
Genetic Epidemiology of Tuberculosis Susceptibility: Impact of Study Design
Several candidate gene studies have provided evidence for a role of host genetics in susceptibility to tuberculosis (TB). However, the results of these studies have been very inconsistent, even within a study population. Here, we review the design of these studies from a genetic epidemiological perspective, illustrating important differences in phenotype definition in both cases and controls, consideration of latent M. tuberculosis infection versus active TB disease, population genetic factors such as population substructure and linkage disequilibrium, polymorphism selection, and potential global differences in M. tuberculosis strain. These considerable differences between studies should be accounted for when examining the current literature. Recommendations are made for future studies to further clarify the host genetics of TB.
Published in the journal:
. PLoS Pathog 7(1): e32767. doi:10.1371/journal.ppat.1001189
Category:
Review
doi:
https://doi.org/10.1371/journal.ppat.1001189
Summary
Several candidate gene studies have provided evidence for a role of host genetics in susceptibility to tuberculosis (TB). However, the results of these studies have been very inconsistent, even within a study population. Here, we review the design of these studies from a genetic epidemiological perspective, illustrating important differences in phenotype definition in both cases and controls, consideration of latent M. tuberculosis infection versus active TB disease, population genetic factors such as population substructure and linkage disequilibrium, polymorphism selection, and potential global differences in M. tuberculosis strain. These considerable differences between studies should be accounted for when examining the current literature. Recommendations are made for future studies to further clarify the host genetics of TB.
Introduction
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), is a growing public health problem in the era of the HIV/AIDS pandemic. Among the one-third of the world infected by Mtb [1], almost 8 million new cases of TB occur annually, with 2 million deaths attributed to the disease each year. Only 10% of those individuals infected by Mtb go on to develop clinical disease, and disease presentation itself is heterogeneous, suggesting that host factors play a large role in disease susceptibility and natural history. An increased understanding of the host response to Mtb will facilitate the development of new vaccines and therapeutics [2].
Several studies have suggested a role for host genetics in TB susceptibility. Support for genetic susceptibility to TB in humans was first provided by twin studies [3], [4], animal models [5]–[8], then later segregation analyses [9], [10]. Countless candidate gene studies have been conducted, as well as seven genome-wide linkage scans [11]–[17]. However, there is a great deal of inconsistency across these studies. Among studies of any candidate gene, there are always several reports that provide both positive and negative evidence for an association with TB. Within genome scans, there has been replication of some results across two of the studies [14], [15], but there is very little replication across the remaining papers.
There are a number of key components of the design of these studies that may explain the inconsistency in the literature. The objective of this review is to discuss these issues, illustrated with examples from the TB genetics literature, and propose some approaches for taking a more thorough approach to the study of TB genetics.
Impact of Study Design
Phenotype Definition
The first step in any epidemiological study is to define the criteria used to diagnose disease. Then, one must define what is meant by non-diseased individuals (“controls”). In TB, this is complicated, because the pathogenesis of TB can be thought of as a two-stage process [18]. The first stage consists of latent Mtb infection (LTBI), in which Mtb establishes a productive infection but does not produce symptoms. LTBI is diagnosed by a positive tuberculin skin test (TST) and/or positive interferon-γ response assay (IGRA) in the absence of clinical signs and symptoms of full-blown disease [19], [20]. Definitive diagnosis of pulmonary TB requires the recovery of Mtb from sputum and cultivation in culture or detection of acid-fast bacilli (AFB) on smear [19], [21]. Studies have shown that AFB smear is less sensitive than culture, and that AFB smear grade could reflect differences in disease severity [21]. Smear-negative, culture-positive TB is also a problem in developing countries [21]. Thus, the method used to diagnose TB could affect the comparability of studies, and these differences could reflect variation in disease severity or even potential misclassification of disease status, generating a significant impact on the type I and type II error of studies. Here, we will first review the various diagnostic criteria used for TB disease, then the clinical characterization of study controls, and how these differences in study design may affect the interpretation of results across studies.
As stated by Möller and colleagues [22], studies of TB are “exquisitely sensitive to phenotype definition”. Different criteria have been used to diagnose TB in different study sites. Here, we focus on studies of the NRAMP1 (SLC11A1) gene, which has been studied most extensively (Table 1). To summarize, some studies have used the gold standard definition for TB diagnosis based on growth of Mtb in culture [19], though other studies only diagnosed TB patients based on positive AFB smear. Some studies had heterogeneous diagnostic criteria, classifying together cases diagnosed by smear or culture or symptoms. Other studies have combined pulmonary and extrapulmonary TB cases in the analysis [23]–[27]. Notice that of the 12 studies demonstrating an association between NRAMP1 and TB, only four used culture positivity as their diagnosis method. Could these differences in diagnostic criteria disguise differences in disease severity across populations?

Related to this is the definition of controls. It is unknown in many of these studies whether or not the “controls” were latently infected with Mtb, as evidenced by either a TST or IGRA. Recent studies have suggested some genes may actually be related to LTBI and not progression to TB [15], [28], [29], while other studies have suggested some genes may differentiate between LTBI and active TB disease [30], [31]. This is important in truly understanding the role of these genes in disease pathogenesis and progression. If controls are latently infected, and there is an association seen between a gene and TB, that suggests the gene influences progression from LTBI to TB. However, if controls are uninfected, it is unclear whether an association implies susceptibility for developing active disease or just acquisition of LTBI.
Finally, the selection of controls is not trivial. In a case-control study, controls should be similar to cases in every way possible except for the presence of disease. In studies of TB, this means controls should be exposed to infectious TB cases, so that they have the opportunity to acquire infection and then progress to active TB disease. Some studies conducted in TB-endemic settings assume all individuals are exposed to TB [25], [32]. However, studies have shown individuals may be persistently exposed to Mtb but never develop LTBI [15], [19]. Characterization of controls in TB genetics studies has differed widely (examples in Table 1). Many studies have utilized population controls, similar to the approach taken in recent large genome-wide association studies (GWAS) [33], i.e., by using blood bank donors. The disadvantage of this design is possible misclassification bias [34]—the chance that some of these “controls” may never become affected for TB, which is problematic when the disease is common [35]. By contrast, other studies have utilized unaffected household members [26], [31], [36], [37] or have conducted thorough clinical evaluation with TST in those without disease [27], [30], [38]; in these situations, exposure in unaffected individuals is known, so these are true controls in the epidemiological sense. Note that only one of the NRAMP1 associations was observed in studies where exposure has been quantified (Table 1).
