
Targeting of Heparin-Binding Hemagglutinin to Mitochondria in Macrophages
Mycobacterium tuberculosis heparin-binding hemagglutinin (HBHA), a virulence factor involved in extrapulmonary dissemination and a strong diagnostic antigen against tuberculosis, is both surface-associated and secreted. The role of HBHA in macrophages during M. tuberculosis infection, however, is less well known. Here, we show that recombinant HBHA produced by Mycobacterium smegmatis effectively induces apoptosis in murine macrophages. DNA fragmentation, nuclear condensation, caspase activation, and poly (ADP-ribose) polymerase cleavage were observed in apoptotic macrophages treated with HBHA. Enhanced reactive oxygen species (ROS) production and Bax activation were essential for HBHA-induced apoptosis, as evidenced by a restoration of the viability of macrophages pretreated with N-acetylcysteine, a potent ROS scavenger, or transfected with Bax siRNA. HBHA is targeted to the mitochondrial compartment of HBHA-treated and M. tuberculosis-infected macrophages. Dissipation of the mitochondrial transmembrane potential (ΔΨm) and depletion of cytochrome c also occurred in both macrophages and isolated mitochondria treated with HBHA. Disruption of HBHA gene led to the restoration of ΔΨm impairment in infected macrophages, resulting in reduced apoptosis. Taken together, our data suggest that HBHA may act as a strong pathogenic factor to cause apoptosis of professional phagocytes infected with M. tuberculosis.
Published in the journal:
. PLoS Pathog 7(12): e32767. doi:10.1371/journal.ppat.1002435
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002435
Summary
Mycobacterium tuberculosis heparin-binding hemagglutinin (HBHA), a virulence factor involved in extrapulmonary dissemination and a strong diagnostic antigen against tuberculosis, is both surface-associated and secreted. The role of HBHA in macrophages during M. tuberculosis infection, however, is less well known. Here, we show that recombinant HBHA produced by Mycobacterium smegmatis effectively induces apoptosis in murine macrophages. DNA fragmentation, nuclear condensation, caspase activation, and poly (ADP-ribose) polymerase cleavage were observed in apoptotic macrophages treated with HBHA. Enhanced reactive oxygen species (ROS) production and Bax activation were essential for HBHA-induced apoptosis, as evidenced by a restoration of the viability of macrophages pretreated with N-acetylcysteine, a potent ROS scavenger, or transfected with Bax siRNA. HBHA is targeted to the mitochondrial compartment of HBHA-treated and M. tuberculosis-infected macrophages. Dissipation of the mitochondrial transmembrane potential (ΔΨm) and depletion of cytochrome c also occurred in both macrophages and isolated mitochondria treated with HBHA. Disruption of HBHA gene led to the restoration of ΔΨm impairment in infected macrophages, resulting in reduced apoptosis. Taken together, our data suggest that HBHA may act as a strong pathogenic factor to cause apoptosis of professional phagocytes infected with M. tuberculosis.
Introduction
Tuberculosis remains a serious global problem, although many researchers have made a persistent effort for several decades. Mycobacterium tuberculosis, a major causative agent of pulmonary tuberculosis, is responsible for 1.8 million deaths per year worldwide [1]. Innate immune system plays a critical role in antimicrobial host response during the early stage of M. tuberculosis infection. Alveolar macrophages mediate innate immunity by phagocytosing pathogens and are the chief defense against M. tuberculosis, which can survive and replicate within phagocytes [2]. The course of tuberculosis rests on the outcome of the interaction between the bacterium and host macrophage. Therefore, a better understanding of these complex interactions is critical to controlling mycobacterial infection.
Many bacterial and viral pathogens utilize various strategies to manipulate host machinery to serve their own needs. Apoptotic cell death has been regarded as an innate cellular response to limit the multiplication of intracellular pathogens [3], although the precise mechanism of the direct antimicrobial action in infected macrophages undergoing apoptosis is unclear. Generally, infectious intracellular pathogens tend to prevent host cell apoptosis during an early stage of infection. However, they may also induce host cell apoptosis with a specific aim to subvert the host attack, such as immune and inflammatory response, at later stages [4], [5].
