
Deletion of a Conserved -Element in the Locus Highlights the Role of Acute Histone Acetylation in Modulating Inducible Gene Transcription
Differentiation-dependent regulation of the Ifng cytokine gene locus in T helper (Th) cells has emerged as an excellent model for functional study of distal elements that control lineage-specific gene expression. We previously identified a cis-regulatory element located 22 kb upstream of the Ifng gene (Conserved Non-coding Sequence -22, or CNS-22) that is a site for recruitment of the transcription factors T-bet, Runx3, NF-κB and STAT4, which act to regulate transcription of the Ifng gene in Th1 cells. Here, we report the generation of mice with a conditional deletion of CNS-22 that has enabled us to define the epigenetic and functional consequences of its absence. Deletion of CNS-22 led to a defect in induction of Ifng by the cytokines IL-12 and IL-18, with a more modest effect on induction via T-cell receptor activation. To better understand how CNS-22 and other Ifng CNSs regulated Ifng transcription in response to these distinct stimuli, we examined activation-dependent changes in epigenetic modifications across the extended Ifng locus in CNS-22-deficient T cells. We demonstrate that in response to both cytokine and TCR driven activation signals, CNS-22 and other Ifng CNSs recruit increased activity of histone acetyl transferases (HATs) that transiently enhance levels of histones H3 and H4 acetylation across the extended Ifng locus. We also demonstrate that activation-responsive increases in histone acetylation levels are directly linked to the ability of Ifng CNSs to acutely enhance Pol II recruitment to the Ifng promoter. Finally, we show that impairment in IL-12+IL-18 dependent induction of Ifng stems from the importance of CNS-22 in coordinating locus-wide levels of histone acetylation in response to these cytokines. These findings identify a role for acute histone acetylation in the enhancer function of distal conserved cis-elements that regulate of Ifng gene expression.
Published in the journal:
. PLoS Genet 10(1): e32767. doi:10.1371/journal.pgen.1003969
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003969
Summary
Differentiation-dependent regulation of the Ifng cytokine gene locus in T helper (Th) cells has emerged as an excellent model for functional study of distal elements that control lineage-specific gene expression. We previously identified a cis-regulatory element located 22 kb upstream of the Ifng gene (Conserved Non-coding Sequence -22, or CNS-22) that is a site for recruitment of the transcription factors T-bet, Runx3, NF-κB and STAT4, which act to regulate transcription of the Ifng gene in Th1 cells. Here, we report the generation of mice with a conditional deletion of CNS-22 that has enabled us to define the epigenetic and functional consequences of its absence. Deletion of CNS-22 led to a defect in induction of Ifng by the cytokines IL-12 and IL-18, with a more modest effect on induction via T-cell receptor activation. To better understand how CNS-22 and other Ifng CNSs regulated Ifng transcription in response to these distinct stimuli, we examined activation-dependent changes in epigenetic modifications across the extended Ifng locus in CNS-22-deficient T cells. We demonstrate that in response to both cytokine and TCR driven activation signals, CNS-22 and other Ifng CNSs recruit increased activity of histone acetyl transferases (HATs) that transiently enhance levels of histones H3 and H4 acetylation across the extended Ifng locus. We also demonstrate that activation-responsive increases in histone acetylation levels are directly linked to the ability of Ifng CNSs to acutely enhance Pol II recruitment to the Ifng promoter. Finally, we show that impairment in IL-12+IL-18 dependent induction of Ifng stems from the importance of CNS-22 in coordinating locus-wide levels of histone acetylation in response to these cytokines. These findings identify a role for acute histone acetylation in the enhancer function of distal conserved cis-elements that regulate of Ifng gene expression.
Introduction
Distal regulatory elements including locus control regions, enhancers, silencers and boundary elements play important roles in regulating cell lineage-specific activation and repression of genes [1], [2], [3], [4], [5], [6]. In addition to genome-wide studies to document and classify putative distal regulatory sites, studies on individual gene loci have been instrumental in shaping our understanding of cis element function [7], [8], [9]. Although genes expressed in several cell types including embryonic stem cells (Hox genes), B-lineage cells (immunoglobulin genes) and erythroid cells (globin genes) have emerged as important models to understand eukaryotic transcription, cytokine genes expressed in T-helper cells are particularly attractive models to study lineage specific regulation. Primary human and murine naïve Th cells can be readily isolated in large numbers and be differentiated into functionally and transcriptionally distinct Th cells as exemplified by Th1, Th2, Th17, and T-regulatory (Treg) cell subsets [10], [11], [12]. In particular, genes that encode Th2 cytokines, comprised of the Il4, Il13 and Il5 genes and the Ifng gene transcribed in Th1 cells have emerged as key models to the study lineage-appropriate gene expression [8], [12] [13], [14].
The importance of distal elements in regulating expression of human and mouse genes that encode IFN-γ was first recognized in mice transgenic for a bacterial artificial chromosome (BAC) that encompassed ∼190 kb flanking the human IFNG gene, which, unlike transgenes that contained more limited flanking sequence, conferred lineage-specific expression of human IFN-γ in mouse Th1 cells [15], [16]. Subsequently, we reported a murine Ifng-Thy1.1 BAC reporter transgene that spanned ∼160 kb surrounding Ifng, which also demonstrated lineage - and activation-specific expression [17], [18], suggesting that distal elements required for lineage specific expression of Ifng were contained in this region. Based on recruitment of CTCF and Rad21 (a cohesin), the IFNG and Ifng loci are predicted to extend from −63 to +119 kb [19] and −70 kb to +66 kb [20], respectively. Within these boundary elements, at least nine conserved non-coding sequences (CNS) have been identified based on the high degree of sequence conservation at these sites in multiple mammalian species [2], [3].
Using ChIP-qPCR and promoter-reporter assays, a subset of these CNSs was probed for trans-factor binding and histone modifications in early studies [18], [21], [22], [23], [24]. More recently, DNase-chip [25] and DNase-seq [20] have been employed to map chromatin conformation of the extended Ifng locus in multiple T cell lineages. In parallel, analyses of trans factor recruitment to these cis elements have facilitated their further functional mapping. T-bet [18], [22], [23], STAT4 [26], [27], Runx3 [28] and members of the NF-κB [29] and NFAT [23] families of transcription factors have been demonstrated to interact with cis elements across the Ifng locus [13].