Epidemiological Study Design
The vast majority of genetic epidemiological studies, not just for TB but for other complex traits as well, tend to be case-control studies. Such studies are easier to conduct because they do not require cooperation of the entire family, and a greater number of cases can be recruited. One major advantage of family-based designs for the study of infectious diseases is the characterization of exposure in the “controls”, as discussed above. Individuals living in the same household have a high likelihood of exposure to an infectious TB case, thereby influencing the probability that they too will develop TB [39]–[41]. As described above, epidemiological characterization of exposure is important in order to construct a valid case-control study.
Another advantage of family-based studies is the ability to account for population substructure. Hidden population stratification may result in bias (false positive results) [42] or false negative results [43]. Studies of TB genetics have been conducted in many admixed populations, including African Americans [26], [27], [44]–[46], Mexicans [30], and South African “Coloureds” [11], [14], [32], [47], [48]. Some of these studies [11], [26], [45], [46] have employed family-based designs. Other studies have examined potential population substructure by analyzing genomic control markers: one study in South Africa utilized ∼25 markers [47], [48], and another study utilized >200 markers [49]. Marchini et al. [43] point out genomic control markers will not adequately correct for population substructure if too few markers are used, but it is difficult to enumerate a sufficient number of markers in populations of African descent. It is unclear if other studies were able to account for population substructure. It may be impossible for existing study cohorts to incorporate family-based designs or retrospectively evaluate population stratification, but this clearly may explain some of the heterogeneity among studies.
Population Differences—More Than Just Geography
A typical explanation for differing results by population is population differentiation [22], [50], including genetic heterogeneity or inestimable polygenic effects. Another important genetic difference between populations is in linkage disequilibrium (LD).
Early studies of TB genetics were restricted to well-characterized markers within genes (studies of SLC11A1/NRAMP1 in Table 1 are examples). Often these markers were exonic or restriction fragment length polymorphisms. The underlying assumption of the power and design of such studies is that the polymorphism being analyzed is the causal polymorphism.
There are millions of single nucleotide polymorphisms (SNPs) throughout the genome [51], [52]. Because of the LD structure in the genome, certain SNPs can be used to “tag” haplotypes, such that one or a few SNPs capture information about LD structure [53]. Many trait-associated SNPs (>40%) are intergenic or intronic, suggesting an important role for non-coding SNPs in complex disease [54]. This serves as a reminder that disease risk alleles may actually be in LD with genotyped markers, which serve as “tags” for haplotypes on which the causal allele may reside. This is illustrated by Figure 1, where we consider an underlying disease allele that is not directly genotyped but surrounded by flanking markers. The ability to detect association with the region where the disease allele resides depends entirely on the strength of LD between the unobserved risk allele and flanking markers. As patterns of LD differ between study populations, the specific trait-associated SNPs will consequently differ.
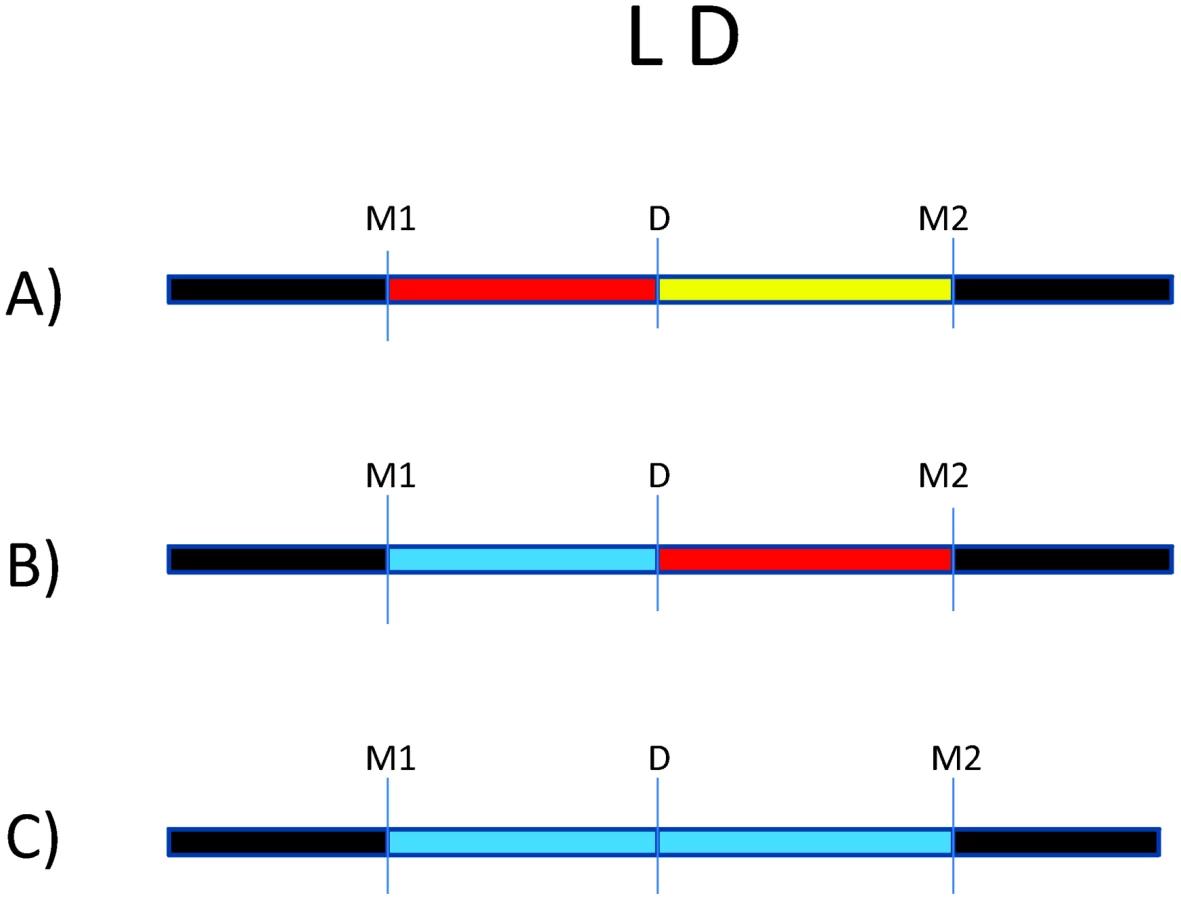
The impact of LD differences between study populations is further illustrated in Figure 2. Here, LD patterns in NRAMP1 were plotted using HapMap reference populations representative of those populations where NRAMP1 has been studied: Caucasians in Utah, United States (CEU), Yoruba in Nigeria (YRI), Maasai in Kenya (MKK), Han Chinese (CHN), and African Americans in the US Southwest (ASW). These LD plots were generated using default parameters in the Genome Variation Server (http://gvs.gs.washington.edu/GVS/), with no minor allele frequency cutoff. African populations (YRI and MKK) have very little LD because they are older populations, and their LD patterns differ. Newer populations (CEU and CHN) have much greater LD, and recently admixed populations (ASW) also exhibit LD between SNPs, but there are differences. Also note that the SNPs themselves (rs numbers) differ between populations, illustrating how different polymorphisms exist within the same genes across world populations. A perfect illustration of this phenomenon is provided by Velez et al. [26], who analyzed a number of SNPs within NRAMP1. Though they did not observe statistical association with the markers that were examined in early studies, they did observe association with intronic and exonic SNPs. If they had not conducted such extensive genotyping, they may have missed these associations.