A number of reports have indicated that M. tuberculosis does indeed inhibit host cell apoptosis, while at the same time it induces pro-apoptotic signals. Recent studies showed that only virulent mycobacterial species can inhibit apoptosis induction in primary human alveolar macrophages [6], THP-1 [7], [8], and J774 macrophage cell lines [9]. Virulent M. tuberculosis reportedly induced the apoptotic death of host cells. For example, enhanced apoptotic response was detected in alveolar macrophages recovered from patients with pulmonary tuberculosis [10], [11]. Extensive apoptosis was also observed in caseating granulomas from lung tissue samples obtained from patients with tuberculosis [12], [13]. Several apoptosis-inducing factors of M. tuberculosis, such as 19-kDa glycolipoprotein (Rv3763) [14], PE_PGRS33 (Rv1818c) [15], ESAT6 (Rv3875) [16], and 38-kDa lipoprotein (Rv0934) [17] are reported.
Heparin-binding hemagglutinin adhesin (HBHA) is a 28-kDa multifunctional protein found on the surface and culture filtrates of mycobacteria. It has hemagglutination activity and binds to sulfated glycoconjugates such as heparin and dextran sulfate [18]. HBHA interacts specifically with non-phagocytic cells and is essential for the infection of lung epithelial cells and extrapulmonary dissemination of M. tuberculosis [18], [19]. Protective immunity induced by HBHA is observed in M. tuberculosis-infected mouse models, indicating that HBHA is a protective antigen [20]. Recent studies suggest that HBHA is a useful diagnostic marker for tuberculosis [21]. We also identified and characterized HBHA as a serologically active mycobacterial antigen in a previous study, whereby HBHA binds strongly to the immunoglobulin M of patients with tuberculosis [22]. Although HBHA function in mycobacterial pathogenesis has been extensively studied, the role of HBHA on professional phagocytes, such as macrophages, is still poorly understood.
The aim of the present study was to characterize the biological effects of M. tuberculosis HBHA on macrophages. We found that HBHA induced apoptosis in murine macrophages and investigated its underlying mechanism. Here, we show that HBHA treatment caused a loss of mitochondrial transmembrane potential (ΔΨm) and the release of cytochrome c from purified mitochondria in vitro, as well as mitochondria of intact cells, and HBHA was efficiently targeted to mitochondria of macrophages.
Results
HBHA induces macrophage apoptosis via caspase activation
We first sought to determine whether HBHA could induce macrophage apoptosis. Apoptosis was assessed by quantifying DNA fragmentation, which is considered a hallmark of apoptosis, in the cytoplasmic fractions of dying cells using a commercially available ELISA kit. The incubation of RAW 264.7 cells with HBHA resulted in a significant increase in the release of oligonucleosomal fragments into the cytoplasm in both dose - and time-dependent manners as compared to compared to control cells (Figure 1A and 1B). Cell death was significantly greater in cells treated with HBHA as compared to buffer-treated control cells. As lactate dehydrogenase was not detected in the cell culture supernatant during HBHA treatment, the possibility that HBHA-induced death is necrosis was excluded (Figure S1). We used native antigen 85 complex (Ag85) as an unrelated control protein. The Ag85 of M. tuberculosis is the major secreted protein and fibronectin-binding protein, and shows strong immunoreactivity [23], [24]. Similar results were observed in bone marrow-derived macrophages (BMDMs); like PBS-treated BMDMs DNA fragmentation was not detected in Ag85-treated cells, whereas dramatic DNA fragmentation was observed in HBHA-treated cells (Figure 1C). HBHA-induced apoptosis was further confirmed by examining the nuclear morphology of dying cells using a fluorescent DNA-binding agent, 4′-6-diamidino-2-phenylindole (DAPI). As shown in Figure 1D, control cells treated with buffer had intact nuclei. In contrast, within 48 h of HBHA treatment, RAW 264.7 cells clearly exhibited condensed or fragmented nuclei indicative of apoptotic cell death. We further analyzed the caspase dependency of HBHA-induced apoptosis. Western blot analysis showed that the cleavage of caspase-3, caspase-9, and poly(ADP-ribose) polymerase (PARP) was evident in cells incubated with HBHA for 48 h (Figure 1E). Inhibition of caspases by a pan-caspase inhibitor, zVAD-fmk, attenuated the HBHA-induced DNA fragmentation, indicating that HBHA induces caspase-dependent apoptosis (Figure 1F). These results suggest that macrophages treated with HBHA undergo caspase-dependent apoptosis.
