in Rotterdam, the Netherlands; subjects born between 2002 and 2006 (2,851 subjects). The Northern Finland Birth Cohort 1966 (NFBC1966) is a prospective pregnancy/birth cohort with expected deliveries in 1966 in the two northernmost provinces of Finland (3,707 subjects). The Raine Study (RAINE) is a prospective pregnancy cohort set up in 1989, which recruited pregnant women from Perth, Western Australia for ultrasound imaging (1,106 subjects). The Uppsala Family Study Study (UFS) is a multigenerational study set up in 1995 in Uppsala, Sweden (594 subjects).
In all studies, weight and height were measured during routine visits at community health centres or research centres. All subjects (or their parents/guardians) gave informed consent and each study obtained ethical approval from the local ethical review board. For further details please see Text S1.
Genotyping and quality control
Genotyping of the rs9939609 was performed directly in all eight cohorts. DNA was isolated either from buccal swabs, blood or cord blood. Further details of the studies and of genotyping undertaken in them can be seen in the Text S1.
Analytical strategy and statistical analysis
Cross-sectional analyses
BMI was defined as weight [kg]/(height [m])2. For the cross-sectional meta-analysis, growth measurements were grouped into ten strata: 0.01 to <0.5 years (i.e. excluding birth); 0.5-; 1.5-; 2.5-; 3.5-; 4.5-; 5.5-; 7-; 9-; and 11 to <13 years. To approximate normality in each stratum BMI was natural log transformed before analysis, and effects may be expressed as percentage changes through multiplication by 100 [61]. To remove outliers stratum-specific Z-scores were created using the “zscore” package in STATA (version 11, Stata Corp. Texas, USA) and values exceeding ±3 were excluded (height and weight were cleaned similarly). To examine the association between FTO genotype and BMI within each stratum multivariable linear regression was used [62]. Study-specific effect estimates within each stratum were created assuming an additive genetic model. These models were adjusted for age because age varied within each age-stratum and sex. No further adjustment was done as variation at rs9939609 is unrelated to birth weight or gestational age [2] and the distribution of genotypes is assumed to be unrelated to possible environmental confounders [63]. To take into account known correlations between BMI and height at early ages, a sensitivity analysis was done using weight/heightp (p ranging from 1.7 to 2.8), with estimated stratum - and sex-specific powers p for each study [60]. Basic analyses were performed in Stata 11 (Stata corp.).
Cross-sectional cohort-specific results were meta-analyzed within each stratum using a random effects model to account for the existence of heterogeneity between studies. Analyses were performed using the “metan” package in STATA (version 11, Stata Corp. Texas, USA).
Curves of median BMI by age were estimated for each of the three genotypes, using the LMS method to adjust for age-specific heteroscedasticity and skewness [64]. This was implemented with the “gamlss” package in R (version 2.11.1) [65]. The median (or M) curve was modelled as a penalized B-spline in log age with 9 degrees of freedom, with corresponding curves for the coefficient of variation (S) with 5 d.f. and skewness (L) with 3. Study (as an eight-level factor) and sex were also adjusted for. Data points with age <0.1 years were excluded to improve model convergence. To illustrate the effect on the curves of a k% age scaling per A allele, scaled age was defined as , where n(A) is the number of A minor alleles (0, 1 or 2), and C is such that mean age(k) = mean age. This ensured that the age scale was foreshortened by k% in the AA group and stretched by k% in the TT group, each relative to TA, while maintaining the same mean age.
It is not possible to estimate the optimal age scaling explicitly by linear regression. Instead a series of six LMS models was fitted with age scaled by k = 0, 1 … 5%, each with a single median BMI curve fitted as before as a penalized B-spline curve in log age(k) with 9 d.f., adjusted for study and sex, and S and L curves with respectively 5 and 3 d.f. The fitted deviance dev(k) for each model was plotted against k and a quadratic in k fitted such that . By differentiation the optimal scaling was given by with 95% confidence interval where and [66]. The model was refitted at for confirmation. As a further stage this age-scaled analysis was repeated including an additive log age(k) by minor allele interaction, and the value of re-estimated.