Only a few other studies have accounted for global variations in LD by analyzing several SNPs within candidate genes of interest. Some studies have selected tag SNPs based on relevant HapMap reference populations [26], [28], [31], [46]. Other studies have sequenced genes of interest first to identify novel SNPs within the gene(s), then analyzed association with those SNPs [55]–[58]. Though other studies did not utilize LD in their selection of SNPs, they later estimated LD between markers in their dataset, and used this analysis to guide haplotype analysis [48], [49]. Since LD patterns differ by population, it should not be surprising that genetic association results differ, especially given the limited number of markers analyzed per gene. There are many implications of this variation. Differences in the strength of LD between the actual disease locus and genotyped markers will affect the power to detect association to markers (Figure 1). In populations with weaker LD such as African populations, denser SNP observed maps are necessary to detect association effects with untyped disease loci. Thus, variation in number of polymorphisms analyzed, differences in LD in the reference population, and existence of still-unknown risk alleles all complicate replication across studies.
Another controversial issue is the study of common versus rare genetic variants. The common disease–common variant (CDCV) hypothesis posits that genetic risk for common diseases will often be due to common risk alleles [59]. This is in contrast to the common disease rare variant (CDRV) hypothesis, which states that a significant proportion of common chronic diseases are influenced by the summation of effects of multiple low frequency variants in the same gene, where tagging SNPs will not be useful in identifying a single haplotype because no single haplotype exists [60]. Most candidate gene studies assume the CDCV hypothesis. Recent sequencing studies [61], [62] have detected rare SNPs in the TLR family of genes; these could be important, but massive studies will be needed in order to detect disease associations at a statistically significant threshold. In addition, copy number variants (CNVs) have recently attracted attention in their association with complex traits, such as HIV acquisition and progression and autoimmune diseases [63]. These are also considered rare variants, so we are again faced with all of the challenges of testing the CDRV hypothesis.
The above discussion focuses on population genetics of humans. Another related issue is variation in Mtb strains. Researchers have categorized Mtb into six main bacterial strain lineages that are associated with particular geographical regions [64], as well as differences in clinical presentation [65] and rate of progression to active TB disease [66]. So, not only do different diagnostic criteria, as discussed above, potentially reflect differences in disease severity, but specific Mtb strains may also influence disease severity. A recent study suggests a host genotype x Mtb genotype interaction, whereby the TLR2 genotype is associated with TB caused by the Beijing strain [67]. Very few studies have the capacity to examine this potential host by Mtb interaction, but it could easily be a potential explanation for differences between studies.
Complex Genetic Effects
Complex traits such as TB are likely influenced by several factors, including gene–gene interaction and gene–environment interaction. Few studies have investigated gene-gene interactions in the context of human TB. Many gene products (e.g., Toll-like receptors [TLRs]) are known to interact biologically [68], and interaction effects have been demonstrated in mouse models of TB [69]. A recent study identified interactions between the NOS2A gene and IFNGR1 and TLR4 [45]. Interestingly, both IFNGR1 and TLR4 showed no evidence of significant main effects in this analysis. Another study by the same research group found interaction between NRAMP1 and TLR2, but TLR2 did not itself have a significant main effect [26]. This suggests many important genes may influence TB in combination with other genes, but this could be overlooked because their individual effects did not meet criteria for statistical significance. Motsinger-Reif et al. used multifactor dimensionality reduction to identify a potential gene–gene interaction between TLR4 and the TNF-α gene (TNF) [70]. In addition, it is well known that HIV influences the pathogenesis of TB, but most genetic epidemiological studies have been restricted to HIV seronegative individuals. Our work [31] showed an interaction between HIV and the TNF receptor 1 gene. Because many studies have excluded HIV-positive individuals, this hypothesis remains relatively unexplored. Similar to the TNF-α pathway, the type I and II interferon pathways have been associated with both TB and HIV pathogenesis [71], and so should also be considered for future studies of gene–HIV interactions. The challenge of examining interaction effects is the requirement of even larger sample sizes, as discussed by Velez et al. [45].
Conclusions, Recommendations, and Future Directions
As reviewed recently by Möller et al. [22], the body of work showing statistical associations between candidate genes and TB continues to grow. This does not include potential unpublished studies that failed to find significant associations and are not readily available due to publication bias [22]. Even in the published body of literature, however, there is a great deal of inconsistency between marker-trait associations, so we are far from reaching a consensus regarding genes involved in TB risk.