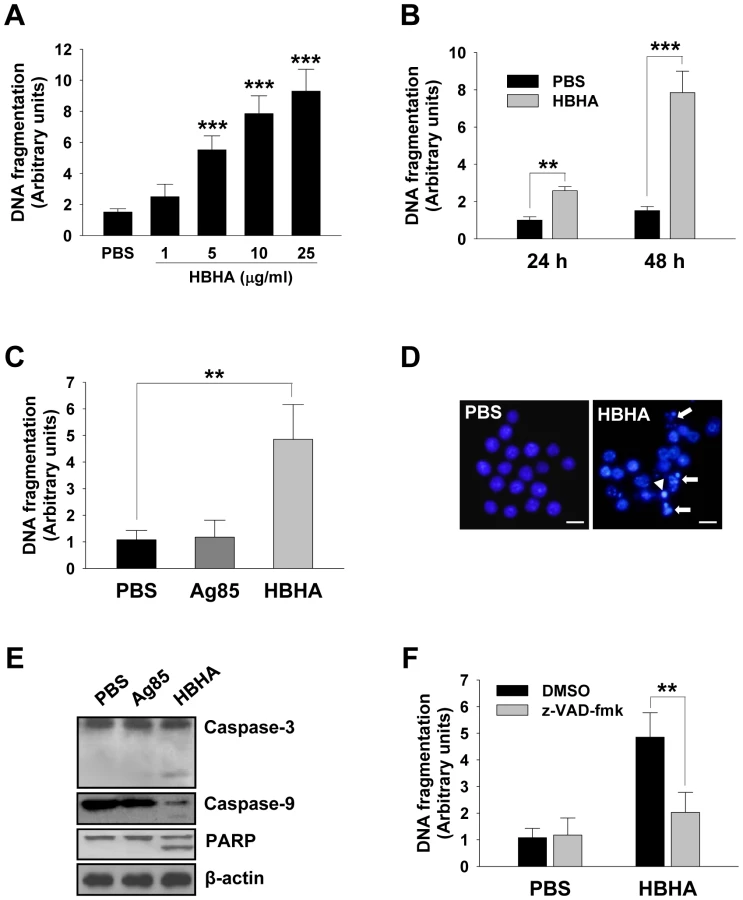
HBHA causes a decrease in ΔΨm
The mitochondrion acts as a central executioner in response to apoptotic stimuli, allowing signals from various inputs to converge [25]. We investigated whether HBHA treatment affected the structural and biochemical integrity of mitochondria. Mitochondrial damage was assessed by examining mitochondrial ΔΨm, which was determined by staining cells with 3,3′-Dihexyloxacarbocyanine (DiOC6), a dye that incorporates into mitochondria with intact membrane potential [26], for flow cytometric analysis. As shown in Figure 2A, a significant loss of ΔΨm was observed in RAW 264.7 cells incubated with HBHA as indicated by a decrease in DiOC6 intensity. Analysis of the time course for examination of ΔΨm onset showed a noticeable dissipation of ΔΨm after 18 h of HBHA treatment, which further decreased with time. A similar result was obtained in BMDMs incubated with HBHA (Figure 2B). These results suggest that mitochondrial damage appears as a subsequent event in the intracellular action of HBHA.

HBHA induces Bax translocation to mitochondria and releases cytochrome c from mitochondria to the cytosol
Apoptosis at the mitochondrial level involves the oligomerization of the pro-apoptotic protein Bax [27], leading to permeabilization of the outer mitochondrial membrane (MOMP) and release of cytochrome c [28]. We performed immunocytochemistry to detect Bax translocation and cytochrome c release. An antibody recognizing the Bax N-terminus, which is exposed by the activation of Bax and its insertion into the mitochondrial membrane, was used. Figure 3A shows the translocation of Bax distributed evenly in the cytoplasm to the mitochondria in macrophages as evident by the colocalization of Bax with Mitotracker, a potential-sensitive dye specific for mitochondria. In PBS-treated cells, cytochrome c showed a punctate pattern that colocalizes with Mitotracker, whereas the faint signal for cytochrome c and the decreased colocalization with Mitotracker were detected in HBHA-treated cells, indicating cytochrome c release. These results were confirmed by performing subcellular fractionation and Western blot analysis (Figure 3B). HBHA caused a decrease in cytochrome c immunoreactivity in the mitochondrial fraction with a concomitant increase in the cytosolic fraction and vice versa for Bax immunoreactivity. Collectively, these findings suggest that the apoptotic effect of HBHA on macrophages is associated with cytochrome c release and Bax translocation. To determine whether Bax activation is necessary for HBHA-induced apoptosis, we knocked down the level of Bax by transfecting RAW 264.7 cells with Bax siRNA. The Bax protein level was significantly reduced in cells transfected with Bax siRNA; Bax protein in control siRNA-transfected cells was unchanged (Figure 3C). We then determined the effect of knockdown Bax on HBHA-induced apoptosis in RAW 264.7 cells. As shown in Figure 3D and 3E, HBHA-induced increase in DNA fragmentation was blocked and ΔΨm loss was restored by Bax knockdown, suggesting that Bax activation is required for HBHA-induced macrophage apoptosis.

