
Whole-Genome Comparison Reveals Novel Genetic Elements That Characterize the Genome of Industrial Strains of
Human intervention has subjected the yeast Saccharomyces cerevisiae to multiple rounds of independent domestication and thousands of generations of artificial selection. As a result, this species comprises a genetically diverse collection of natural isolates as well as domesticated strains that are used in specific industrial applications. However the scope of genetic diversity that was captured during the domesticated evolution of the industrial representatives of this important organism remains to be determined. To begin to address this, we have produced whole-genome assemblies of six commercial strains of S. cerevisiae (four wine and two brewing strains). These represent the first genome assemblies produced from S. cerevisiae strains in their industrially-used forms and the first high-quality assemblies for S. cerevisiae strains used in brewing. By comparing these sequences to six existing high-coverage S. cerevisiae genome assemblies, clear signatures were found that defined each industrial class of yeast. This genetic variation was comprised of both single nucleotide polymorphisms and large-scale insertions and deletions, with the latter often being associated with ORF heterogeneity between strains. This included the discovery of more than twenty probable genes that had not been identified previously in the S. cerevisiae genome. Comparison of this large number of S. cerevisiae strains also enabled the characterization of a cluster of five ORFs that have integrated into the genomes of the wine and bioethanol strains on multiple occasions and at diverse genomic locations via what appears to involve the resolution of a circular DNA intermediate. This work suggests that, despite the scrutiny that has been directed at the yeast genome, there remains a significant reservoir of ORFs and novel modes of genetic transmission that may have significant phenotypic impact in this important model and industrial species.
Published in the journal:
. PLoS Genet 7(2): e32767. doi:10.1371/journal.pgen.1001287
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001287
Summary
Human intervention has subjected the yeast Saccharomyces cerevisiae to multiple rounds of independent domestication and thousands of generations of artificial selection. As a result, this species comprises a genetically diverse collection of natural isolates as well as domesticated strains that are used in specific industrial applications. However the scope of genetic diversity that was captured during the domesticated evolution of the industrial representatives of this important organism remains to be determined. To begin to address this, we have produced whole-genome assemblies of six commercial strains of S. cerevisiae (four wine and two brewing strains). These represent the first genome assemblies produced from S. cerevisiae strains in their industrially-used forms and the first high-quality assemblies for S. cerevisiae strains used in brewing. By comparing these sequences to six existing high-coverage S. cerevisiae genome assemblies, clear signatures were found that defined each industrial class of yeast. This genetic variation was comprised of both single nucleotide polymorphisms and large-scale insertions and deletions, with the latter often being associated with ORF heterogeneity between strains. This included the discovery of more than twenty probable genes that had not been identified previously in the S. cerevisiae genome. Comparison of this large number of S. cerevisiae strains also enabled the characterization of a cluster of five ORFs that have integrated into the genomes of the wine and bioethanol strains on multiple occasions and at diverse genomic locations via what appears to involve the resolution of a circular DNA intermediate. This work suggests that, despite the scrutiny that has been directed at the yeast genome, there remains a significant reservoir of ORFs and novel modes of genetic transmission that may have significant phenotypic impact in this important model and industrial species.
Introduction
During its long history of association with human activity, the genomic makeup of the yeast S. cerevisiae is thought to have been shaped through the action of multiple independent rounds of wild yeast domestication combined with thousands of generations of artificial selection. As the evolutionary constraints that were applied to the S. cerevisiae genome during these domestication events were ultimately dependent on the desired function of the yeast (e.g baking, brewing, wine or bioethanol production), these multitude of selective schemes have produced large numbers of S. cerevisiae strains, with highly specialized phenotypes that suit specific applications [1], [2]. As a result, the study of industrial strains of S. cerevisiae provides an excellent model of how reproductive isolation and divergent selective pressures can shape the genomic content of a species.
Despite their diverse roles, industrial yeast strains all share the general ability to grow and function under the concerted influences of a multitude of environmental stressors, which include low pH, poor nutrient availability, high ethanol concentrations and fluctuating temperatures. In comparison, non-industrial isolates such as laboratory strains, have been selected for rapid and consistent growth in nutrient rich laboratory media, thereby producing markedly different phenotypic outcomes when compared to their industrial relatives [3]. The outcomes of these very different selection pressures are therefore most evident when comparing industrial and non-industrial yeasts. As an example, laboratory strains of S. cerevisiae, such as S288c, are unable to grow in the low pH and high osmolarity of most grape juices and therefore cannot be used to make wine. This is a clear difference between industrial and non-industrial strains of S. cerevisiae, however there are numerous subtle differences not only between industrial strains, but also between strains used within the same industry [4], [5], highlighting the overall genetic diversity found in this species.