This review focused on methodological reasons for inconsistency across studies. One important factor is the diagnostic criteria for TB disease, which have differed dramatically across studies. Resources available for TB diagnosis differ by country, which is confounded when there has been conflict [72]. Differences in diagnostic criteria across studies can reflect differences in TB severity and may lead to misclassification of cases as controls; this would have a significant impact on the type I and type II error of studies. It is impossible to standardize the diagnostic definitions used across all study sites, but researchers should be mindful of such differences when interpreting their findings. We strongly recommend that researchers characterize the level of exposure to Mtb in individuals without disease, which should include TST/IGRA and careful epidemiological characterization. New studies could utilize the household contact design, which facilitates the characterization of all stages of Mtb exposure, infection, and disease [41]. When the household contact study design is not feasible, spousal controls are also ideal because of persistent and prolonged exposure.
Recall that TB follows two stages of pathogenesis, and LTBI precedes TB disease. Recent studies suggest that LTBI may have unique genetic influences [15], [28], [29]. Persons with LTBI constitute a major impediment to TB control efforts [73]. Since many ongoing vaccine development efforts will focus either preventing LTBI or progression to TB, it is important to understand host factors that influence containment of Mtb infection. However, the study of the genetics of LTBI is also not trivial. Indication of T cell memory response via positive TST and/or IGRA does not necessarily imply the presence of viable Mtb bacilli. In the US as well as other public health systems, individuals with positive TST are treated as though there are viable organisms present, adding further confusion to this phenotype. According to Parrish et al., there is a 2%–23% lifetime probability of developing TB after acquisition of Mtb infection (LTBI) [73]. This illustrates the heterogeneity in this clinical group, since the risk of progression to active TB may depend on a variety of known and unknown risk factors. Furthermore, prophylaxis of LTBI with isoniazid (INH) is the standard of care in many research settings, so that many individuals with “LTBI” based on positive TST/IGRA, genetically predisposed to develop TB, may not. One way to investigate the role of host genetics in LTBI would be to compare TST (or IGRA) positive individuals that develop incident TB to those that do not. Ideally, such a study would not include individuals on INH prophylaxis, though that is unethical in many settings. For these reasons, some may argue that it is more relevant to study TB genetics, and not LTBI, from a public health standpoint.
Thus, it is essential to take a multidisciplinary approach [74] to develop an all-encompassing picture of the natural history of Mtb infection and disease. Few studies have examined the genetics of TB immunology [15], [31], [75]–[77]. Gene expression studies using microarrays may also shed light on host responses to Mtb [78]. Proteomic studies will further elucidate host factors involved in pathogenesis. These various approaches should be analyzed together to hopefully identify more meaningful clinical groups. For example, genomic, proteomic, and immunologic data, collectively, may better capture the heterogeneity in latently infected individuals.
Additional complicating factors in comparing geographically diverse studies are potential population substructure and LD differences among populations. We recommend that future studies analyze enough SNPs to capture LD in their study population. Analyses of a few markers within a gene no longer advance the field, particularly in light of LD differences between populations. Even with advances in genotyping, many studies of “old” markers continue to be published. The choice of a reference population for tag SNP selection is not trivial [62]; thus, dense SNP mapping may be necessary, particularly in studies of African populations. If it is impossible to rigorously examine genes in this way, publishing the LD patterns in the study data [28], [45], [48], [49] is a good start. Furthermore, studies in admixed populations should attempt to examine population substructure to minimize this source of bias. Populations also differ in the Mtb strain lineage that caused TB; future studies examining host gene by Mtb gene interaction are warranted. Finally, as in all genetic epidemiological studies of complex traits, genes may act in complex ways. Genes may interact with other genes and/or epidemiological factors; these potential relationships should not be overlooked. Furthermore, too many researchers (authors and journal reviewers alike) focus too much on p-values. All p-values must be reported, even if greater than 0.05. Markers with p-values greater than 0.05 may still be important in their interaction with other markers or environmental factors. Researchers should collect sufficient data to explore these meaningful biological effects.
There are GWAS of TB forthcoming. Given the issues discussed in this review, we must interpret the findings of those GWAS cautiously. Will these studies be underpowered due to the heterogeneity among TB cases and controls? A recent summary analysis of published GWAS found the reported SNP–trait associations attaining significance (p<10−5) had a median odds ratio of 1.33, with an interquartile range of 1.20–1.61 [54]; thus, the effect sizes of SNPs identified through GWAS are relatively small. Furthermore, the proportion of heritability explained by these variants ranges between 1% and 50% [79]. TB GWAS may provide new clues into the host biology of TB pathogenesis, but the overall clinical relevance of these SNPs will be limited. In addition, GWAS of other complex traits have often merged data across ongoing research studies. Because of the dramatic heterogeneity among studies described in this review, meta-analyses of TB genetic association studies should be conducted with care.
In sum, we have barely scratched the surface in understanding the genetic determinants of TB pathogenesis. Because of the significant public health impact of TB, additional studies are necessary, and should be multidisciplinary in nature. Future studies should carefully consider phenotype definition and genetic epidemiological principles when designing, analyzing, and interpreting findings. Ideally, culture confirmation for pulmonary TB should be conducted where feasible, thorough epidemiological data should be collected in individuals without TB to better understand LTBI and risk of progression to TB, and population genetic factors should be carefully characterized and considered in the analysis.
Accession Numbers for Genes Mentioned in This Paper (GeneIDs from EntrezGene)
TLR2 (7097); SLC11A1, aka NRAMP1 (6556); IFNGR1 (3459); TLR4 (7099); TNF (7124); TNFSF1A, aka TNF receptor 1 (7132); NOS2A (4843).