Reactive oxygen species (ROS) are required for apoptosis induced by HBHA
Enhanced reactive oxygen species (ROS) production, characteristic of early apoptotic events, can be both a cause and a consequence of changes in ΔΨm [26], [29]. To examine the involvement of ROS generation on HBHA effects in macrophages, the oxidation of DCF was monitored by flow cytometry and fluorescent microscopy (Figure 4A). Compared to PBS or Ag85, HBHA significantly induced the increase of intracellular hydroperoxide in macrophages. To determine the requirement of ROS increase in HBHA-induced apoptosis, the effect of HBHA alone or in combination with N-acetylcysteine (NAC), a general ROS scavenger, on DNA fragmentation was assessed. NAC pretreatment effectively inhibited HBHA-induced DNA fragmentation (Figure 4B) as well as ROS production, suggesting that ROS increase is essential for the apoptotic response caused by HBHA.

HBHA is targeted to the mitochondria
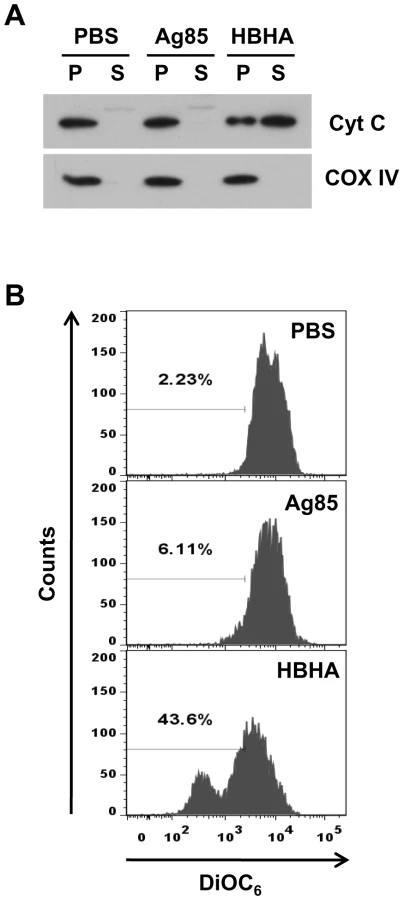
Studies have suggested that some infectious intracellular pathogens regulate apoptosis of their host cells by targeting proteins to mitochondrial membranes that either induce or inhibit MMP [30]. We addressed the question of where HBHA is localized in mitochondria of HBHA-treated cells. Therefore, the possibility that HBHA interacts with the mitochondrial compartment was examined. Confocal microscopic analysis revealed the presence of HBHA in the mitochondria of HBHA-treated cells, as evidenced by a significant overlap between HBHA and Mitotracker (Figure 5A). Subcellular fractionation and Western blot analysis consistently showed that large amounts of HBHA were detected in the mitochondrial fraction, but not in the cytosolic fraction, where little HBHA was observed (Figure 5B). In contrast, the minimum of Ag85 were detected in cytoplasmic fraction of macrophage treated with Ag85, suggesting that it is not able to pass through plasma membrane. Furthermore, to determine whether HBHA was imported into mitochondria, we isolated mitochondria from cells treated with HBHA. The purified mitochondria were subsequently digested with proteinase K. As shown in Figure 5C, HBHA disappeared in mitochondria digested with proteinase K, indicating that HBHA adheres to the outer membrane of the mitochondria.

HBHA induces the release of cytochrome c and the loss of ΔΨm in isolated mitochondria
We next determined whether HBHA induced cytochrome c release from isolated mitochondria. As shown in Figure 6A, isolated mitochondria from RAW 264.7 cells released cytochrome c after HBHA treatment, whereas the buffer control or Ag85 did not stimulate this release in a cell-free assay. We also examined the effect of HBHA on the collapse of membrane potential in purified mitochondria. For this, mitochondria incubated with HBHA were stained with DiOC6, and the fluorescence intensity was monitored by flow cytometry (Figure 6B). A significant shift to a lower intensity was observed in mitochondria treated with HBHA as compared to buffer control or Ag85, indicating the decrease in ΔΨm. These data provide evidence that similar to the event that occurs in macrophages, HBHA can solely induce mitochondrial damage in a cell-free system, indicating that Bax translocation to mitochondria is not essential for of ΔΨm loss and cytochrome c release.