There have been several attempts to characterize the genomes of industrial strains of S. cerevisiae which have uncovered differences that included single nucleotide polymorphisms (SNPs), strain-specific ORFs and localized variations in genomic copy number [6]–[14]. However, the type and scope of genomic variation documented by these studies were limited either by technology constraints (e.g arrayCGH relying on the laboratory strain as a “reference” genome), or by the resources required for the production of high-quality genomic assemblies which has limited the scope and number of whole-genome sequences available for comparison. In addition, to limit genomic complexity to a manageable level, previously published whole-genome sequencing studies on industrial strains used haploid representations of diploid, and often heterozygous, commercial and environmental strains [9]–[13].
We sought to address these shortcomings by sequencing the genomes of four wine and two brewing strains of S. cerevisiae in their industrially-used forms. The industries of winemaking and brewing were targeted for this work as they have the longest association with S. cerevisiae (measured in the thousands of years) and each industry has accumulated large numbers of phenotypically distinct strains for which genetic comparisons can be made. This study demonstrates that industrial yeasts display significant genotypic heterogeneity both between strains, but also between alleles present within strains (i.e. heterozygosity). This variation was manifest as SNPs, small insertions and deletions, and as novel, strain and allele-specific ORFs, many of which had not been found previously in the S. cerevisiae genome and may provide the basis for novel phenotypic characteristics. Interestingly, several ORFs were shown to comprise a gene cluster that was present in multiple copies and at a variety of genomic loci in a subset of the strains examined. Furthermore, this cluster appears to have integrated into genomic locations by a novel circular intermediate, but without employing classical transposition or homologous recombination, which we believe represents the first time such an element has been characterized in S. cerevisiae.
Overall, this work suggests that, despite the scrutiny that has been directed at the yeast genome, there remains a significant reservoir of ORFs and novel modes of genetic transmission which may have significant phenotypic impact in this important model and industrial species.
Results
Six industrial yeasts were chosen for genomic analysis, comprising four commercial wine strains and two brewing strains used for the production of ales (ale strains are primarily S. cerevisiae, while lager-style brewing strains are S. pastorianus, a hybrid of S. cerevisiae and S. bayanus [15], [16]). These six strains were sequenced to an average coverage of 20 fold with a combination of shotgun and paired-end methods using the GS FLX Titanium series chemistry [17], which resulted in six high quality genomic assemblies (Table 1).

Large chromosomal variations in industrial yeast strains
Rather than being strictly diploid, many industrial yeast strains display chromosomal copy number variation (CNV) [18]. In order to catalogue CNV in the industrial yeast genomes, the depth of sequencing coverage determined for each sequence contig were calculated such that areas of CNV could be detected as localized variations in that coverage (Figure 1). There were several large areas of increased copy number across the strains including six potential whole-chromosome amplifications (chrI of AWRI796, chrVIII of VL3, chrIII of FostersO and chrIII, V and XV of FostersB) and one potential reduction in chromosomal copy number (chrXIV of FostersO). There were also several partial chromosomal CNVs, including amplification of 200 kb of chrXIV in AWRI796, 600 kb of chrII and 200 kb of chrX in FostersO and a 400 kb reduction from chrVII of FostersO (Figure 1). However, while the ale strains had a higher number of large CNVs than wine strains, the overall fold change of these CNVs was generally reduced. This reduction can be most easily explained by the brewing strains having a polyploid genetic base while the wine strains are diploid, an observation which has been seen previously in these industrial yeasts [18].