Longitudinal analyses
Modelling BMI longitudinally is complex due to the adiposity peak in infancy and the increasing population variance in BMI throughout childhood. For this reason the data were split into two age windows: 2 weeks to 18 months (infancy) and 18 months to 13 years (childhood), using the studies with the most data in these age windows (BCG, GENR, NFBC1966 and UFS in the first and ALSPAC, RAINE, NFBC1966 and Uppsala in the second). BMI measurements in the first two weeks of life were excluded to avoid the period of weight loss after birth. The change point between infancy and childhood was set at 18 months primarily on statistical grounds to lie roughly mid-way between AP and AR.
Derivation of age and BMI at adiposity peak (AP) and adiposity rebound (AR)
Age and BMI at AP and AR were derived from cubic models in age for the two age groups separately, with random effects for the intercept and slope terms. Sex was also adjusted for, but rs9939609 genotype was ignored in order to estimate the outcomes for individuals and to then relate them to FTO. Subsequently, sex interactions with linear and quadratic age were added to the childhood model (both interactions p<0.01 in ALSPAC and NFBC1966). The models are written as:
-
Infancy model:
-
Childhood model:
For each participant, predicted BMI at AP and AR (on a grid of every 0.05 years in infancy and every 0.1 years in childhood) was calculated using the estimated fixed and random coefficients. Age at AP was defined as the age at maximum BMI between 0.25 and 1.25 years, and age at AR as the age at minimum BMI between 2.5 and 8.5 years. These cut-off points were chosen based on descriptive analysis of growth curves in the NFBC1966. The associations between rs9939609 genotype and these growth parameters were analyzed using both general and additive genetic models. To account for uncertainty in the derived parameters, each person's data were weighted by the number of measurements within the age window, and those with fewer than three were excluded. Sensitivity analyses with gestational age as a further adjustment in the AP models made no substantive difference to the results (performed in the NFBC1966). Age at AP and age at AR were analysed without transformation, but are presented as percentages for comparison with the cross-sectional results. BMI at AP and AR was log-transformed due to right skewness, and association results are reported as percentage differences in BMI between genotypes by multiplying the log differences by 100. Study-specific association results between each growth parameter and FTO were meta-analyzed using random effects models to account for the existence of heterogeneity between studies using the “metan” package in STATA (version 11, Stata Corp. Texas, USA).
Supporting Information
Zdroje
1. DinaC
MeyreD
GallinaS
DurandE
KornerA
2007 Variation in FTO contributes to childhood obesity and severe adult obesity. Nature Genetics 39 724 726
2. FraylingTM
TimpsonNJ
WeedonMN
ZegginiE
FreathyRM
2007 A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316 889 894
3. HinneyA
NguyenTT
ScheragA
FriedelS
BronnerG
2007 Genome wide association (GWA) study for early onset extreme obesity supports the role of fat mass and obesity associated gene (FTO) variants. PLoS ONE 2 e1361 doi:10.1371/journal.pone.0001361
4. PriceRA
LiWD
ZhaoH
PriceRA
LiW-D
2008 FTO gene SNPs associated with extreme obesity in cases, controls and extremely discordant sister pairs. BMC Medical Genetics 9 4
5. ScuteriA
SannaS
ChenW
UdaM
AlbaiG
2007 Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet 20 e115 doi:10.1371/journal.pgen.0030115
6. WillerK
2009 Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nature Genetics 41 25 34 for the GIANT Consortium
7. CecilJE
TavendaleR
WattP
HetheringtonMM
PalmerCNA
2008 An obesity-associated FTO gene variant and increased energy intake in children. New England Journal of Medicine 359 2558 2566
8. SpeakmanJR
RanceKA
JohnstoneAM