HBHA partially localizes to mitochondria during M. tuberculosis infection and impacts viability of infected macrophages
HBHA is a secreted protein in M. tuberculosis as well as a surface-associated protein [18]. To examine whether HBHA is also transported to mitochondria during M. tuberculosis infection, BMDMs were infected with H37Rv wild type and mutant disrupted in hbhA. Immunofluorescence microscopy of infected cells revealed that a part of HBHA colocalized with mitochondria (Figure 7A). Purified mitochondrial fraction of these cells contained a considerable amount of HBHA protein, although a large portion of HBHA were observed in cytosolic fraction (Figure 7B). These findings demonstrate that HBHA is efficiently transported to mitochondria of infected macrophages. To analyze the effects of HBHA on macrophages in the context of the bacterium as a whole, we compared the relative ability of M. tuberculosis H37Rv wild type and mutant disrupted in hbhA to induce apoptosis and ΔΨm collapse in macrophages. A reduced DNA fragmentation and an increased intact mitochondria were observed in BMDMs infected with mutant strain compared to its parent (Figure 7C and 7D), which was noticeable when macrophages were infected at MOIs of 5 and 10 but not at an MOI of 25 (Figure S2A). On the other hand, there was no significant difference in LDH release between cells infected with two strains (Figure S2B). Similarly, more significant DNA fragmentation and ΔΨm loss were detected in cells infected with M. smegmatis ectopically expressing HBHA compared to cells infected with the M. smegmatis control.

HBHA has no influence on the viability of A549 cells
HBHA is involved in the interaction of mycobacteria with alveolar epithelial cells [19]. To determine whether these cells exposed to HBHA undergo apoptosis, human type II A549 pneumocytes were treated with purified HBHA for 48 h. As shown in Figure 8A and 8B, neither DNA fragmentation nor ΔΨm collapse was observed in HBHA-treated A549 cells. Immunofluorescent microscopy showed that a very faint green signal was detected in A549 cells incubated with HBHA, indicating that HBHA enters A549 cells much less efficiently (Figure 8C, upper panels). To confirm this issue, A549 cells were infected with M. tuberculosis wild type and hbhA deficient strains. Like experiments conducted in macrophages, M. tuberculosis infection led to severe ΔΨm dissipation, accompanied by the partial presence of HBHA in mitochondrial compartments (Figure 8B and 8C, lower panels). In contrast, a decrease in the percentage of cells displaying loss of ΔΨm was observed in A549 cells infected with the hbhA deficient strain (Figure 8B). These data suggest that cell entry and targeting to mitochondria of HBHA are essential for ΔΨm loss and apoptotic response.

Discussion
Programmed cell death is emerging as a major effect of bacterial pathogenesis. Numerous studies have shown that M. tuberculosis infection can increase the rate of macrophage apoptosis [31], [32]. Pro-apoptotic activities of a growing number of mycobacterial components have recently been described [14]–[17]. Nevertheless, data regarding the identities of the mycobacterial molecules involved and the underlying apoptotic mechanism are still scarce. We showed here that intracellular HBHA is targeted to mitochondria in murine macrophages, which leads to ΔΨm dissipation and eventual apoptosis. Although the possibility that HBHA may interact with cytosolic molecules or other cell compartments cannot be ruled out completely, these connections clearly appear to be insignificant. To our knowledge, the present study is the first description of a mycobacteria-encoded protein stimulating apoptotic cell death via a mitochondria-dependent pathway in macrophages.
M. tuberculosis HBHA is a protein that is both surface-associated and secreted. HBHA is involved in the binding of M. tuberculosis to type II pneumocytes, but not to professional phagocytes such as macrophages, and is required for the dissemination of tubercle bacilli from the lungs to other tissues [19]. In this respect, its impact on macrophages has received relatively little attention. However, HBHA was recently demonstrated to have the capacity to bind to complement component C3, and recombinant HBHA was found to mediate the attachment of latex beads to murine macrophage-like cells in both C3-dependent and -independent manners [33]. M. tuberculosis can bind to the complement receptors and is subsequently introduced into the phagocytic cell [34]. These results raise the possibility of the interaction between HBHA and macrophages during mycobacterial infection.
Mitochondria are central organelles in which a variety of key events in apoptosis occur, including the release of cytochrome c, changes in electron transport, ΔΨm collapse, altered cellular oxidation–reduction, and participation of pro - and anti-apoptotic Bcl-2 family proteins [26]. Presently, mitochondria are regarded as the targets for the manipulation of many bacterial and viral pathogens determining the fate of infected host cells [35]. Moreover, mitochondrial damage has been suggested to play a critical role in the outcome of macrophage infection with M. tuberculosis [36]. These findings offer the potential of mycobacterial components for the regulation of programmed cell death at the mitochondrial level.