To date, the functions of four Ifng/IFNG CNSs have been examined by deletional analyses in the context of Ifng or IFNG BAC transgenes [18], [30]. Deletion of the human homolog of Ifng CNS-34 (IFNG CNS-30) from a 190 kb human IFNG transgene resulted in impaired expression of IFNG in T cells but not NK cells [30] suggesting that one or more of these regulatory sequences may have lineage-specific functions. In our own studies, deletion of CNS-22 from the Ifng-Thy1.1 reporter transgene led to nearly complete ablation of Thy1.1 reporter expression in both T cells and NK cells [18]. In more recent studies using IFNG BAC transgenes, human homologs of CNSs −6 and +17–19 were also shown to be important enhancers of IFNG transcription [31]. CNS-22 is of particular interest, since it is hypersensitive not only in Th1 cells, but also in naïve precursors, suggesting that CNS-22 might act as a principal site for recruitment of “pioneer” trans factors that initiate remodeling of the Ifng locus [25]. Further, CNS-22 remains hypersensitive to DNase I in Th2 and Th17 cells despite repression of Ifng expression in these lineages, suggesting that CNS-22 may be involved in Ifng transcriptional silencing in addition to its role in transcriptional activation [25], [29]. Taken together with the profound effect of selective CNS-22 deletion on BAC reporter expression, these findings led us to speculate that this element could be an important node for directing chromatin remodeling of the Ifng locus [18].
Therefore, we generated mice targeted for conditional deletion of CNS-22 in the endogenous Ifng loci (Ifng.CNS-22fl/fl) to enable mapping of epigenetic modifications not previously possible using BAC transgenic mice. Here, we report the functional consequences of the deletion of CNS-22 on epigenetic remodeling and gene expression of the Ifng locus in naïve and differentiated T cells. We find that deletion of CNS-22 in Th1 cells results in greater impairment of Ifng transcription in response to IL-12 plus IL-18 than that induced by TCR dependent signaling. This is associated with a defect in the deposition of histone acetylation marks on nucleosomes immediately flanking CNS-22 as well as those distributed distally across the Ifng locus. These findings identify a previously unappreciated role for activation-induced modulation of HAT activity in driving cytokine gene transcription.
Results
Functional characterization of Ifng.CNS-22−/− mice
Ifng.CNS-22fl/fl mice were generated using a targeting construct in which 391 bp corresponding to CNS-22 in the Ifng locus was flanked by loxp sites to enable Cre-mediated excision (Fig. S1A, B) [18], [32], [33]. CNS-22 resides in a broad DNase I hypersensitive (HS) site that encompasses CNS-22 at the 3′ end (Fig. S1C). Cre-mediated excision of this element deletes several evolutionarily conserved trans-factor binding sequences, including sites that recruit T-bet, STAT4, RelA and Runx3 (Fig. 1B) [18], [28], [29]. Ifng.CNS-22 was deleted in the germline by crosses with EIIa.Cre mice, such that all cells, including T cells, were CNS-22–deficient (henceforth referred to as CNS-22−/− mice). Phenotypically, CNS-22−/− mice were indistinguishable from littermate controls. Numbers of CD4+ and CD8+ T cells in the periphery were comparable to wildtype controls (unpublished observations).

To examine the impact of CNS-22 deletion on Ifng gene expression, naïve CD4+ T cells from OT-II transgenic WT and CNS-22−/− mice were differentiated ex vivo in the presence of a low or high concentration of IL-12 and the expression of IFN-γ induced by restimulation with IL-12+IL-18 or TCR signaling was examined (Fig. S2A). Irrespective of the concentration of IL-12 used, Th1 cells generated from CNS-22−/− mice were significantly impaired in their expression of IFN-γ in response to IL-12+IL-18 restimulation, whereas a deficit in IFN-γ expression in response to TCR stimulation was only apparent for cells differentiated with the low concentration of IL-12 (Figs. 2A and S2A). In contrast to cells restimulated with IL-12+IL-18, which showed a similar deficit in IFN-γ expression whether activated on day 3 or day 5 of differentiation, the impact of CNS-22 deletion on impaired TCR-driven induction of IFN-γ was more pronounced in Th1 cells restimulated on day 3 (Figs. 2A and S2A, and data not shown). The impairment of Ifng transcription was not due to alterations in the expression of key trans factors, as there were no significant differences in expression of Tbx21 or Runx3 in CNS-22-deficient T cells (Fig. S2B). IL-12+IL-18 driven induction of Ifng was also considerably impaired in Tc1 cells and in NK cells from in CNS-22−/− mice (Fig. 2B). Impairment of Ifng expression was also observed in vivo for CNS-22-deficient T cells responding to infection with Listeria monocytogenes, an intracellular bacterial pathogen that induces a type 1 immune response (Figs. S3A–C).

By comparing the kinetics of induction of Ifng transcripts in Th1 cells, we found that early transcription of Ifng in response to IL-12+IL-18 was significantly compromised in the absence of CNS-22, while there was no significant impairment in response to TCR signaling (Fig. 2C), reinforcing the observation that induction of transcription via the TCR signaling pathway was less dependent on CNS-22 function. Finally, Th1 cells derived from CNS-22+/− mice showed an intermediate, copy number–dependent impairment in IL-12+IL-18 driven Ifng gene transcription (unpublished observations), demonstrating that CNS-22 plays an obligatory role in integrating activating signals downstream of IL-12 and IL-18 receptors [29]. Taken together with our previous findings that CNS-22 recruits STAT4 and NF-κB downstream of the IL-12 and IL-18 receptors, respectively [13], whereas TCR signaling does not activate STAT4, these results suggest that CNS-22 is more important as a STAT4-dependent enhancer for acute activation of Ifng transcription, although they do not exclude a critical contribution for CNS-22 as node for epigenetic modifications of the Ifng locus during the development of immune cells.
CNS-22 plays a developmental stage-specific role in remodeling of the Ifng locus
Our previous studies demonstrated that deletion of CNS-22 in the context of the Ifng-Thy1.1 reporter transgenic locus resulted in nearly complete ablation of Thy1.1 reporter expression in Th1, Tc1 and NK cells [13]. This led us to hypothesize that CNS-22 was essential for orchestrating remodeling of the Ifng locus in this transgene in addition to its role as a key enhancer (ref. [18], and see Fig. 1A). While we confirm here the importance of CNS-22 as a positive modulator of Ifng transcription (Fig. 2), the less pronounced impairment of Ifng transcription found in CNS-22−/− Th1 cells suggested that deletion of CNS-22 from endogenous Ifng loci might not have as fundamental a role in regulating chromatin accessibility during Th1 cell development as speculated (see below). Nevertheless, IL-12 dependent acquisition of Ifng competence was considerably delayed in CNS-22−/− CD4+ T cells (Fig. 2A). Since we had previously documented that CNS-22 was permissive in naïve CD4+ T cells (ref. [13], and see also Fig. 3A), we hypothesized that CNS-22 might play an obligatory role in epigenetic remodeling the Ifng locus prior to Th1 differentiation.