Heterozygosity in industrial strains
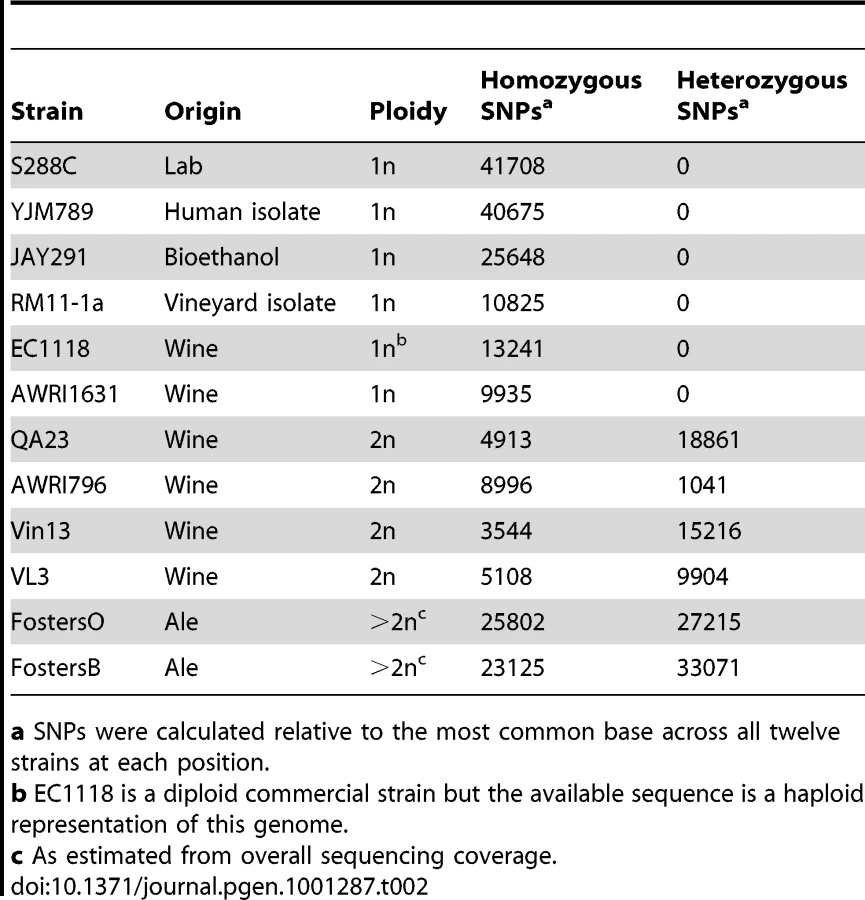
As existing published industrial yeast genome sequences were either generated from haploid derivatives of industrial strains [9]–[12] or had heterozygous regions discarded during analysis [13], the level of genome-wide heterozygosity present in industrial strains remains largely unknown. However, as the assemblies performed in this study retained genomic heterozygosity, it was possible to determine the level of allelic differences within each of these strains (Table 2). While every industrial strain contained heterozygous single nucleotide polymorphisms (SNPs), the proportion of these varied over thirty-fold between wine strain AWRI796 (1041 total heterozygous bp) and the brewing strain FostersB (33071 bp). Heterozygous insertions and deletions (InDels) were also present and ranged from single base pair variants to large InDels of up to 35.3 kb. Strains were also shown to contain heterozygous instances of Ty element insertion, although, due to the repetitive nature of these elements, their presence in the genome could generally only be estimated through paired-end information (data not shown).
Nucleotide variation present in S. cerevisiae
In addition to the intra-strain variation that was present between homologous chromosomes within individual strains, there was also significant nucleotide variation between strains. As seen for the allelic variation, both SNPs and InDels were found between strains, with inter-strain InDels of up to 45 kb being observed. Many of the smaller InDels (both heterozygous and homozygous) were located in regions comprising tandem repeats (Figure 2A, Table S1) and primarily in the expansion and contraction of di - and tri-nucleotide tandem repeats (Figure 2B). Indeed, when using chromosome XVI as an example, over 86% of the instances of di - and tri-nucleotide repeats displayed variable length in at least one of the strains. As the size of tandem repeats has been associated with differences in gene expression [19], this suggests that there are both strain and allele-specific differences in the expression of genes proximal to these repeat-associated InDel events.

SNP variation was also common throughout the strains with a total of 165,913 non-degenerate SNPs (unique points of nucleotide variation) that were present in at least one allele of the twelve strains investigated (∼1.3% of the total genome length). However, given the influence of large, strain-specific InDels (which were filtered out of the SNP analysis) the apparent SNP density is much higher than 1.3%, such that these SNPs were shown to display a median inter-SNP distance of only 37 bp.
By using the number of SNPs separating any two isolates as an estimation of their relatedness (Figure 3A), we were able to show that industrial yeasts are distinct from both the laboratory and human pathogenic strains and were also found to group by industry. This was especially true of the brewing strains which displayed a high degree of genetic distance not only from the laboratory and human isolates, but also from the wine and bioethanol strains. The only exception to this pattern of grouping by industry or environment niche was with the ‘natural’ isolate RM11-1a which grouped closely with wine strains. However, given that it is descended from a strain sourced from a vineyard, RM11-1a may well share genetic origins with those strains used in winemaking.

In order to put the genetic variation observed in these genomic alignments in a larger population context, twelve strains were selected to represent each of the six main S. cerevisiae population groups as proposed by Liti et al [12] for further SNP comparison (Figure 3B). In this broader context, wine strains sequenced in this study were shown to also group tightly with the wine/European strains DBVPG1106 and DBVPG1373, showing that the data produced across these two studies are directly comparable. However, while the ale strains were still shown to be distinct from the wine isolates they were found to be far closer to the wine strains than isolates such as those used in sake production, which display the greatest level of nucleotide diversity when compared to the wine strains. Indeed, when the SNP data from these additional strains in included in the calculations of SNP density, the total number of non-degenerate SNPs increases to 216,207 (∼1.7%) with a median inter-SNP distance of only 27 bp. However, despite comparisons to eighteen other diverse strains of S. cerevisiae 15,576 of these SNPs were found solely in this study (2,501 in more than one strain) and with the vast majority of these SNPs being present in a heterozygous form (only 1,864 novel SNPs were homozygous in at least one strain).