MMP is regulated by endogenous molecules, including Bcl-2 family members such as Bax [37]. The Bax present in the cytosol under normal conditions fosters the loss of ΔΨm and releases cytochrome c and apoptosis-inducing factor (AIF) from mitochondria after its introduction into the mitochondrial compartment [26]. Indeed, mitochondrial translocation of Bax was observed in macrophages treated with HBHA, and the interaction of HBHA with mitochondria resulted in cytochrome c release in murine macrophages. However, Bax translocation may not be essential for mitochondrial dysfunction by HBHA, as evidenced by a mitochondrial cell-free assay in which HBHA caused ΔΨm loss and cytochrome c release in vitro. Not surprisingly, we observed the activation of caspases 3 and 9 and subsequent cleavage of PARP after incubation of macrophages with HBHA. In contrast, we found no evidence of cytosolic or nuclear translocation of AIF induced by HBHA (data not shown), indicating that it is not involved in HBHA-induced cell death. ROS generation with ΔΨm modulation and caspase-9 activation is known to be a major component of the mitochondrial pathway of apoptosis [38]. ROS are predominantly produced in the mitochondria and lead to the modulation of ΔΨm, which finally results in apoptosis [39]. Our results indicate that HBHA induces macrophage apoptosis through ROS generation and ΔΨm collapse, suggesting that these play an essential role in HBHA-induced apoptosis.
Our results indicate that cellular entry is essential for mitochondria-mediated apoptotic effect of HBHA, although the mechanism by which HBHA internalized by host cells remains unresolved. In A549 cells infected with M. tuberculosis but not cells incubated with purified HBHA, the severe ΔΨm collapse and the presence of intracellular HBHA in mitochondrial compartment were observed. There was a significant increase in the percentage of cells with intact ΔΨm, when A549 cells were infected with the mutant strain lacking HBHA gene. We cannot rule out that these results might come from decreased number of mycobacteria in cells, because invasion of A549 cells, but not macrophages, by HBHA-deficient strain compared with parental strain was reduced [19]. Moreover, HBHA induced ΔΨm loss and cytochrome c release in purified mitochondria from not only RAW 264.7 cells but also mouse liver (data not shown). Thus, no impact on viability of epithelial cells treated with HBHA might be due to the absence of intracellular this protein.
What host molecules physically and functionally interact with intracellular HBHA and how do they then induce mitochondrial dysfunction? In the present study, proteinase K digestion in vitro showed that intracellularly inserted HBHA is attached to the mitochondrial surface but is not imported into mitochondria, indicating that HBHA probably interacts with integral outer membrane molecules. Several mitochondria-targeted proteins encoded by pathogens interact with voltage-dependent anion channel (VDAC). The porin B from N. meningitidis is a VDAC-targeted protein [40]. Hepatitis B virus X protein also co-localizes to mitochondria where it interacts with a particular VDAC isoform, HVDAC3 [41]. Anti-apoptotic members of the Bcl-2 family, such as Bcl-2 and Bcl-xL, are located in mitochondrial membranes where they inhibit cytochrome c release from mitochondria and thereby prevent downstream caspase activation. Pro-apoptotic members of the Bcl-2 family, such as Bax, can translocate into mitochondria and induce MMP [42]. These Bcl-2-like proteins can be prominent targets of bacterial proteins [30], [43]. Recombinant HBHA used in the present study was a His-tagged fusion protein. To determine the interaction between HBHA and VDAC or the Bcl-2 family proteins, HBHA and interacting molecules were purified by Ni-NTA affinity chromatography, followed by immunoblotting against them. However, HBHA showed no direct interaction with VDAC or Bcl-2 family members (data not shown). In addition, the possibility of HBHA nonspecific binding to mitochondria cannot be excluded. The C-terminal region of HBHA contains several cationic lysine-rich repeats where methylation can occur [44]. This region may work like natural antibiotic peptides which form cationic residues on one end and interact with anionic molecules such as phospholipids to disrupt negatively charged membranes and result in apoptosis [45].