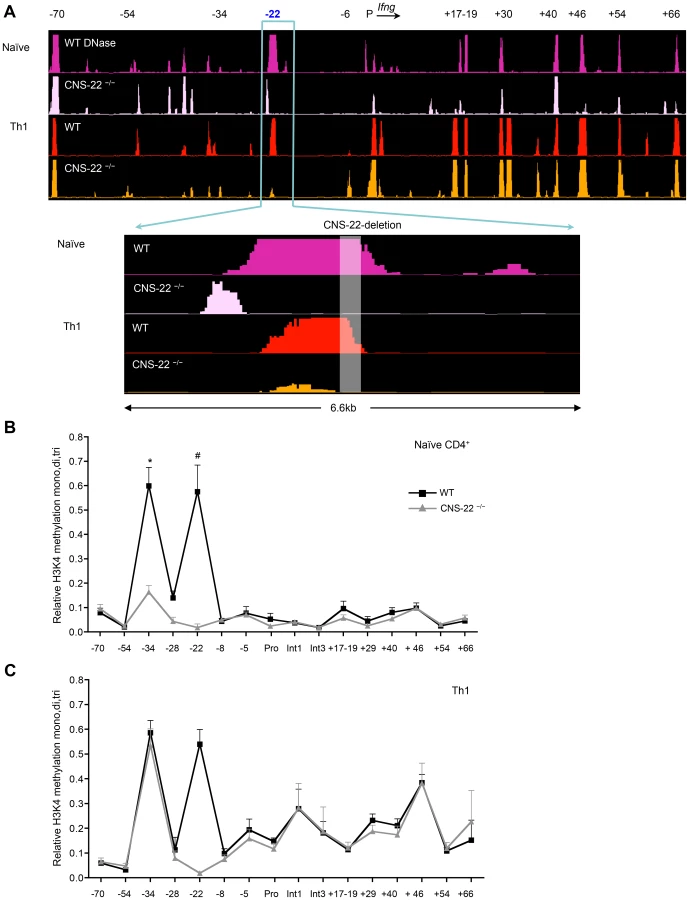
To examine this, we performed DNase-chip and ChIP-qPCR-based analysis of histone 3 lysine 4 methylation (H3K4) to assess the epigenetic status of the extended Ifng locus in CNS-22-deficient naïve CD4+ T cells and Th1 cells (Fig. 3). In agreement with our previous study [25], we found that several key distal elements in the Ifng locus are hypersensitive (HS) to DNase I in naïve WT CD4+ T cells (Fig. 3A). Notably, naïve cells from CNS-22−/− mice showed marked reduction or a lack of hypersensitivity at most sites identified in WT cells, including those at CNSs +17–19, +30, +46, +54 and +66 (Fig. 3A). In contrast, DNase I HS sites at the upstream CTCF binding insulator element (−70) and CNS+40 arise in a CNS-22–independent fashion, indicating that even prior to exposure to lineage-specifying signals, multiple nodes appear poised to initiate reorganization of the extended Ifng locus. We also observed a prominent HS peak immediately upstream of CNS-22 in naïve CNS-22−/− CD4+ T cells (Fig. 3A), although this was much less pronounced following Th1 differentiation. While the basis for this is unclear, it is plausible that some functions of CNS-22 are not compromised by deletion of the core regulatory element.
In accord with the functional analyses of Ifng expression in CNS-22−/− Th1 cells, defects in locus-wide remodeling apparent in naïve CNS-22–deficient CD4+ T cells were largely reversed upon Th1 differentiation (Fig. 3A). Specifically, the pattern of DNAse HS at regulatory elements downstream of the Ifng gene was nearly indistinguishable from WT Th1 cells, indicating that differentiation-dependent remodeling of the Ifng locus is largely independent of CNS-22.
To complement DNase-chip analyses, we also carried out ChIP-qPCR to evaluate H3K4 mono-, di - and tri methylation status is Th1 cells derived from either WT or CNS-22−/− mice (Fig. 3B). In parallel to the DNase I HS results, deposition of permissive H3K4 methylation marks in naïve CD4+ T cells was significantly impaired in the absence of CNS-22, particularly at CNS-34 and non-conserved site −28 (Fig. 3B). However, levels of H3K4 methylation was comparable at both these sites in CNS-22–deficient and WT Th1 cells (Fig. 3C), indicating that CNS-22 was dispensable for differentiation-dependent remodeling of the Ifng locus in Th1 cells. Thus, the effect of CNS-22 deletion in the context of the endogenous Ifng was less pronounced than in the context of an Ifng BAC transgenic locus we reported previously (Figs. 1A and 4A).
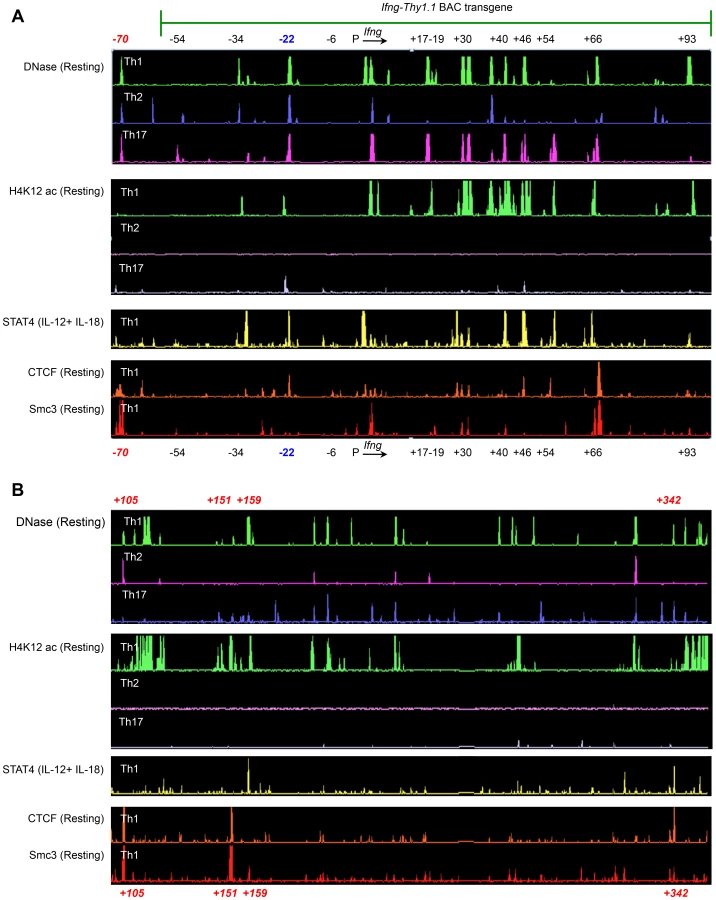
We hypothesized that the observed differences in the contribution of CNS-22 to Ifng transcription from the endogenous or transgenic loci might be attributed to the absence of key distal regulatory sequences in the BAC-transgene. The BAC-transgenic reporter included 60 kb upstream and 100 kb downstream of the Ifng gene and consequently lacked a more recently defined insulator/chromatin looping sequence that is located 70 kb upstream of Ifng (Fig. 4A) [19], [20]. In addition, it was also possible that some distal elements that regulate Ifng lie greater than 100 kb downstream of Ifng. The first clue that this was likely came from ChIP-chip analyses carried out in our previous studies to identify STAT4 and RelA binding enhancer sequences [29]. We discovered at least one prominent STAT4 binding site that was 159 kb downstream of the Ifng gene (Fig. 4B). This site was hypersensitive to DNase I in Th1 cells, but not in Th2 or Th17 cells suggesting that it functioned as a STAT4 responsive module in Th1 cells.