ORF conservation across S. cerevisiae
To determine how inter-specific variation at the nucleotide level translated into protein-coding differences, the predicted coding potential of each strain was compared. ORFs were predicted from each sequence (including the pre-existing whole genome sequences) using Glimmer [20] and compared using a combination of BLAST [21] homology matches and genomic synteny to differentiate instances of orthology from gene duplication (Table S2). When using the laboratory strain S288c as a reference, there was an average of 92% ORF coverage across the strains. The majority of S288c ORFs without a match in other strains were shown to be located in repetitive regions of the S. cerevisiae genome such as in the sub-telomeric zones or the numerous Ty retrotransposons that are present in S288c genome relative to other strains. Due to the repetitive nature of these regions it was often impossible to unambiguously position these sequences in the industrial yeast genome assemblies and they remain within repetitive, unmappable contigs in the various genome assemblies. It therefore appears that, due to its persistent propagation in the laboratory, the genome of S288c may represent a reduced genomic state as it does not appear to contain additional genes that provide unique metabolic or cellular potential outside of those present in other strains. It does however contain a far greater number of Ty transposons relative to all of the other strains suggesting that transposon proliferation occurred on at least one occasion during the development of this laboratory strain.
Novel ORFs
While the laboratory strain S288c is considered the reference for the genomic complement of S. cerevisiae, it is becoming apparent that it lacks a multitude of ORFs which exist in other strains of S. cerevisiae [9]–[13], [22], [23]. This is confirmed n the present study with between 36 (FostersB) and 110 (Lalvin QA23) ORFs lacking significant homology to the S288c genome but for which there were clear matches to sequences in other S. cerevisiae strains or microbial species (Table S2). Orthologs of 102 out of 218 of the non-degenerate set of these ‘non-S288c’ ORFs have been identified previously in S. cerevisiae strains, mainly through whole-genome sequencing of AWRI1631, EC1118 and RM11-1a and YJM789 [8], [9], [13] (Table S2). These include genes encoding proteins such as the Khr1 killer toxin [24] which is found in YJM789, EC1118, Vin13, VL3, FostersB and FostersO and orthologs of the MPR1 stress-resistance gene (which was originally identified in the Sigma 1278b strain[23]) in RM11-1a, EC1118, AWRI1631, JAY291, QA23 and VL3.
Interestingly, in addition to these ORFs there were at least three proteins present in the human pathogen YJM789 and the FostersB and FostersO ale strains but which were lacking from the wine, biofuel and laboratory strains (Figure 4C). These included the YJM-GNAT GCN5-related N-acetyltransferase [8] and a separate gene cluster which is predicted to contain both RTM1, which was identified previously as a distillery-strain specific gene that provides resistance to an inhibitory substance found in molasses [22], and a large ORF of around 2.3 kb which, despite its large size and high-degree of conservation across the brewing and human pathogenic strains, lacks significant homology to any other protein sequences except for six isolates from the large S. cerevisiae population genomic screen which also appear to encode this protein [12] (Figure S1). In addition to these two conserved ORFs, in the ale strains this cluster also appears to encode an invertase that would be expected convert sucrose into the sugars glucose and fructose.

Despite the presence of at least two existing high-coverage wine strain sequences and at least an additional six low coverage genomes, the entire repertoire of ORFs present in wine strains of S. cerevisiae, let alone the species as a whole, is far from complete. In addition to expanding the strain range of previously identified non-S228c proteins, it was possible to identify at least eleven ORFs that lacked homology to existing proteins from S. cerevisiae, in addition to many new paralogs of existing S. cerevisiae genes. These novel ORFs often clustered in large InDels, the largest of which was a 45 kb fragment in the wine strain AWRI796. This novel genomic region is located adjacent to a large repetitive element present on chromosomes XIII, XV and XVI, which hampered initial efforts to assign this region to a specific chromosome. However, through the application of a 20 kb paired-end library, it was possible to bridge the repetitive region and position this novel region at the end of the right arm of chromosome XV. This fragment is predicted to encode nineteen ORFs (Figure 4A), three of which are predicted to encode aryl-alcohol dehydrogenases (AADs). AADs have been extensively characterized in filamentous fungi where they catalyze the reversible reduction of aldehydes and ketones to aromatic alcohols during lignin-degradation [25], [26]. These new AAD homologs are phylogenetically distinct from other AAD enzymes that have been identified, including the seven predicted AADs that are present in the S288c genome [27], [28] (Figure 4B).