SpeakmanJR
RanceKA
2008 Polymorphisms of the FTO gene are associated with variation in energy intake, but not energy expenditure. Obesity 16 1961 1965
9. WardleJ
LlewellynC
SandersonS
PlominR
2009 The FTO gene and measured food intake in children. International Journal of Obesity 33 42 45
10. FischerJ
KochL
EmmerlingC
VierkottenJ
PetersT
2009 Inactivation of the Fto gene protects from obesity. Nature 458 894 898
11. GerkenT
GirardCA
TungYL
WebbyCJ
SaudekV
2007 The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science 318 1469 1472
12. StratigopoulosG
PadillaSL
LeDucCA
WatsonE
HattersleyAT
2008 Regulation of Fto/Ftm gene expression in mice and humans. AJP - Regulatory, Integrative and Comparative Physiology 294 R1185 1196
13. TimpsonNJ
EmmettP
FraylingTM
RogersI
HattersleyAT
2008 The FTO/obesity associated locus and dietary intake in children. American Journal of Clinical Nutrition 88 971 978
14. ChurchC
LeeS
BaggEA
McTaggartJS
DeaconR
2009 A mouse model for the metabolic effects of the human fat mass and obesity associated FTO gene. PLoS Genet 5 e1000599 doi:10.1371/journal.pgen.1000599
15. TungYC
AyusoE
ShanX
BoschF
O'RahillyS
2010 Hypothalamic-specific manipulation of Fto, the ortholog of the human obesity gene FTO, affects food intake in rats. PLoS ONE 5 e8771 doi:10.1371/journal.pone.0008771
16. Villalobos ComparanM
Flores-DorantesMT
Villarreal-MolinaMT
2008 The FTO gene is associated with adulthood obesity in the Mexican population. Obesity 16 2296 2301
17. WahlenK
SjolinE
HoffstedtJ
2008 The common rs9939609 gene variant of the fat mass - and obesity-associated gene FTO is related to fat cell lipolysis. Journal of Lipid Research 49 607 611
18. ZabenaC
González-SánchezJL
Martínez-LarradMT
Torres-GarcíaA
Alvarez-Fernández-RepresaJ
2009 The FTO obesity gene. Genotyping and gene expression analysis in morbidly obese patients. Obesity Surgery 19 87 95
19. ChurchC
MoirL
McMurrayF
ChristopheG
BanksGT
2010 Overexpression of Fto leads to increased food intake and results in obesity. Nature Genetics 42 1086 1093
20. LeinES
HawrylyczMJ
AoN
AyresM
BensingerA
2007 Genome-wide atlas of gene expression in the adult mouse brain. Nature 445 168 176
21. OlszewskiPK
FredrikssonR
OlszewskaAM
StephanssonO
Alsi
2009 Hypothalamic FTO is associated with the regulation of energy intake not feeding reward. BMC Neuroscience 10 129
22. Lopez-BermejoA
PetryCJ
DiazM
SebastianiG
de ZegherF
2008 The association between the FTO gene and fat mass in humans develops by the postnatal age of two weeks. Journal of Clinical Endocrinology & Metabolism 93 1501 1505
23. QiL
KangK
ZhangC
van DamRM
KraftP
2008 Fat mass-and obesity-associated (FTO) gene variant is associated with obesity: longitudinal analyses in two cohort studies and functional test. Diabetes 57 3145 3151
24. HardyR
WillsAK
WongA
ElksCE
WarehamNJ
2010 Life course variations in the associations between FTO and MC4R gene variants and body size. Human Molecular Genetics 19 545 552
25. HaworthCM
CarnellS
MeaburnEL
DavisOS
PlominR
2008 Increasing heritability of BMI and stronger associations with the FTO gene over childhood. Obesity 16 2663 2668
26. JessT
ZimmermannE
KringSI
BerentzenT
HolstC
2008 Impact on weight dynamics and general growth of the common FTO rs9939609: a longitudinal Danish cohort study. International Journal of Obesity 32 1388 1394
27. LajunenHR
KaprioJ
Keski-RahkonenA
RoseRJ
PulkkinenL
2009 Genetic and environmental effects on body mass index during adolescence: a prospective study among Finnish twins. Int J Obes 33 559 567
28. TannerJM
1989 Foetus into man: physical growth from conception to maturity Ware (UK) Castlemead Publications
29. ColeTJ
DonaldsonMDC
Ben ShlomoY
2010 SITAR – a useful instrument for growth curve analysis. International Jounal of Epidemiology Submitted
30. Rolland-CacheraMF
DeheegerM
BellisleF
SempeM
Guilloud-BatailleM
1984 Adiposity rebound in children: a simple indicator for predicting obesity. American Journal of Clinical Nutrition 39 129 135