To further explore the possibility that potential regulatory elements may reside greater than 100 kb downstream of Ifng and therefore would be excluded from the transgene, we carried out ChIP-chip analyses to map recruitment of CTCF and a cohesion family member Smc3. We identified at least three prominent CTCF and Smc3 binding sites +105, +151 and +342 kb downstream of Ifng (Fig. 4B). At all three sites, we observed Th1-specific acquisition of histone H4 Lysine-12 acetylation (H4K12ac) marks indicating that these elements are functional in Th1 cells (Fig. 4B). Therefore, we speculate that exclusion of these downstream regulatory sequences and the CTCF binding site located −70 kb upstream of Ifng in our previous BAC-transgenic studies may have magnified the role of CNS-22 in coordinating Ifng expression. Nonetheless, by deleting CNS-22 at the endogenous locus, we demonstrate here that CNS-22 is an obligate enhancer that is primarily important in regulating IL-12+IL-18 dependent induction of Ifng in Th1, Tc1 and NK cells.
CNS-22 modulates both proximal and distal increases in activation-driven histone acetylation
Th1/Tc1 specific transcriptional competence of the Ifng gene is marked not only by acquisition of H3K4 mono, di and tri - methylation marks, but also acetylation of several lysine residues on histones H3 and H4 across the extended Ifng locus [18]. Acetylation of histone residues H4K16, H3K4 and H4K12 is present not only at promoters, but also throughout the extended loci of transcribed genes [34]. More recently, acetylation of H3K27 has been correlated with lineage-specific activity of enhancers [35]. We initially examined these modifications and others (unpublished observations), and chose to focus on H4K12 acetylation due to the greater efficiency, specificity and dynamic range of chromatin immunoprecipitation observed for this particular histone modification. Acquisition of H4K12ac marks at CNS-22 and globally across the extended Ifng locus was specific to differentiated Th1 cells (Fig. 4) [18], [26]. Similarly, acquisition of lineage-specific H4K12ac marks correlated with transcriptional competence at other T lineage-specific gene loci, including the Il17a/Il17f and Il4-Il13-Il5 gene clusters in differentiated Th17 and Th2 cells, respectively (Fig. S4).
In a previous study we noted that H4 acetylation at multiple distal sites in the Ifng locus, including CNS-22, was increased by signals that led to acute induction of Ifng transcription in both Th1 and Tc1 cells [18]. Notably, the activation dependent change in H4 acetylation at these non-conserved sites was much greater than changes observed at Ifng CNSs [18]. We therefore speculated that inducible acquisition of histone acetylation marks at these distal non-conserved sites might be linked to activation-induced transcription of Ifng, and that CNS-22 and other CNSs in the Ifng locus might act as nucleation sites for enhanced recruitment or activation of HATs that decorate the histones of neighboring nucleosomes. If true, induction of Ifng transcription in Th1 and Tc1 cells would be predicted to correlate with acute increases in histone acetylation at sites immediately flanking CNSs across the Ifng locus. Accordingly, deletion of CNS-22 would be predicted to impair acquisition of these inducible acetylation marks.
To test this, we evaluated levels of histone H4 acetylation in the immediate vicinity of CNS-22 in resting and activated Th1 cells. In response to both IL-12+IL-18 and TCR driven activation signals, a prominent increase in levels of H4 acetylation at sites within ∼1 kb flanking CNS-22 (−22.9, −22.4, −21.7) was observed (Fig. S5). We also observed similar activation-induced acetylation at Ifng CNSs −34 and +46 (see Fig. 5, below, and data not shown). We subsequently performed ChIP-qPCR experiments in Th1 cells derived from CNS-22−/− mice to examine whether activation-driven acquisition of acetylation marks at −22.9, −22.4 and −21.7 was dependent on CNS-22. While resting levels of H4 acetylation at −22.9, −22.4 and −21.7 were unaffected by deletion of CNS-22, acute hyperacetylation at these sites in response to both IL-12+IL-18 and TCR signaling was significantly impaired (Figs. S5B, C). These results led us to speculate that the ability of CNS-22 to acutely recruit HATs in response to external stimuli is linked to its ability to enhance Ifng transcription. This also led us to ask two further questions. Firstly, is activation-driven acquisition of H4 acetylation marks unique to CNS-22, or is this a common mechanism employed by multiple Ifng enhancers? Secondly, since Th1 cells generated from CNS-22−/− mice show a more prominent defect in IL-12+IL-18 driven activation signals, but acquisition of H4 acetylation near CNS-22 was significantly impaired in response to both IL-12+IL-18 and TCR reactivation signals, does CNS-22 regulate histone acetylation only within its immediate vicinity or does it influence histone acetylation levels at more distal sites as well? To address these two questions, we have carried out ChIP-chip experiments to examine H4K12 acetylation in resting and activated Th1 cells generated from WT and CNS-22−/− mice.