Characterization of a novel, and potentially transmissible, gene cluster
One particularly curious feature of many of the industrial yeast strains analyzed in this study, was a cluster of five conserved ORFs that was present in all of the wine strains, RM11-1a and the bioethanol strain JAY291, and potentially in at least four of the strains present in the Liti et al [12] study (Figure 3). This cluster is predicted to encode two potential transcription factors (one zinc-cluster, one C6 type), a cell surface flocullin, a nicotinic acid permease and a 5-oxo-L-prolinase, and has been suggested to be horizontally acquired by S. cerevisiae from Zygosacharomyces spp [13]. In this study we have been able to show that while the sequences of the individual genes within this cluster are highly conserved between strains, the cluster itself is actually highly diverse with respect to copy number, genomic location and overall gene order (Figure 5, Table S3). The cluster was present in one to at least three copies across strains, with individual clusters being located in at least seven different genomic loci (Figure 5A). For example, wine strain Lalvin QA23 was shown to contain at least three copies of the cluster, found in three different genomic loci and with at least two copies being heterozygous. However, despite this diversity, the sequence of the ORFs and intergenic regions of the cluster were highly conserved, with only fifteen nucleotide substitutions (0.01%) recorded across the eleven known copies of the cluster (Figure 5B, Figure S2).

In addition to the differences in copy number and location, the exact order of the ORFs within the cluster differed in a location dependent manner (Figure 5B, 5C). However, all of these different ORF arrangements could be resolved into a syntenically-conserved order if the linear genomic copy of each cluster resulted from the differential resolution of a common circular intermediate, with a unique breakpoint in this circular arrangement being observed for each genomic location (Figure 5B–5D). However, despite the differential location of these clusters these integration events appear to select for functional conservation of the genes with the majority of the breakpoints being located within intergenic regions (Figure 5B). Of the two exceptions to this, one of these events occurs at the extreme 3′ end (∼100 bp from the predicted stop codon) of one ORF such that a functional protein is likely to still be produced from this gene.
Adding further interest to the mode of transfer of this cluster, its integration into the genome appears to occur without the production of the terminal repeated sequences that would be expected if integration of this element occurred by either homologous recombination or classical mobilization via a transposon-like mechanism. In fact, for at least three of the seven different integration events characterized in this study, integration of the cluster has occurred between two directly adjacent, conserved nucleotides, with a further two events showing only single nucleotide indels at the junction between the cluster and the flanking genomic sequences (Figure 5E).
Discussion
While S. cerevisiae is one of the most intensively studied biological model organisms and economically-important industrial microorganisms, many characteristics of its genome remain unknown, especially in strains other than the laboratory reference S288c. Through the analysis of six industrial strains, it was possible to show that the industrial members of this species are distinct, with wine and brewing strains being almost as distantly related at the DNA level as they are to either the laboratory or human pathogenic strains. This suggests that despite their roles in performing industrial fermentations, the two groups comprise genetically separate S. cerevisiae lineages. While this is a situation similar to that proposed previously for wine and sake strains of S. cerevisiae [2], the wine and ale strains were much more closely related to each other than to strains with origins outside of Europe [12], and this may reflect a distant common European-type ancestor. The bioethanol strain JAY291 displays an intermediate level of sequence relatedness to the wine strains (compared to ale strains) and also contains the five-gene cluster, suggesting that this strain shares at least some of its genomic origins with the wine isolates. With the relatively recent development of the bioethanol industry, it is not entirely unexpected that yeasts used in this process may well have their origins in commercial strains used in established ethanologenic industries. Wine strains would therefore make a logical choice for this starting point given their highly efficient production of ethanol and relatively high tolerance to a variety inhibitory substances, such as ethanol or polyphenols, that also exist in bioethanol fermentations [29].
In addition to mapping the relationships between these strains, this study uncovered a number of genetic elements not previously identified in the S. cerevisiae genome, as well as expanding the range of several strain specific elements that had been identified previously. This highlights the fact that the genetic variation that underlies the phenotypic diversity of S. cerevisiae goes well beyond that of SNPs or small InDels and is similar to the situation observed with many bacterial species where the pan (species-wide) genome is larger than that observed in any single strain [30]. As for the situation observed with single nucleotide variation, several of these genetic elements link strains to specific industries (e.g. the RTM1 cluster in the ale strains and the five-gene cluster in the wine strains). It would therefore be expected that these ORFs provide selective advantage within specific industries that have favored their retention. For some of these ORFs, such as the RTM1 cluster, the phenotypic benefits that they have historically provided in one industry may be advantageous in modern incarnations of others. For example, modern wine production generally makes use of inoculated commercial strains (rather than the historical use of wild yeast), which are produced on a large scale using molasses as a feedstock. Genes such as the RTM1 cluster may therefore provide advantages in the production of modern commercial wine yeast, but which are lacking from the genomic complement of this group of strains due to the historical practices of winemaking.