31. SiervogelRM
RocheAF
GuoSM
MukherjeeD
ChumleaWC
1991 Patterns of change in weight/stature2 from 2 to 18 years: findings from long-term serial data for children in the Fels longitudinal growth study. International Journal of Obesity 15 479 485
32. ProkopecM
BellisleF
1993 Adiposity in Czech children followed from 1 month of age to adulthood: analysis of individual BMI patterns. Annals of Human Biology 20 517 525
33. WilliamsS
DavieG
LamF
1999 Predicting BMI in young adults from childhood data using two approaches to modelling adiposity rebound. International Journal of Obesity & Related Metabolic Disorders: Journal of the International Association for the Study of Obesity 23 348 354
34. WhitakerRC
PepeMS
WrightJA
SeidelKD
DietzWH
1998 Early adiposity rebounf and the risk of adult obesity. Paediatrics 101 E5
35. WilliamsSM
GouldingA
WilliamsSM
GouldingA
2009 Patterns of growth associated with the timing of adiposity rebound. Obesity 17 335 341
36. Rolland-CacheraMF
DeheegerM
MaillotM
BellisleF
2006 Early adiposity rebound: causes and consequences for obesity in children and adults. International Journal of Obesity 30 S11 S17
37. BarkerDJ
OsmondC
ForsenTJ
KajantieE
ErikssonJG
2005 Trajectories of growth among children who have coronary events as adults. New England Journal of Medicine 353 1802 1809
38. PowerC
LiL
HertzmanC
2006 Associations of early growth and adult adiposity with patterns of salivary cortisol in adulthood. Journal of Clinical Endocrinology & Metabolism 91 4264 4270
39. GuoSS
ChumleaWC
1999 Tracking of body mass index in children in relation to overweight in adulthood. American Journal of Clinical Nutrition 70 supp 148S 148S
40. GuoSS
RocheAF
ChumleaWC
GardnerJD
SiervogelRM
1994 The predictive value of childhood body mass index values for overweight at age 35 y. American Journal of Clinical Nutrition 59 810 819
41. DeheegerM
Rolland-CacheraMF
FontveilleAM
1997 Physical activity and body composition in 10-year-old French children: linkages with nutritional intake? Int J Obes Relat Metab Disord 21 372 379
42. Rolland-CacheraMF
DeheegerM
AkroutM
BellisleF
1995 Influence of macronutrients on adiposity development: a follow up study of nutrition and growth from 10 months to 8 years of age. International Journal of Obesity & Related Metabolic Disorders: Journal of the International Association for the Study of Obesity 19 573 578
43. ReillyJJ
ArmstrongJ
DorostyAR
EmmettPM
NessAR
2005 Early life risk factors for obesity in childhood: cohort study. British Medical Journal 330 1357
44. TzoulakiI
SovioU
PillasD
HartikainenA-L
PoutaA
2010 Relation of Immediate Postnatal Growth With Obesity and Related Metabolic Risk Factors in Adulthood: The Northern Finland Birth Cohort 1966 Study. Am J Epidemiol 171 989 998
45. SinghalA
LucasA
2004 Early origins of cardiovascular disease: is there a unifying hypothesis? Lancet 363 1642 1645
46. ErikssonJG
2006 Early growth, and coronary heart disease and type 2 diabetes: experiences from the Helsinki birth cohort studies. International Journal of Obesity 30 S18 S22
47. ErikssonJG
ForsenT
TuomilehtoJ
OsmondC
BarkerDJ
2003 Early adiposity rebound in children and risk of type 2 diabetes in adult life. Diabetologia 46 190 194
48. PowerC
LakeJK
ColeTJ
1997 Body mass index and height from childhood to adulthood in the 1958 British born cohort. American Journal of Clinical Nutrition 66 1094 1101
49. SandhuJ
Ben-ShlomoY
ColeT
HollyJ
Davey SmithG
2006 The impact of childhood body mass index on timing of puberty, adult stature an obesity: A follow-up study based on adolescent anthropometry recorded at Christ's Hospital (1936–1964). International Journal of Obesity 30 14 22
50. TaylorPD
PostonL
2007 Developmental programming of obesity in mammals. Experimental Physiology 92 287 298
51. EhrenbergHM
MercerBM
CatalanoPM
EhrenbergHM
MercerBM
2004 The influence of obesity and diabetes on the prevalence of macrosomia. American Journal of Obstetrics & Gynecology 191 964 968