In resting Th1 cells, H4K12ac marks were confined to the regions in immediate proximity to the CNSs themselves and deletion of CNS-22 did not significantly alter acquisition of these marks (Fig. 5A). Upon activation with either IL-12+IL-18 or with anti-CD3+anti-CD28, there was robust, extensive deposition of acute acetylation marks that extended beyond the CNSs to more distal, non-conserved sequences (Fig. 5A). As predicted by conventional ChIP-qPCR, acquisition of acetylation marks in the immediate vicinity of CNS-22 was absolutely dependent upon CNS-22 irrespective of the mode of activation (Fig. S5). However, normalization and comparison on H4K12ac levels between WT and CNS-22−/− Th1 cells revealed a much more prominent defect in IL-12+IL-18 dependent acquisition of H4K12ac marks extending across the Ifng locus (Fig. 5B). Specifically, in response to TCR driven acquisition of H4K12ac marks, deletion of CNS-22 impaired acquisition of H4K12ac in proximity to CNS-22 as well as in enhancers upstream of CNS-22 (CNSs −54 and −34), but had little to no effect on deposition of acetylation marks at CNSs downstream of the Ifng gene (Fig. 5B). In contrast, in CNS-22–deficient Th1 cells IL-12+IL-18 dependent acquisition of H4K12ac was globally altered across the Ifng locus such that defects in acquisition of this epigenetic mark were evident at multiple CNSs downstream of the Ifng gene (Fig. 5B). Therefore, the greater defect in acute acquisition of H4K12ac marks in response to IL-12+IL-18 might account for the greater impairment of IL-12+IL-18 dependent induction of Ifng compared to that observed following TCR driven induction.
Acute, activation-dependent histone acetylation regulates inducible recruitment of RNA Pol II to the Ifng gene
The positive correlation between accumulation of histone acetylation marks that accompany lineage specification and induction of gene transcription is well established. More recently, the recruitment of bromodomain-containing HATs, particularly p300 and CBP, to distal regulatory sequences has been used to identify lineage-specific enhancers [36], [37]. In view of the role played by CNS-22 in the HAT-mediated modification of the Ifng locus, we interrogated its association with p300 (Fig. S6). Binding of p300 to CNS-22 and other CNSs across the Ifng locus was evident in resting and activated Th1 cells, and with few exceptions the level of p300 binding — whether to CNS-22 or other CNSs across the locus — was not substantially altered by activation. This finding, coupled with the marked impairment in acquisition of H4K12 acetylation in CNS-22–deficient Th1 cells following IL-12+IL-18 stimulation, led us to speculate that activation-induced enhancement of HAT activity at CNS-22 resulted in increased recruitment of RNA Pol II to the Ifng promoter. To examine this, we compared recruitment of RNA Pol II to the Ifng gene in resting Th1 cells and Th1 cells activated by IL-12+IL-18 or TCR stimulation (Fig. 5C). In response to IL-12+IL-18, recruitment of RNA Pol II to the Ifng promoter, first exon and first intron was significantly impaired in the absence of CNS-22, suggesting that the ability of CNS-22 to enhance Ifng transcription is linked to its ability to up-regulate HAT activity. In contrast, while CNS-22–deficient Th1 cells showed a modest decrement in Pol II recruitment to the promoter and first exon of Ifng in response to TCR signaling, this did not achieve statistical significance (Fig. 5C). Together, these results suggest that trans-factor recruitment to CNS-22 downstream of IL-12+IL-18 signaling induced increased acetylation that was largely independent of enhanced p300 recruitment. These results further suggest that inducible acetylation at distal enhancers that regulate Ifng transcription is directly linked to their ability to modulate recruitment of RNA Pol II to the Ifng gene in response to external stimuli.
Discussion
CNS-22 was previously identified as an important cis-regulatory element hypothesized to have a central role in epigenetic remodeling of the Ifng locus during lineage-specific Th1, Tc1 and NK cell differentiation [13], [18]. In the current study, CNS-22–deficient mice were generated to enable study of the consequences of deletion of this element for epigenetic remodeling of the endogenous Ifng locus. Here we identify CNS-22 as critical element for early remodeling of the Ifng locus in naïve T cells and establish its importance as an enhancer for optimal Ifng transcription in Th1, Tc1 and NK cells. However, we find that the epigenetic consequences of the deletion of CNS-22 during lineage specification are limited to circumscribed effects on critical upstream regulatory elements surrounding CNS-22, such that remodeling of regulatory elements downstream of the Ifng gene occurs largely independently of CNS-22 in Th1 or Tc1 cells. Unexpectedly, fine mapping of the epigenetic consequences of CNS-22 deletion led to the finding that distal cis-acting elements activate inducible gene transcription through hyperacetylation of nucleosomes that flank trans-factor binding core elements of enhancers. This supports a model of eukaryotic enhancer function wherein the activity and/or composition of HAT complexes loaded onto core enhancer elements during lineage-specific differentiation are rapidly modulated in concert with activation-induced trans-factor recruitment to effect increased gene transcription.
Speculation that CNS-22 is a central node for permissive remodeling of the extended Ifng locus arose from our previous studies wherein deletion of CNS-22 from an Ifng-Thy1.1 BAC reporter transgene caused nearly complete ablation of reporter expression in Th1, Tc1 and NK cells [13], [18]. In the present study, deletion of CNS-22 from the endogenous Ifng locus led to a substantial defect in induction of Ifng, albeit less pronounced than the same deletion from the BAC transgene. Although several factors could account for the disparities observed, the exclusion from our Ifng BAC transgene of boundary elements located −70 kb upstream and the new potential boundary element identified herein located 342 kb downstream of the Ifng gene leads us to speculate that absence of these architectural elements might have compromised the efficiency of approximation of the CNS-22 enhancer to the core promoter mediated by a CTCF-cohesin–dependent mechanism. This might well have resulted in an exaggerated loss of Ifng transcription upon deletion of CNS-22 from the BAC transgene [17], [18]. Also missing from the BAC transgene were hypersensitive sites identified herein located +105, +151 and +159 kb downstream of the transcriptional state site of the Ifng gene. All three of these elements are likely to be functional, as the first two co-recruit CTCF and Smc3 while the site at +159 recruits STAT4. Any or all of these could contribute to the exaggerated expression defect observed on deletion of CNS-22 in from the BAC-transgene. Finally, it should be noted that Tmevp3, a gene that encodes a long intergenic non-coding RNA (lincRNA) that maps to ∼+60 kb to +120 kb downstream of the Ifng gene was excluded in the Ifng-Thy1.1 BAC transgene [38]. Although two recent studies document that the lincRNA encoded by Tmevp3 acts in trans to recruit H3K4 methyltransferases to the Ifng locus, it is possible, although we think it unlikely, that exclusion of Tmevp3 in the Ifng-Thy1.1 transgene might have compromised transcriptional activation upon deletion of CNS-22 in this context [39], [40]. Suffice it to say that further studies will be required to precisely define the basis for the observed expression disparities and should be informative.