While other strain-specific ORFs were shown to have much narrower strain ranges (often single strains), it was possible to predict industrially-relevant roles for some of these genes. For example, the novel AAD proteins that were identified in the wine strain AWRI796 may have a direct impact on the range of volatile aromas produced during fermentation, as the aromatic alcohols produced through the action of the AAD enzymes can present very different aromas profiles to their corresponding aldehydes and ketones [31]. The presence of these AADs in specific industrial yeasts may therefore alter the profile of volatile aromas produced during winemaking or brewing, contributing to strain-specific aroma characteristics that are vitally important to many flavor and aroma-based industrial applications.
The role of ORFs such as those present in the wine yeast five-gene cluster are less clear but, given the potential regulatory role for at least two of these proteins, they could produce significant phenotypic effects. The generally similar characteristics of high sugar and ethanol tolerance of Zygosacharomyces spp and the wine and bioethanol strains of S. cerevisiae [29], [32], may provide a selective advantage for growth under these conditions. However, understanding the function of individual ORFs is overshadowed by questions regarding the origins of this novel cluster in addition to its effect on genome structure and dynamics. It was recently proposed that this cluster entered the S. cerevisiae genome from Zygosacharomyces spp [13]. Our data suggests that if this is the case, the transfer has either occurred on multiple occasions via a conserved circular intermediate that has integrated randomly into different genomic loci, or the fragment has entered the S. cerevisiae genome on a single occasion but has subsequently mobilized to new genomic locations via a circular intermediate (Figure S3). Alternatively, this cluster is a mobile feature of the S. cerevisiae genome that has been lost from many strains and was transferred to Zygosacharomyces spp. Regardless of the direction or precise mode of transfer it appears that this genetic cluster may mobilize throughout the genome via a method which has yet to be characterized in yeast and therefore provides an entirely new mechanism for the generation of variation in the S. cerevisiae genome.
A thorough understanding of the scope of plasticity of the yeast genome is a vital prerequisite for the systematic understanding of yeast biology or for the development of the next generation of yeasts for industrial applications. As more S. cerevisiae strains are sequenced, the suitability of S288c as a “reference” strain for this species is becoming less clear, especially as it appears to lack a large numbers of ORFs found in many other S. cerevisiae strains while containing an abnormally high number of Ty transposable elements [8], [9]. Given the ubiquitous nature of the S288c genome for the design of ‘omics experiments, these novel elements have generally not been considered when studying strains other than S288c. Thus, little data exists regarding the functional contributions of these proteins. As such, they represent a significant knowledge gap with respect to cellular and metabolic modeling strategies. This is especially true for proteins such as the ORF located next to RTM1 which is large (∼800 amino acids) and highly conserved but has no significant homologs outside of a small subset of S. cerevisiae strains on which a function can be based. Fortunately, the continued development of next generation sequencing, such as that applied in this work, have provided the means to now characterize large numbers of yeast strains to provide this information and outline the true scope and variability of this species.
Materials and Methods
Yeast strains
Each commercial strain was obtained from the original mother cultures from the supplier. Genomic DNA was prepared by zymolase digestion and standard phenol-chloroform extraction.
Sequencing and assembly
Library construction and sequencing was performed at 454 Life Sciences, A Roche Company (Branford, CT) using a pre-release development version of the GS FLX Titanium series shotgun and 3 kb paired-end protocols. Sequences were assembled using MIRA (http://sourceforge.net/apps/mediawiki/mira-assembler/index.php?title=Main_Page) and manually-edited using Seqman Pro (DNAstar).
Regions of chromosomal CNV were determined by calculating the per-base sequencing coverage across each sequencing contig with median smoothing (1001 bp window, 100 bp step size). The ratio between the coverage at each genomic location and the overall median genomic coverage was the calculated to determine the level of over-representation for each location. Large-scale chromosomal aneuploidies were detected by screening for regions in which median ratio for a contiguous stretch of at least 101 individual segments differed from the overall genomic median by either 1.25 (5∶4 ratio representing at least 1 extra genomic copy in a tetraploid) or 1.4 fold (3∶2 ratio representing at least 1 extra genomic copy in a diploid).