Virulent M. tuberculosis induces necrosis of the infected macrophages by inhibiting the repair process of plasma membrane; this leads to cellular lysis and reinforces the spreading to the adjacent infection sites [46]–[48]. Recent reports suggested that high intracellular burden of virulent M. tuberculosis induces host cell death via a new caspase-independent apoptotic pathway involved in the bacterial escape and extracellular replication [49], [50]. Because gain of function mutation in HBHA enhanced the apoptogenic potency of M. smegmatis (Figure 7C), it is plausible that HBHA may be the factor that allows M. tuberculosis to escape from the infected macrophages at high intracellular burden. However, similar levels of apoptosis were observed between macrophages infected with M. tuberculosis H37Rv wild type and mutant disrupted in hbhA at an MOI of 25 but not low MOIs (Figure S2A). Further, HBHA deficiency had no influence on the macrophage necrosis caused by M. tuberculosis at both low and high MOIs (Figure S2B), indicating no involvement of HBHA in bacterial escape from the macrophages at an early stage of infection.
Studies on the comparison of virulent and attenuated mycobacterial strains have demonstrated that the latter has much stronger apoptotic activity in macrophages. This concept is supported by the identification of genes that inhibit apoptosis of host cells [51]–[53]. In this sense, our claims that HBHA targets to the mitochondria of host cells in the induction of apoptosis may be confusing. However, there is cumulative evidence suggesting that virulent M. tuberculosis induces host cell apoptosis. Furthermore, the transcriptional profiling of cells infected with virulent M. tuberculosis showed increases in the expression of both pro - and anti-apoptotic genes [54], [55]. Collectively, it is highly likely that M. tuberculosis infection results in pro - and anti-apoptotic response of host cells. The final outcome may depend on the nature and activation status of the host cell. Although the pro-apoptotic response is inarguably beneficial to the host, it may provide a favorable circumstance for the induction of necrotic cell death and subsequent bacterial escape to the adjacent cells, which may provide a clue for HBHA function during M. tuberculosis infection [46], [49], [50].
Taken together, the present study suggests the possibility that the M. tuberculosis HBHA may be an apoptosis-inducing factor of mycobacteria, although the molecular mechanism by which HBHA causes loss of ΔΨm remains unknown. Future work should focus on the exploration of host targets of HBHA and the mechanism by which HBHA modulates ΔΨm and cytochrome c release in detail, as well as identification of the HBHA domain essential for its activity in mitochondrial dysfunction.
Materials and Methods
Ethics statement
All animal procedures were approved by the Institutional Animal Care and Use Committees of Chungnam National University (Permit Number: 2010-3-9). All animal experiments were performed in accordance with Korean Food and Drug Administration (KFDA) guidelines.
Reagents and antibodies
Antibodies against caspase-3, caspase-9, and VDAC were purchased from Cell Signaling Technology Inc (Beverly, MA). The anti-PARP and anti-β-actin, anti-Bax, and anti-Tom40 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against cytochrome c (for immunofluorescence, clone 6H2.B4; for Western blot analysis, clone 7H8.2C12) were acquired from BD Pharmingen (San Diego, CA), and the anti-cytochrome oxidase subunit IV (COX IV) antibody was purchased from Abcam (Cambridge, UK). Dichlorodihydrofluorescein diacetate (H2DCFDA), DAPI, and DiOC6 were obtained from Molecular Probes (Eugene, OR) and zVAD-fmk and NAC were purchased from Calbiochem (San Diego, CA).
Recombinant HBHA protein, Native Ag85 protein, anti-HBHA, and mycobacterial strains
Mycobacterium smegmatis strains, recombinant HBHA protein from M. smegmatis, and antiserum to HBHA were produced and prepared as described previously [22]. Ag85 was purified from the culture filtrate protein of M. tuberculosis H37Rv (ATCC 27294), as previously described by Lim et al [24]. Parental and mutant (hbhA deletion) Mycobacterium tuberculosis 103 were kindly provided by Dr. Camille Locht (Institut Pasteur de Lille, Lille, France) [19]. HBHA proteins were used in experiments after lipopolysaccharide (LPS) inactivation with polymyxin B (Invivogen, San Diego, CA), a known pharmacological antagonist of LPS.
Cell culture
RAW 264.7 murine macrophage cell line and A549 human alveolar epithelial cell line were cultured in Dulbecco's modified Eagle's medium (DMEM; Lonza, Walkersville, MD) supplemented with 10% fetal bovine serum (FBS; Hyclone, Logan, UT), 1% HEPES, and 1% l-glutamine at 37°C with 5% CO2. BMDMs were obtained from 6–8-week-old female C57BL/6 mice. Briefly, bone marrow cells from the femur and tibia were cultured in DMEM that contained 2 mM l-glutamine, 100 U/mL penicillin, 100 µg/mL streptomycin, 10% FBS, and 25 ng/mL recombinant mouse M-CSF (R&D system, Minneapolis, MN) at 37°C with 5% CO2. After 4 days, non-adherent cells were removed and differentiated macrophages were incubated in antibiotic-free DMEM until use.