Importantly, deletion of CNS-22 from endogenous Ifng alleles enabled analyses of developmental stage-specific effects on chromatin organization not previously possible using the BAC transgenic model. Thus, while CNS-22−/− naïve CD4+ T cells demonstrated compromised development of several HS sites present in WT cells, including those at CNSs +17–19 and +46, as well as H3K4 methylation of CNS-34, Th1 differentiation largely overrode these defects. This indicates that developmentally regulated HS sites that dictate low-level Ifng expression competence characteristic of naïve CD4+ T cells are dependent upon the presence CNS-22, exposing an important role for CNS-22 in early remodeling of the locus. However, differentiation that confers high-level Ifng transcriptional activity in Th1 and Tc1 cells proceeds such that a number of key HS sites, primarily those downstream of the Ifng gene, can be remodeled independently of CNS-22. Thus, although CNS-22 remains an important element for differentiation-dependent remodeling of the extended Ifng locus, its influence is not global, rather it is more local, consistent with the previously proposed organization of the Ifng locus into upstream and downstream regulatory domains coordinated by recruitment of CTCF boundary elements to the intronic CTCF element within the Ifng gene [19], [20].
CNS-22 recruits at least four key transcription factors to activate Ifng transcription: STAT4 [29], T-bet [18], Runx3 [28] and RelA [29]. We predict that defects in Ifng induction in CNS-22−/− T and NK cells stems from deletion of the STAT4 binding site in CNS-22. STAT4 is activated downstream of the IL-12 receptor and plays an essential role in IL-12 dependent polarization of naïve CD4+ T cells to IFN-γ-competent Th1 cells [41], [42]. IL-12+IL-18 dependent induction of Ifng is also absolutely dependent on activation of STAT4 [43]. In addition to the impaired IL-12+IL-18 driven induction of Ifng in CNS-22−/− Th1 cells, we also document that acquisition of competency of the Ifng locus is considerably delayed in the absence of CNS-22. At least three STAT4 recruiting modules, CNSs −22, +40 and +46 are accessible in naïve CD4+ T cells [25]. During the course of Th1 differentiation, additional STAT4 recruiting modules at the Ifng promoter, CNSs −34, +30 and +54 also become accessible [29]. It is likely that sustained IL-12 signaling combined with CNS+40 - and +46-dependent remodeling of other STAT4 binding modules compensate for the absence of CNS-22. Nonetheless, subsequent deficiency in IL-12+IL-18 dependent induction of Ifng highlights the importance of the STAT4 binding site within CNS-22. Further studies to mutate and evaluate individual transcription factor binding sites within CNS-22 would provide new insights into the roles of individual trans factor binding sites within CNS-22.
An important finding of this study is the identification of a link between activation-dependent recruitment of HAT activity and enhancer function. Several previous studies have linked acetylation of histones to differentiation-dependent activation of gene transcription, including the recently discovered association between H3K27ac and enhancer actions [35]. In addition, at least two bromodomain-containing HATs — p300 [36], [37] and CBP [36] — have been demonstrated to regulate lineage-specific activation of enhancers. Here, we establish for the first time a clear relationship between enhancer actions and acute increases in HAT activity by demonstrating that CNS-22 not only dictates activation-induced acquisition of H4K12ac marks in its immediate vicinity, but also regulates acquisition of H4K12ac marks at multiple distal sites. Together, these results suggest that CNSs across the Ifng locus interact with each other and coordinate activation-driven recruitment of HATs via long-range interactions. Moreover, by evaluating IL-12+IL-18 dependent RNA Pol II recruitment in CNS-22−/− Th1 cells, we demonstrate that impaired hyperacetylation compromises the ability of CNS-22 deficient Th1 cells to recruit RNA Pol II to the proximal Ifng promoter in response to IL-12+IL-18. Notably, the marked CNS-22–dependent increases in histone acetylation that followed IL-12+IL-18 signaling, and to a lesser extent TCR-mediated signaling, proceeded without substantive changes in the levels of p300 binding at these elements. This is consistent with a role for acute trans-factor-dependent recruitment of other HATs and/or activation of pre-loaded p300 HAT complexes to increase histone acetylation and Pol II loading on the promoter, and will require further study.
In summary, studies herein have delineated a role for CNS-22 in regulating epigenetic changes that control chromatin remodeling and transcriptional competence of the Ifng locus prior to, during and subsequent to lineage specification. CNS-22, while essential for locus remodeling in naïve T cells and optimal Ifng transcription in mature effector T cells, has a more restricted function in developmentally driven remodeling of the Ifng locus than previously thought, with its principal effects being exerted on regulatory elements bounded by the upstream and intronic CTCF elements thought to be important in approximating these upstream elements to the proximal promoter. Further, analysis of T cells from CNS-22−/− mice has uncovered a previously unappreciated role of HATs in modulating actions of eukaryotic enhancers for induction of high-level transcription that is characteristic of cytokine genes. Our findings suggest that recruitment and activation of HATs at CNS-22 and other distal enhancers in the Ifng locus occurs in two stages: the first, an initial wave to induce basal acetylation of enhancer-associated histones that confers receptiveness to a second, subsequent wave of trans-factor–induced hyperacetylation that is linked to high-level Ifng transcription. Although further studies will be necessary, these findings support a model in which a conserved regulatory module, such as CNS-22, contains a core enhancer that includes sites for trans-factor binding contingent upon differentiation-dependent nucleosomal clearing or remodeling, whereas the flanking nucleosomes constitute part of an extended enhanceosome that is less evolutionarily constrained. While whole genome analyses of the epigenome have rapidly advanced our understanding of differentiation-dependent alterations in chromatin remodeling and identified important epigenetic markers that have enabled identification of potential enhancers, detailed locus-specific analyses of the type exemplified herein for the Ifng locus will be important to complement our understanding of cis-element function moving forward.
Materials and Methods
Ethics statement
All animal studies were conducted in accordance with guidelines and oversight of the institutional animal use and care committee of University of Alabama at Birmingham.