SNP prediction
Chromosomal scaffolds from each yeast strain were aligned using FSA [33]. Diploid sequences were assigned into two haploid alleles by converting any degenerate bases into their non-degenerate pairs. Heterozygous regions were divided into both an insertion and deletion allele. A chromosomal consensus was computed for the alignment based upon the most frequent allele at each position in the alignment. Nucleotides that varied from the consensus in each strain were scored as sequence variants and were subsequently divided into SNPs (nucleotide substitution) or InDels (nucleotide insertion or deletion). To enable the comparison to strains with low coverage sequences [12], SNPs that were calculated for each strain relative to S288c (imputed SNPs) were used to create synthetic S288c-based genome sequences that contain the SNPs present in these strains. The genetic relationship between the strains was calculated by editing and concatenating the nucleotide alignments of all sixteen chromosomes using Seaview [34] followed by calculating the distance tree using the NJ algorithm of Clustalw (ignoring gapped regions in the alignment). Tandem repeats were predicted from the chromosomal alignment of all twelve yeast strains using Tandem Repeats Finder [35] using default parameters (match weight, 2; mismatch, 7; indel, 7; pM, 0.80; pI, 0.10; minimum alignment score, 50; maximum period size, 500). Individual repeats were then scored as either being variable if the specific tandem repeat region contained strain - or allele - specific InDels.
ORF prediction and comparison
ORFs were predicted using Glimmer [20] with the predicted ORFs of S288c being used to build the prediction model (See Datasets S1, S2, S3, S4, S5, S6, S7, S8, S9, S10, S11 for actual CDS sequences for each strain). Initial ORF designations were made by identifying the best sequence match for each ORF when compared to S288c using BLASTn [21]. Glimmer was also used to predict ORFs from the sequence of S288c (Accession numbers NC001133-NC001148) to correct for false-negatives in the predictions when compared to existing ORF designations in S288c. ORFs with no match to S288c were searched against the full list of non-redundant Genbank proteins to identify a closest existing homology match. ORFs from each strain were then arranged in syntenic order (Table S2 for a full list of ordered ORFs). For protein sequence comparisons, predicted protein sequences were aligned using Clustalw [36] (http://align.genome.jp).
Supporting Information
Zdroje
1. QuerolA
BellochC
Fernandez-EspinarMT
BarrioE
2003 Molecular evolution in yeast of biotechnological interest. Int Microbiol 6 201 205
2. FayJC
BenavidesJA
2005 Evidence for domesticated and wild populations of Saccharomyces cerevisiae. PLoS Genet 1 e5 doi:10.1371/journal.pgen.0010005
3. MortimerRK
JohnstonJR
1986 Genealogy of principal strains of the yeast genetic stock center. Genetics 113 35 43
4. LambrechtsMG
PretoriusIS
2000 Yeast and its importance to wine aroma - a review. Sth Afr J Enol Vitic 21 97 129
5. SwiegersJH
PretoriusIS
2005 Yeast modulation of wine flavor. Adv Appl Microbiol 57 131 175
6. GoffeauA
BarrellBG
BusseyH
DavisRW
DujonB
1996 Life with 6000 genes. Science 274 New York, , NY 546, 563 547
7. DunnB
LevineRP
SherlockG
2005 Microarray karyotyping of commercial wine yeast strains reveals shared, as well as unique, genomic signatures. BMC Genomics 6 53
8. WeiW
McCuskerJH
HymanRW
JonesT
NingY
2007 Genome sequencing and comparative analysis of Saccharomyces cerevisiae strain YJM789. Proc Natl Acad Sci USA 104 12825 12830
9. BornemanAR
ForganAH
PretoriusIS
ChambersPJ
2008 Comparative genome analysis of a Saccharomyces cerevisiae wine strain. FEMS Yeast Res 8 1185 1195
10. DonigerSW
KimHS
SwainD
CorcueraD
WilliamsM
2008 A catalog of neutral and deleterious polymorphism in yeast. PLoS Genet 4 e1000183 doi:10.1371/journal.pgen.1000183
11. ArguesoJL
CarazzolleMF
MieczkowskiPA
DuarteFM
NettoOV
2009 Genome structure of a Saccharomyces cerevisiae strain widely used in bioethanol production. Genome Res 19 2258 2270
12. LitiG
CarterDM
MosesAM
WarringerJ
PartsL
2009 Population genomics of domestic and wild yeasts. Nature 458 337 341