Zdroje
1. RaviglioneM
SniderD
KochiA
1995 Global epidemiology of tuberculosis: morbidity and mortality of a worldwide epidemic. JAMA 273 220 226
2. KaufmannSH
BaumannS
NasserEA
2006 Exploiting immunology and molecular genetics for rational vaccine design against tuberculosis. Int J Tuberc Lung Dis 10 1068 1079
3. ComstockG
1978 Tuberculosis in twins: a re-analysis of the Prophit Study. Am Rev Resp Dis 117 621 624
4. KallmannF
ReisnerD
1943 Twin studies on the significance of genetic factors in tuberculosis. Am Rev Tuberculosis 47 549 574
5. BlackwellJ
BartonC
WhiteJ
SearleS
BakerA
WilliamsH
ShawM
2004 Genomic organizaton and sequence of the human NRAMP gene: identification and mapping of a promotor region polymorphism. Mol Med 1 194 205
6. FlynnJ
GoldsteinM
ChanJ
TrieboldK
PfefferK
LowensteinC
SchreiberR
MakT
BloomB
1995 Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 2 561 572
7. KramnikI
DietrickW
DemantP
BloomB
2000 Genetic control of resistance to experimental infection with virulent Mycobacterium tuberculosis. Proc Natl Acad Sci 97 8560 8565
8. LurieM
1941 Heredity, constitution and tuberculosis: An experimental study. Am Rev Tuberc 44 1 125
9. ShawMA
CollinsA
PeacockCS
MillerEN
BlackGF
SibthorpeD
Lins-LainsonZ
ShawJJ
RamosF
SilveiraF
BlackwellJM
1997 Evidence that genetic susceptibility to Mycobacterium tuberculosis in a Brazilian population is under oligogenic control: linkage study of the candidate genes NRAMP1 and TNFA. Tuberc Lung Dis 78 35 45
10. SteinCM
NshutiL
ChiundaAB
BoomWH
ElstonRC
MugerwaRD
IyengarSK
WhalenCC
2005 Evidence for a major gene influence on tumor necrosis factor-alpha expression in tuberculosis: path and segregation analysis. Hum Hered 60 109 118
11. BellamyR
BeyersN
McAdamK
RuwendeC
GieR
SamaaiP
BesterD
MeyerM
CorrahT
CollinM
CamidgeD
WilkinsonD
Hoal-van HeldenE
WhittleH
AmosW
van HeldenP
HillA
2000 Genetic susceptibility to tuberculosis in Africans: A genome-wide scan. Proc Natl Acad Sci 97 8005 8009
12. JamiesonS
MillerE
BlackG
PeacockC
CordellH
HowsonJ
ShawM
BurgnerD
XuW
Lins-LainsonZ
RamosF
SilveiraF
BlackwellJ
2004 Evidence for a cluster of genes on chromosome 17q11-q21 controlling susceptibility to tuberculosis and leprosy in Brazilians. Genes and Immunity 5 46 57
13. MillerE
JamiesonS
JobertyC
FakiolaM
HudsonD
PeacockC
CordellH
ShawM
Lins-LainsonZ
RamosF
SilveiraF
BlackwellJ
2004 Genome-wide scans for leprosy and tuberculosis susceptibility genes in Brazilians. Genes Immun 5 63 67
14. CookeGS
CampbellSJ
BennettS
LienhardtC
McAdamKP
SowO
GustafsonP
MwanguluF
vanHP
FineP
HoalEG
HillAV
2008 Mapping of a Novel Susceptibility Locus Suggests a Role for MC3R and CTSZ in Human Tuberculosis. Am J Respir Crit Care Med 178 203 207
15. SteinCM
ZalwangoS
MaloneLL
WonS
Mayanja-KizzaH
MugerwaRD
LeontievDV
ThompsonCL
CartierKC
ElstonRC
IyengarSK
BoomWH
WhalenCC
2008 Genome scan of M. tuberculosis infection and disease in Ugandans. PLoS ONE 3 e4094 doi:10.1371/journal.pone.0004094
16. BaghdadiJE
OrlovaM
AlterA
RanqueB
ChentoufiM
LazrakF
ArchaneMI
CasanovaJL
BenslimaneA
SchurrE
AbelL
2006 An autosomal dominant major gene confers predisposition to pulmonary tuberculosis in adults. J Exp Med 203 1679 1684
17. MahasirimongkolS
YanaiH
NishidaN
RidruechaiC
MatsushitaI
OhashiJ
SummanapanS
YamadaN
MoolphateS
ChuchotawornC
ChaiprasertA
ManosuthiW
KantipongP
KanitwittayaS
SuraT
KhusmithS
TokunagaK
SawanpanyalertP
KeichoN
2009 Genome-wide SNP-based linkage analysis of tuberculosis in Thais. Genes Immun 10 77 83
18. ComstockG
1982 Epidemiology of tuberculosis. Am Rev Resp Dis 125 8 15
19. [No authors listed] 2000 Diagnostic Standards and Classification of Tuberculosis in Adults and Children. This official statement of the American Thoracic Society and the Centers for Disease Control and Prevention was adopted by the ATS Board of Directors, July 1999. This statement was endorsed by the Council of the Infectious Disease Society of America, September 1999. Am J Respir Crit Care Med 161 1376 1395