52. GillmanMW
2004 A lifecourse approach to obesity.
KuhD
Ben ShlomoY
Oxford OUP
53. OkenE
GillmanMW
2003 Fetal origins of obesity. Obesity Research 11 496 506
54. KirchengastS
HartmannB
1998 Maternal prepregnancy weight status and pregnancy weight gain as major determinants for newborn weight and size. Annals of Human Biology 25 17 28
55. MohantyC
PrasadR
Srikanth ReddyA
GhoshJK
SinghTB
2006 Maternal anthropometry as predictors of low birth weight. Journal of Tropical Pediatrics 52 24 29
56. HullHR
DingerMK
KnehansAW
ThompsonDM
FieldsDA
2008 Impact of maternal body mass index on neonate birthweight and body composition. American Journal of Obstetrics & Gynecology 198 416.e411 416
57. SurkanPJ
HsiehCC
JohanssonAL
DickmanPW
CnattingiusS
2004 Reasons for increasing trends in large for gestational age births. Obstetrics & Gynecology 104 720 726
58. KramerMS
MorinI
YangH
PlattRW
UsherR
2002 Why are babies getting bigger? Temporal trends in fetal growth and its determinants. Journal of Pediatrics 141 538 542
59. EllisKJ
AbramsSA
WongWW
1999 Monitoring childhood obesity: assessment of the weight/height index. American Journal of Epidemiology 150 939 946
60. ColeTJ
1986 Weight/heightp compared to weight/height2 for assessing adiposity in childhood: influence of age and bone age on p during puberty. Annals of Human Biology 13 433 451
61. ColeTJ
2000 Sympercents: symmetric percentage differences on the 100 loge scale simplify the presentation of log transformed data. Statistics in Medicine 19 3109 3125
62. McCullaghP
NelderJ
1989 Generalised Linear Models London Chapman and Hall
63. Davey SmithG
LawlorDA
HarbordR
TimpsonN
DayI
2007 Clustered environments and randomized genes: a fundamental distinction between conventional and genetic epidemiology. PLoS Med 4 e352 doi:10.1371/journal.pmed.0040352
64. ColeTJ
GreenPJ
1992 Smoothing reference centile curves: the LMS method and penalized likelihood. Statistics in Medicine 11 1305 1319
65. RigbyRA
StasinopoulosDM
2005 Generalized additive models for location, scale and shape (with discussion). Applied Statistics 54
66. EdwardsAWF
1972 Likelihood Cambridge CUP
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 2
Nejčtenější v tomto čísle
- Meta-Analysis of Genome-Wide Association Studies in Celiac Disease and Rheumatoid Arthritis Identifies Fourteen Non-HLA Shared Loci
- MiRNA Control of Vegetative Phase Change in Trees
- Risk Alleles for Systemic Lupus Erythematosus in a Large Case-Control Collection and Associations with Clinical Subphenotypes
- The Cardiac Transcription Network Modulated by Gata4, Mef2a, Nkx2.5, Srf, Histone Modifications, and MicroRNAs