13. NovoM
BigeyF
BeyneE
GaleoteV
GavoryF
2009 Eukaryote-to-eukaryote gene transfer events revealed by the genome sequence of the wine yeast Saccharomyces cerevisiae EC1118. Proc Natl Acad Sci USA 106 16333 16338
14. StambukBU
DunnB
AlvesSLJr
DuvalEH
SherlockG
2009 Industrial fuel ethanol yeasts contain adaptive copy number changes in genes involved in vitamin B1 and B6 biosynthesis. Genome Res 19 2271 2278
15. TamaiY
MommaT
YoshimotoH
KanekoY
1998 Co-existence of two types of chromosome in the bottom fermenting yeast, Saccharomyces pastorianus. Yeast 14 923 933
16. DunnB
SherlockG
2008 Reconstruction of the genome origins and evolution of the hybrid lager yeast Saccharomyces pastorianus. Genome Res 18 1610 1623
17. MarguliesM
EgholmM
AltmanWE
AttiyaS
BaderJS
2005 Genome sequencing in microfabricated high-density picolitre reactors. Nature 437 376 380
18. MortimerRK
2000 Evolution and variation of the yeast (Saccharomyces) genome. Genome Res 10 403 409
19. VincesMD
LegendreM
CaldaraM
HagiharaM
VerstrepenKJ
2009 Unstable tandem repeats in promoters confer transcriptional evolvability. Science 324 1213 1216
20. DelcherAL
BratkeKA
PowersEC
SalzbergSL
2007 Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23 673 679
21. AltschulSF
MaddenTL
SchafferAA
ZhangJ
ZhangZ
1997 Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. NAR 25 3389 3402
22. NessF
AigleM
1995 RTM1: a member of a new family of telomeric repeated genes in yeast. Genetics 140 945 956
23. TakagiH
ShichiriM
TakemuraM
MohriM
NakamoriS
2000 Saccharomyces cerevisiae sigma 1278b has novel genes of the N-acetyltransferase gene superfamily required for L-proline analogue resistance. J Bacteriol 182 4249 4256
24. GotoK
IwaseT
KichiseK
KitanoK
TotukaA
1990 Isolation and properties of a chromosome-dependent KHR killer toxin in Saccharomyces cerevisiae. Agric Biol Chem 54 505 509
25. ConstamD
MuheimA
ZimmermannW
FiechterA
1991 Purification and Partial Characterization of an Intracellular NADH - Quinone Oxidoreductase from Phanerochaete chrysosporium. J Gen Microbiol 137 2209 2214
26. ReiserJ
MuheimA
HardeggerM
FrankG
FiechterA
1994 Aryl-alcohol dehydrogenase from the white-rot fungus Phanerochaete chrysosporium. Gene cloning, sequence analysis, expression, and purification of the recombinant enzyme. J Biol Chem 269 28152 28159
27. DelneriD
GardnerDC
BruschiCV
OliverSG
1999 Disruption of seven hypothetical aryl alcohol dehydrogenase genes from Saccharomyces cerevisiae and construction of a multiple knock-out strain. Yeast 15 1681 1689
28. DelneriD
GardnerDC
OliverSG
1999 Analysis of the seven-member AAD gene set demonstrates that genetic redundancy in yeast may be more apparent than real. Genetics 153 1591 1600
29. PretoriusIS
2000 Tailoring wine yeast for the new millennium: novel approaches to the ancient art of winemaking. Yeast 16 675 729
30. LefebureT
StanhopeMJ
2007 Evolution of the core and pan-genome of Streptococcus: positive selection, recombination, and genome composition. Genome Biol 8 R71
31. UglianoM
HenschkePA
2009 Yeasts and Wine Flavour.
Moreno-ArribasMV
PoloMC
Wine Chemistry and Biochemistry New York Springer 313 392
32. SponholzW
1993 Wine spoilage by microorganisms.
FleetG
Wine Microbiology and Biotechnology London Taylor and Francis 395 420
33. BradleyRK
RobertsA
SmootM
JuvekarS
DoJ
2009 Fast statistical alignment. PLoS Comput Biol 5 e1000392 doi:10.1371/journal.pcbi.1000392
34. GouyM
GuindonS
GascuelO
SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27 221 224
35. BensonG
1999 Tandem repeats finder: a program to analyze DNA sequences. NAR 27 573 580
36. ThompsonJD
HigginsDG
GibsonTJ
1994 CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. NAR 22 4673 4680
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 2
Nejčtenější v tomto čísle
- Meta-Analysis of Genome-Wide Association Studies in Celiac Disease and Rheumatoid Arthritis Identifies Fourteen Non-HLA Shared Loci
- MiRNA Control of Vegetative Phase Change in Trees
- Risk Alleles for Systemic Lupus Erythematosus in a Large Case-Control Collection and Associations with Clinical Subphenotypes
- The Cardiac Transcription Network Modulated by Gata4, Mef2a, Nkx2.5, Srf, Histone Modifications, and MicroRNAs