20. NyendakM
LewinsohnDA
LewinsohnD
2008 The use of interferon-gamma release assays in clinical practice.
DaviesP
BarnesPF
GordonSB
Clinical tuberculosis London Hodder Arnold
21. GarayS
2004 Pulmonary tuberculosis.
RomWN
GaraySM
Tuberculosis Philadelphia Lippincott Williams & Wilkins 345 394
22. MollerM
de WitE
HoalEG
2010 Past, present and future directions in human genetic susceptibility to tuberculosis. FEMS Immunol Med Microbiol 58 3 26
23. MaX
ReichR
WrightJ
TookerH
TeeterL
MusserJ
GravissE
2003 Association between Interleukin-8 gene alleles and human susceptbility to tuberculosis disease. J Infect Dis 188 349 355
24. RossouwM
NelHJ
CookeGS
van HeldenPD
HoalEG
2003 Association between tuberculosis and a polymorphic NFκB binding site in the interferon γ gene. Lancet 361 1871 1872
25. TaypeCA
CastroJC
AccinelliRA
Herrera-VelitP
ShawMA
EspinozaJR
2006 Association between SLC11A1 polymorphisms and susceptibility to different clinical forms of tuberculosis in the Peruvian population. Infect Genet Evol 6 361 367
26. VelezDR
HulmeWF
MyersJL
StryjewskiME
AbbateE
EstevanR
PatilloSG
GilbertJR
HamiltonCD
ScottWK
2009 Association of SLC11A1 with tuberculosis and interactions with NOS2A and TLR2 in African-Americans and Caucasians. Int J Tuberc Lung Dis 13 1068 1076
27. Motsinger-ReifAA
AntasPR
OkiNO
LevyS
HollandSM
SterlingTR
2010 Polymorphisms in IL-1beta, vitamin D receptor Fok1, and Toll-like receptor 2 are associated with extrapulmonary tuberculosis. BMC Med Genet 11 37
28. ThyeT
BrowneEN
ChinbuahMA
GyapongJ
OseiI
Owusu-DaboE
BrattigNW
NiemannS
Rusch-GerdesS
HorstmannRD
MeyerCG
2009 IL10 haplotype associated with tuberculin skin test response but not with pulmonary TB. PLoS ONE 4 e5420 doi:10.1371/journal.pone.0005420
29. CobatA
GallantCJ
SimkinL
BlackGF
StanleyK
HughesJ
DohertyTM
HanekomWA
EleyB
JaisJP
Boland-AugeA
vanHP
CasanovaJL
AbelL
HoalEG
SchurrE
AlcaisA
2009 Two loci control tuberculin skin test reactivity in an area hyperendemic for tuberculosis. J Exp Med 206 2583 2591
30. Flores-VillanuevaPO
Ruiz-MoralesJA
SongCH
FloresLM
JoEK
MontanoM
BarnesPF
SelmanM
GranadosJ
2005 A functional promoter polymorphism in monocyte chemoattractant protein-1 is associated with increased susceptibility to pulmonary tuberculosis. J Exp Med 202 1649 1658
31. SteinCM
ZalwangoS
ChiundaAB
MillardC
LeontievDV
HorvathAL
CartierKC
ChervenakK
BoomWH
ElstonRC
MugerwaRD
WhalenCC
IyengarSK
2007 Linkage and association analysis of candidate genes for TB and TNFalpha cytokine expression: evidence for association with IFNGR1, IL-10, and TNF receptor 1 genes. Hum Genet 121 663 673
32. HoalEG
LewisLA
JamiesonSE
TanzerF
RossouwM
VictorT
HillermanR
BeyersN
BlackwellJM
Van HeldenPD
2004 SLC11A1 (NRAMP1) but not SLC11A2 (NRAMP2) polymorphisms are associated with susceptibility to tuberculosis in a high-incidence community in South Africa. Int J Tuberc Lung Dis 8 1464 1471
33. 2007 Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447 661 678
34. EdwardsBJ
HaynesC
LevenstienMA
FinchSJ
GordonD
2005 Power and sample size calculations in the presence of phenotype errors for case/control genetic association studies. BMC Genet 6 18
35. McCarthyMI
AbecasisGR
CardonLR
GoldsteinDB
LittleJ
IoannidisJPA
HirschhornJN
2008 Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet 9 356 369
36. CervinoA
LakissS
SowO
HillA
2000 Allelic association between the NRAMP1 gene and susceptibility to tuberculosis in Guinea-Conakry. Ann Hum Genet 64 507 512
37. El BaghdadiJ
RemusN
BenslinaneA
El AnnazH
ChentoufiM
AbelL
SchurrE
2004 Variants of the human NRAMP1 gene and susceptibility to tuberculosis in Morocco. Int J Tuberc Lung Dis 7 599 602
38. DubaniewiczA
JamiesonSE
Dubaniewicz-WybieralskaM
FakiolaM
NancyME
BlackwellJM
2005 Association between SLC11A1 (formerly NRAMP1) and the risk of sarcoidosis in Poland. Eur J Hum Genet 13 829 834
39. ChapmanJS
DyerlyMD
1964 Social and other factors in intrafamilial transmission of tuberculosis. Am Rev Respir Dis 90 48 60
40. EnarsonD
ChiangC-Y
MurrayJ
2004 Global epidemiology of tuberculosis.
RomWN
GaraySM
Tuberculosis Philadelphia Lippincott Williams & Wilkins 13 30
41. GuwattudeD
NakakeetoM
Jones-LopezE
MagandaA
ChiundaA
MugerwaR
EllnerJ
BukenyaG
WhalenC
2003 Tuberculosis in household contacts of infectious cases in Kampala, Uganda. Am J Epidemiol 158 887 898
42. SattenGA
FlandersWD
YangQ
2001 Accounting for unmeasured population substructure in case-control studies of genetic association using a novel latent-class model. Am J Hum Genet 68 466 477
43. MarchiniJ
CardonLR
PhillipsMS
DonnellyP
2004 The effects of human population structure on large genetic association studies. Nat Genet 36 512 517
44. MaX
ReichR
GonzálezA
PanX
FothergillA
StarkeJ
TeeterL
MusserJ
GravissE
2003 No evidence for association between the polymorphism in the 3′ untranslated region of interleukin-12B and human susceptibility to tuberculosis. J Infect Dis 188 1116 1118
45. VelezDR
HulmeWF
MyersJL
WeinbergJB
LevesqueMC
StryjewskiME
AbbateE
EstevanR
PatilloSG
GilbertJR
HamiltonCD
ScottWK
2009 NOS2A, TLR4, and IFNGR1 interactions influence pulmonary tuberculosis susceptibility in African-Americans. Hum Genet 126 643 653
46. VelezDR
WejseC
StryjewskiME
AbbateE
HulmeWF
MyersJL
EstevanR
PatilloSG
OlesenR
TacconelliA
SirugoG
GilbertJR