Mice, antibodies and reagents
C57BL/6 and OT-II TCR transgenic mice were purchased from Jackson Laboratory and/or bred at the University of Alabama at Birmingham. Generation of CNS-22−/− mice is described in the supplement. Primers used for PCR-based screening have been previously described [18]. Antibodies against H3K4me1,2,3 (04-791), H3K4me2 (17-677), H3K4me3 (17-678), Pan H4 (08-858), H4 acetyl (06-598), H3K9ac (17-658) and H4K12ac (07-595) and RNA polymerase II (05-952) were purchased from Millipore (Billlerica, MA). Antibodies against p300 (sc-584, sc-585) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). All primers and probes were synthesized by IDT (Coralville, IA).
Naïve cell sorting and generation of Th1, Th2, Th17 and Tc1 cells
FSClo, CD62Lhi, CD44lo cells were purified from CD4+ T cells isolated by positive selection from spleen and pooled lymph nodes. For generation of Th cells, CD4+ T cells were isolated by positive selection from spleen and pooled lymph nodes. Differentiation of Th1, Th2, Th17 and Tc1 cells was performed as previously described [25], [29], [44], [45]. Positive selection was carried out using CD4-DYNAL beads (Invitrogen) or anti-PE microbeads (Miltenyi). Reactivation of cells for ChIP has been previously described [29]. Briefly, Th1, Tc1 or NK cells were activated with 10 ng/ml rIL-12 and 25 ng/ml rIL-18. For TCR restimulation, anti-CD3 antibody was diluted to 10 µg/ml in phosphate buffered saline (PBS, 150 mM NaCl, 0.02M Phosphate) and coated overnight at 4°C. The following day the plates were was washed twice with PBS and the media was supplemented with 5 µg/ml of anti-CD28 antibody.
Intracellular cytokine staining
Cells were reactivated for 4 hours in the presence of GolgiStop (BD Biosciences; San Jose, CA) as per manufacturer's recommendations and stained with fluorescent-labeled antibodies against CD4, CD8, IL-4 and IFN-γ using the Cytofix/Cytperm kit (BD Biosciences). Dead cells were excluded by staining with LIVE/DEAD fixable stain kits (Invitrogen; Carlsbad, CA). Samples were acquired on an LSRII flow cytometer and analyzed using FlowJo software (Treestar Inc.; Ashland, OR).
Chromatin immunoprecipitation, DNase-chip and ChIP-chip
Protocols employed for ChIP, ChIP-chip DNase-chip have been previously described [25], [29]. Primer sequences that were not previously published [25] are available upon request. We employed a slightly modified ChIP protocol for p300 ChIP. Cells were dounced in 25 mM Hepes (pH 7.8), 1.5 mM MgCl2, 10 mM KCl, 0.1% NP-40, 1× complete protease inhibitor cocktail (Roche). Nuclei were isolated, resuspended in 0.1× SDS lysis buffer (Millipore) diluted in 1× ChIP dilution buffer (Millipore). Samples were sonicated in and subject to ChIP as previously described using ChIP assay kit (Millipore). For ChIP-chip, samples and inputs were amplified using WGA2 kit (Sigma), labeled and hybridized to custom-designed microarrays (Roche-Nimblegen). A previously described algorithm, ACME was employed for peak calling and identifying enriched regions in the ChIP-chip datasets [46].
Transcript analyses
RNA was isolated using RNeasy kit (Qiagen), subject to DNAfree treatment (Applied Biosystems) to remove any contaminating DNA. RNA was then reverse-transcribed using Superscript III cDNA synthesis kit (Invitrogen) and transcript levels were normalized against housekeeping gene β2-microglobulin.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. OngCT, CorcesVG (2011) Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat Rev Genet 12 : 283–293.
2. FrazerKA, PachterL, PoliakovA, RubinEM, DubchakI (2004) VISTA: computational tools for comparative genomics. Nucleic Acids Res 32: W273–279.
3. LootsGG, OvcharenkoI, PachterL, DubchakI, RubinEM (2002) rVista for comparative sequence-based discovery of functional transcription factor binding sites. Genome Res 12 : 832–839.
4. BoyleAP, DavisS, ShulhaHP, MeltzerP, MarguliesEH, et al. (2008) High-resolution mapping and characterization of open chromatin across the genome. Cell 132 : 311–322.
5. CrawfordGE, HoltIE, WhittleJ, WebbBD, TaiD, et al. (2006) Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Res 16 : 123–131.
6. BarskiA, ZhaoK (2009) Genomic location analysis by ChIP-Seq. J Cell Biochem 107 : 11–18.
7. KieferCM, HouC, LittleJA, DeanA (2008) Epigenetics of beta-globin gene regulation. Mutation research 647 : 68–76.
8. AnselKM, DjureticI, TanasaB, RaoA (2006) Regulation of Th2 differentiation and Il4 locus accessibility. Annu Rev Immunol 24 : 607–656.
9. SproulD, GilbertN, BickmoreWA (2005) The role of chromatin structure in regulating the expression of clustered genes. Nature reviews Genetics 6 : 775–781.
10. ZhuJ, YamaneH, PaulWE (2010) Differentiation of effector CD4 T cell populations. Annu Rev Immunol 28 : 445–489.
11. RowellE, MerkenschlagerM, WilsonCB (2008) Long-range regulation of cytokine gene expression. Curr Opin Immunol 20 : 272–280.
12. LeeGR, KimST, SpilianakisCG, FieldsPE, FlavellRA (2006) T helper cell differentiation: regulation by cis elements and epigenetics. Immunity 24 : 369–379.
13. BalasubramaniA, MukasaR, HattonRD, WeaverCT (2010) Regulation of the Ifng locus in the context of T-lineage specification and plasticity. Immunol Rev 238 : 216–232.
14. WilsonCB, RowellE, SekimataM (2009) Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol 9 : 91–105.
15. SouttoM, ZhangF, EnersonB, TongY, BoothbyM, et al. (2002) A minimal IFN-γ promoter confers Th1 selective expression. J Immunol 169 : 4205–4212.
16. SouttoM, ZhouW, AuneTM (2002) Cutting edge: distal regulatory elements are required to achieve selective expression of IFN-γ in Th1/Tc1 effector cells. J Immunol 169 : 6664–6667.
17. HarringtonLE, JanowskiKM, OliverJR, ZajacAJ, WeaverCT (2008) Memory CD4 T cells emerge from effector T-cell progenitors. Nature 452 : 356–360.
18. HattonRD, HarringtonLE, LutherRJ, WakefieldT, JanowskiKM, et al. (2006) A distal conserved sequence element controls Ifng gene expression by T cells and NK cells. Immunity 25 : 717–729.