HamiltonCD
ScottWK
2010 Variants in toll-like receptors 2 and 9 influence susceptibility to pulmonary tuberculosis in Caucasians, African-Americans, and West Africans. Hum Genet 127 65 73
47. BarreiroLB
NeyrollesO
BabbCL
TailleuxL
QuachH
McElreaveyK
HeldenPD
HoalEG
GicquelB
Quintana-MurciL
2006 Promoter variation in the DC-SIGN-encoding gene CD209 is associated with tuberculosis. PLoS Med 3 e20 doi:10.1371/journal.pmed.0030020
48. MollerM
NebelA
ValentonyteR
Van HeldenPD
SchreiberS
HoalEG
2009 Investigation of chromosome 17 candidate genes in susceptibility to TB in a South African population. Tuberculosis (Edinb) 89 189 194
49. DavilaS
HibberdML
HariDR
WongHE
SahiratmadjaE
BonnardC
AlisjahbanaB
SzeszkoJS
BalabanovaY
DrobniewskiF
van CrevelR
van de VosseE
NejentsevS
OttenhoffTHM
SeielstadM
2008 Genetic association and expression studies indicate a role of toll-like receptor 8 in pulmonary tuberculosis. PLoS Genet 4 e1000218 doi:10.1371/journal.pgen.1000218
50. DengHW
2001 Population admixture may appear to mask, change or reverse genetic effects of genes underlying complex traits. Genetics 159 1319 1323
51. International HapMap Consortium 2003 The International HapMap Project. Nature 426 789 796
52. International HapMap Consortium 2005 A haplotype map of the human genome. Nature 437 1299 1320
53. GabrielSB
SchaffnerSF
NguyenH
MooreJM
RoyJ
BlumenstielB
HigginsJ
DefeliceM
LochnerA
FaggartM
Liu-CorderoSN
RotimiC
AdeyemoA
CooperR
WardR
LanderES
DalyMJ
AltshulerD
2002 The structure of haplotype blocks in the human genome. Science 296 2225 2229
54. HindorffLA
SethupathyP
JunkinsHA
RamosEM
MehtaJP
CollinsFS
ManolioTA
2009 Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A 106 9362 9367
55. TangNL
FanHP
ChangKC
ChingJK
KongKP
YewWW
KamKM
LeungCC
TamCM
BlackwellJ
ChanCY
2009 Genetic association between a chemokine gene CXCL-10 (IP-10, interferon gamma inducible protein 10) and susceptibility to tuberculosis. Clin Chim Acta 406 98 102
56. DissanayekeSR
LevinS
PienaarS
WoodK
EleyB
BeattyD
HendersonH
AndersonS
LevinM
2009 Polymorphic variation in TIRAP is not associated with susceptibility to childhood TB but may determine susceptibility to TBM in some ethnic groups. PLoS ONE 4 e6698 doi:10.1371/journal.pone.0006698
57. HawnTR
DunstanSJ
ThwaitesGE
SimmonsCP
ThuongNT
LanNT
QuyHT
ChauTT
HieuNT
RodriguesS
JanerM
ZhaoLP
HienTT
FarrarJJ
AderemA
2006 A polymorphism in Toll-interleukin 1 receptor domain containing adaptor protein is associated with susceptibility to meningeal tuberculosis. J Infect Dis 194 1127 1134
58. ThuongNT
HawnTR
ThwaitesGE
ChauTT
LanNT
QuyHT
HieuNT
AderemA
HienTT
FarrarJJ
DunstanSJ
2007 A polymorphism in human TLR2 is associated with increased susceptibility to tuberculous meningitis. Genes Immun 8 422 428
59. ReichDE
LanderES
2001 On the allelic spectrum of human disease. Trends Genet 17 502 510
60. BodmerW
BonillaC
2008 Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet 40 695 701
61. MaX
LiuY
GowenBB
GravissEA
ClarkAG
MusserJM
2007 Full-exon resequencing reveals toll-like receptor variants contribute to human susceptibility to tuberculosis disease. PLoS ONE 2 e1318 doi:10.1371/journal.pone.0001318
62. BakerA
RandhawaA
SheyM
de KockM
KaplanG
AdamsM
HanekomWA
BoomWH
HawnTR
SteinCM
2009 Comparison of genotype frequencies in Toll-like receptor genes in Ugandans, South Africans, and African HapMap populations. Presented at the American Society of Human Genetics 59th annual meeting; 20–24 October 2009; Honolulu, Hawaii, United States of America. 1808/T/Poster Board #357
63. McCarrollSA
AltshulerDM
2007 Copy-number variation and association studies of human disease. Nat Genet 39 S37 S42
64. GagneuxS
SmallPM
2007 Global phylogeography of Mycobacterium tuberculosis and implications for tuberculosis product development. Lancet Infect Dis 7 328 337
65. ThwaitesG
CawsM
ChauTT
D'SaA
LanNT
2008 The relationship between Mycobacterium tuberculosis genotype and the clinical phenotype of pulmonary and meningeal tuberculosis. J Clin Microbiol 46 1363 1368
66. de JongBC
HillPC
AikenA
AwineT
AntonioM
AdetifaIM
Jackson-SillahDJ
FoxA
DeriemerK
GagneuxS
BorgdorffMW
McAdamKP
CorrahT
SmallPM
AdegbolaRA
2008 Progression to active tuberculosis, but not transmission, varies by Mycobacterium tuberculosis lineage in The Gambia. J Infect Dis 198 1037 1043
67. CawsM
ThwaitesG
DunstanS
HawnTR
LanNT
ThuongNT
StepniewskaK
HuyenMN
BangND
LocTH
GagneuxS
VanSD
KremerK
van derSM
SmallP
AnhPT
ChinhNT
QuyHT
DuyenNT
ThoDQ
HieuNT
TorokE
HienTT
DungNH
NhuNT
DuyPM
van VinhCN
FarrarJ
2008 The influence of host and bacterial genotype on the development of disseminated disease with Mycobacterium tuberculosis. PLoS Pathog 4 e1000034 doi:10.1371/journal.ppat.1000034
68. BerringtonWR
HawnTR
2007 Mycobacterium tuberculosis, macrophages, and the innate immune response: does common variation matter? Immunol Rev 219 167 186
69. BaficaA
ScangaCA
FengCG
LeiferC
CheeverA
SherA
2005 TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J Exp Med 202 1715 1724
70. GomezLM
AnayaJM
VilchezJR
CadenaJ
HinojosaR
VelezL
Lopez-NevotMA
MartinJ
2007 A polymorphism in the inducible nitric oxide synthase gene is associated with tuberculosis. Tuberculosis (Edinb) 87 288 294
71. GiacominiE
RemoliME
GafaV
PardiniM
FattoriniL
CocciaEM
2009 IFN-beta improves BCG immunogenicity by acting on DC maturation. J Leukoc Biol 85 462 468
72. GustafsonP
GomesVF
VieiraCS
JensenH
SengR
NorbergR
SambB
NauclerA
AabyP
2001 Tuberculosis mortality during a civil war in Guinea-Bissau. JAMA 286 599 603
73. ParrishNM
DickJD
BishaiWR
1998 Mechanisms of latency in Mycobacterium tuberculosis. Trends Microbiol 6 107 112
74. ComasI
GagneuxS
2009 The past and future of tuberculosis research. PLoS Pathog 5 e1000600 doi:10.1371/journal.ppat.1000600
75. WheelerE
MillerEN
PeacockCS
DonaldsonIJ
ShawMA
JamiesonSE
BlackwellJM
CordellHJ
2006 Genome-wide scan for loci influencing quantitative immune response traits in the Belem family study: comparison of methods and summary of results. Ann Hum Genet 70 78 97
76. HawnTR
MischEA
DunstanSJ
ThwaitesGE
LanNT
2007 A common human TLR1 polymorphism regulates the innate immune response to lipopeptides. Eur J Immunol 37 2280 2289
77. SheyMS
RandhawaAK
BowmakerM
SmithE
ScribaTJ
de KockM
MahomedH
HusseyG
HawnTR
HanekomWA
2010 Single nucleotide polymorphisms in toll-like receptor 6 are associated with altered lipopeptide - and mycobacteria-induced interleukin-6 secretion. Genes Immun 11 561 572
78. ThuongNT
DunstanSJ
ChauTT
ThorssonV
SimmonsCP
QuyenNT
ThwaitesGE
Thi Ngoc LanN
HibberdM
TeoYY
SeielstadM
AderemA
FarrarJJ
HawnTR
2008 Identification of tuberculosis susceptibility genes with human macrophage gene expression profiles. PLoS Pathog 4 e1000229 doi:10.1371/journal.ppat.1000229
79. ManolioTA
CollinsFS
CoxNJ
GoldsteinDB
HindorffLA
HunterDJ
McCarthyMI
RamosEM
CardonLR
ChakravartiA
ChoJH
GuttmacherAE
KongA
KruglyakL
MardisE
RotimiCN
SlatkinM
ValleD
WhittemoreAS
BoehnkeM
ClarkAG
EichlerEE
GibsonG
HainesJL
MackayTF
McCarrollSA
VisscherPM
2009 Finding the missing heritability of complex diseases. Nature 461 747 753
80. BellamyR
RuwendeC
CorrahT
McAdamK
WhittleH
HillA
1998 Variations in the NRAMP1 gene and susceptibility to tuberculosis in West Africans. N Engl J Med 338 640 644
81. AwomoyiA
MarchantA
HowsonJ
McAdamK
BlackwellJ
NewportM
2002 Interleukin-10, Polymorphism in SLC11A1 (formerly NRAMP1), and susceptibility to tuberculosis. J Infect Dis 186 1808 1814
82. FitnessJ
FloydS
WarndorffD
SichaliL
MalemaS
CrampinA
FineP
HillA
2004 Large-scale candidate gene study of tuberculosis susceptibility in the Karonga district of Nothern Malawi. Am J Trop Med Hyg 71 341 349
83. SoborgC
AndersenAB
RangeN
MalenganishoW
FriisH
MagnussenP
TemuMM
ChangaluchaJ
MadsenHO
GarredP
2007 Influence of candidate susceptibility genes on tuberculosis in a high endemic region. Mol Immunol 44 2213 2220
84. MaX
WrightJ
ReichR
TeeterL
El SahlyH
AweR
MusserJ
GravissE
2002 5′ dinucleotide repeat polymorphism of NRAMP1 and susceptibility to tuberculosis among Caucasian patients in Houston, Texas. Int J Tuberc Lung Dis 6 818 823
85. DelgadoJ
BaenaA
ThimS
GoldfeldA
2002 Ethnic-specific genetic associations with pulmonary tuberculosis. J Infect Dis 186 1463 1468
86. LiuW
CaoW
ZhangC
TianL
WuX
HabbemaJ
ZhaoQ
ZhangP
XinZ-T
LiC
YangH
2004 VDR and NRAMP1 gene polymorphisms in susceptibility to pulmonary tuberculosis among the Chinese Han population: a case-control study. Int J Tuberc Lung Dis 8 428 434
87. AbeT
IinumaY
AndoM
YokoyamaT
YamamotoT
NakashimaK
TakagiN
BabaH
HasegawaY
ShimokataK
2003 NRAMP1 polymorphisms, susceptibility and clinical features of tuberculosis. J Infect 46 215 220
88. GaoP-S
FujishimaS
MaoX-Q
RemusN
KandaM
EnomotoT
DakeY
BottiniN
TabuchiM
HasegawaN
YamaguchiK
TiemessenC
HopkinJ
ShirakawaT
KishiF
2000 Genetic variants of NRAMP1 and active tuberculosis in Japanese populations. Clin Genet 58 74 76
89. LiawY-S
WuJ-JT
WuC-H
HungC-C
LeeC-N
YangP-C
LuhK-T
KuoS-H
2002 Variations in the NRAMP1 gene and susceptibility of tuberculosis in Taiwanese. Int J Tuberc Lung Dis 6 454 460
90. AkahoshiM
IshiharaM
RemusN
UnoK
MiyakeK
HirotaT
NakashimaK
MatsudaA
KandaM
EnomotoT
OhnoS
NakashimaH
CasanovaJL
HopkinJM
TamariM
MaoXQ
ShirakawaT
2004 Association between IFNA genotype and the risk of sarcoidosis. Hum Genet 114 503 509
91. VejbaesyaS
ChierakulN
LuangtrakoolP
SermduangprateepC
2007 NRAMP1 and TNF-alpha polymorphisms and susceptibility to tuberculosis in Thais. Respirology 12 202 206
92. LeungKH
YipSP
WongWS
YiuLS
ChanKK
LaiWM
ChowEY
LinCK
YamWC
ChanKS
2007 Sex - and age-dependent association of SLC11A1 polymorphisms with tuberculosis in Chinese: a case control study. BMC Infect Dis 7 19
93. RyuS
ParkY-K
BaiG-H
KimS-J
ParkS-N
KangS
2000 3′UTR polymorphisms in the NRAMP1 gene are associated with susceptibility to tuberculosis in Koreans. Int J Tuberc Lung Dis 4 577 580
94. KusuharaK
YamamotoK
OkadaK
MizunoY
HaraT
2007 Association of IL12RB1 polymorphisms with susceptibility to and severity of tuberculosis in Japanese: a gene-based association analysis of 21 candidate genes. Int J Immunogenet 34 35 44
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 1
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Dual-Use Research and Technological Diffusion: Reconsidering the Bioterrorism Threat Spectrum
- Pathogenesis of the 1918 Pandemic Influenza Virus
- Critical Role of IRF-5 in the Development of T helper 1 responses to infection
- A Cardinal Role for Cathepsin D in Co-Ordinating the Host-Mediated Apoptosis of Macrophages and Killing of Pneumococci