19. HadjurS, WilliamsLM, RyanNK, CobbBS, SextonT, et al. (2009) Cohesins form chromosomal cis-interactions at the developmentally regulated IFNG locus. Nature 460 : 410–413.
20. SekimataM, Perez-MelgosaM, MillerSA, WeinmannAS, SaboPJ, et al. (2009) CCCTC-binding factor and the transcription factor T-bet orchestrate T helper 1 cell-specific structure and function at the interferon-γ locus. Immunity 31 : 551–564.
21. SchoenbornJR, DorschnerMO, SekimataM, SanterDM, ShnyrevaM, et al. (2007) Comprehensive epigenetic profiling identifies multiple distal regulatory elements directing transcription of the gene encoding interferon-γ. Nat Immunol 8 : 732–742.
22. ShnyrevaM, WeaverWM, BlanchetteM, TaylorSL, TompaM, et al. (2004) Evolutionarily conserved sequence elements that positively regulate IFN-γ expression in T cells. Proc Natl Acad Sci U S A 101 : 12622–12627.
23. LeeDU, AvniO, ChenL, RaoA (2004) A distal enhancer in the interferon-γ (IFN-γ) locus revealed by genome sequence comparison. J Biol Chem 279 : 4802–4810.
24. ChangS, AuneTM (2007) Dynamic changes in histone-methylation ‘marks’ across the locus encoding interferon-γ during the differentiation of T helper type 2 cells. Nat Immunol 8 : 723–731.
25. MukasaR, BalasubramaniA, LeeYK, WhitleySK, WeaverBT, et al. (2010) Epigenetic instability of cytokine and transcription factor gene loci underlies plasticity of the T helper 17 cell lineage. Immunity 32 : 616–627.
26. ChangS, AuneTM (2005) Histone hyperacetylated domains across the Ifng gene region in natural killer cells and T cells. Proc Natl Acad Sci U S A 102 : 17095–17100.
27. WeiL, VahediG, SunHW, WatfordWT, TakatoriH, et al. (2010) Discrete roles of STAT4 and STAT6 transcription factors in tuning epigenetic modifications and transcription during T helper cell differentiation. Immunity 32 : 840–851.
28. YagiR, JunttilaIS, WeiG, UrbanJFJr, ZhaoK, et al. (2010) The transcription factor GATA3 actively represses RUNX3 protein-regulated production of interferon-gamma. Immunity 32 : 507–517.
29. BalasubramaniA, ShibataY, CrawfordGE, BaldwinAS, HattonRD, et al. (2010) Modular utilization of distal cis-regulatory elements controls Ifng gene expression in T cells activated by distinct stimuli. Immunity 33 : 35–47.
30. CollinsPL, ChangS, HendersonM, SouttoM, DavisGM, et al. (2010) Distal regions of the human IFNG locus direct cell type-specific expression. J Immunol 185 : 1492–1501.
31. CollinsPL, HendersonMA, AuneTM (2012) Diverse functions of distal regulatory elements at the IFNG locus. Journal of immunology 188 : 1726–1733.
32. LiuP, JenkinsNA, CopelandNG (2003) A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res 13 : 476–484.
33. WarmingS, CostantinoN, CourtDL, JenkinsNA, CopelandNG (2005) Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33: e36.
34. WangZ, ZangC, RosenfeldJA, SchonesDE, BarskiA, et al. (2008) Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 40 : 897–903.
35. CreyghtonMP, ChengAW, WelsteadGG, KooistraT, CareyBW, et al. (2010) Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA 107 : 21931–21936.
36. GhislettiS, BarozziI, MiettonF, PollettiS, De SantaF, et al. (2010) Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity 32 : 317–328.
37. ViselA, BlowMJ, LiZ, ZhangT, AkiyamaJA, et al. (2009) ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457 : 854–858.
38. VigneauS, RohrlichPS, BrahicM, BureauJF (2003) Tmevpg1, a candidate gene for the control of Theiler's virus persistence, could be implicated in the regulation of gamma interferon. J Virol 77 : 5632–5638.
39. GomezJA, WapinskiOL, YangYW, BureauJF, GopinathS, et al. (2013) The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-γ locus. Cell 152 : 743–754.
40. CollierSP, CollinsPL, WilliamsCL, BoothbyMR, AuneTM (2012) Cutting edge: influence of Tmevpg1, a long intergenic noncoding RNA, on the expression of Ifng by Th1 cells. J Immunol 189 : 2084–2088.
41. KaplanMH, SunYL, HoeyT, GrusbyMJ (1996) Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature 382 : 174–177.
42. JacobsonNG, SzaboSJ, Weber-NordtRM, ZhongZ, SchreiberRD, et al. (1995) Interleukin 12 signaling in T helper type 1 (Th1) cells involves tyrosine phosphorylation of signal transducer and activator of transcription (Stat)3 and Stat4. J Exp Med 181 : 1755–1762.
43. RobinsonD, ShibuyaK, MuiA, ZoninF, MurphyE, et al. (1997) IGIF does not drive Th1 development but synergizes with IL-12 for interferon-γ production and activates IRAK and NFκB. Immunity 7 : 571–581.
44. LeeYK, TurnerH, MaynardCL, OliverJR, ChenD, et al. (2009) Late developmental plasticity in the T helper 17 lineage. Immunity 30 : 92–107.
45. HarringtonLE, HattonRD, ManganPR, TurnerH, MurphyTL, et al. (2005) Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 6 : 1123–1132.
46. ScacheriPC, CrawfordGE, DavisS (2006) Statistics for ChIP-chip and DNase hypersensitivity experiments on NimbleGen arrays. Methods Enzymol 411 : 270–282.
47. ScacheriPC, CrawfordGE, DavisS (2006) Statistics for ChIP-chip and DNase hypersensitivity experiments on NimbleGen arrays. Meth Enzymol 411 : 270–282.
48. NicolJW, HeltGA, BlanchardSG, RajaA, LoraineAE (2009) The Integrated Genome Browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics 25 : 2730–2731.
49. CrawfordGE, DavisS, ScacheriPC, RenaudG, HalawiMJ, et al. (2006) DNase-chip: a high-resolution method to identify DNase I hypersensitive sites using tiled microarrays. Nat Methods 3 : 503–509.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 1
Nejčtenější v tomto čísle
- GATA6 Is a Crucial Regulator of Shh in the Limb Bud
- Large Inverted Duplications in the Human Genome Form via a Fold-Back Mechanism
- Differential Effects of Collagen Prolyl 3-Hydroxylation on Skeletal Tissues
- Affects Plant Architecture by Regulating Local Auxin Biosynthesis