Bax siRNA transfection
One day before transfection, RAW 264.7 cells were plated and grown at 37°C to 70% confluency in complete medium without antibiotics in 6 well plates. One micrograms of a Bax siRNA (Bioneer, Deajeon, Korea, sense: CCGGCGAAUUGGAGAUGAA; anti-sense: UUCAUCUCCAAUUGGCCGG) or a noncomplementary siRNA were transiently transfected into RAW 264.7 using Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instructions.
DNA fragmentation assay (Apoptosis ELISA)
Cells were seeded in 96-well flat-bottom culture plates. After incubation with recombinant HBHA proteins, cells were collected, washed with PBS, and processed for quantification of cytoplasmic histone-associated DNA fragments formed during apoptosis using an enzyme-linked immunosorbent assay (Cell Death Detection ELISA PLUS; Roche Diagnostic) according to the manufacturer's instructions.
Lactate dehydrogenase (LDH) assay
The release of LDH from RAW 264.7 cells incubated with recombinant HBHA or from BMDMs infected with M. tuberculosis was measured using a Cytotoxicity Detection Kit plus (Roche, Indianapolis, IN) according to the manufacturer's protocol. Relative cytotoxicity was calculated using the following equation: Cytotoxicity (%) = % of LDH released from the infected cells/maximum LDH released.
Assessment of ΔΨm
ΔΨm was assessed by measuring retention of the lipophilic cationic dye DiOC6 in mitochondria. Cells were harvested and incubated in a DiOC6 solution (10 nM in fresh medium) for 20 min at 37°C in the dark. The cells were then washed and resuspended in PBS. Immediately after PBS washing, ΔΨm was measured by sorting the cells using FACSCanto (BD Biosciences). Dead cells were excluded by forward and side-scatter gating. Data were acquired by analyzing an average population of 10 000 cells using CELLQuest software (BD Biosciences).
Immunofluorescence microscopy
Cells were seeded onto glass coverslips in 12-well plates. Nuclear changes were analyzed by DAPI staining. After cells were incubated with HBHA for the indicated times, they were fixed with 4% paraformaldehyde and incubated with DAPI (10 µg/mL) for 10 min in the dark. The nuclei of stained cells were visualized using an Olympus BX50 fluorescence microscope (Olympus Optical Co., Hamburg, Germany). To determine the localization of cytochrome c or Bax, cells treated with HBHA were incubated in pre-warmed medium containing 100 nM of Mitotracker Red (Molecular Probes), fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and then stained with anti-cytochrome c or anti-Bax and Alexa-488-conjugated secondary antibody (Jackson Immuno Research Laboratories) before confocal microscopy. The subcellular localization of HBHA was analyzed using a confocal microscope (LSM510 META; Carl Zeiss). The cells incubated with Mitotracker Red were fixed, permeabilized, and stained with an anti-HBHA antibody followed by a fluorophore-conjugated antibody (anti-mouse IgG Alexa-488). After DAPI staining, cells were imaged with a confocal microscope.
Mitochondrial and cytosolic fractionation
Subcellular fractionation was performed as previously described [56]. Briefly, cells were incubated on ice for 5 min in 100 µL of ice cold CLAMI buffer (200 mM sucrose, 70 mM KCl, 200 µg/mL digitonin in PBS) and centrifuged at 1,000 × g for 5 min at 4°C. The supernatants (cytosolic fractions) were stored at −80°C and the pellets were resuspended in 50 µL of IP buffer (50 mM Tris-Cl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 0.2% Triton X-100, 0.3% NP-40) containing protease inhibitor cocktail (Roche Diagnostics Corporation, Indianapolis, IN) and incubated on ice for 10 min. The samples were centrifuged at 10 000 × g for 5 min at 4°C and the supernatants (mitochondrial fractions) were stored at −80°C until use in further experiments.
Immunoblot analysis
Cells were detached, centrifuged, and lysed in lysis buffer (10 mM Tris, pH 7.4, 5 mM EDTA, 150 mM NaCl, 1% Triton X-100, 1 mM PMSF, protease inhibitor cocktail). Protein concentrations were determined with the Bradford assay and 30 µg of protein was separated with SDS-PAGE, followed by electrotransfer to a nitrocellulose membrane (Hybond-ECL; Amersham Pharmacia Biotech). The blots were probed with primary antibodies at optimized concentrations followed by horseradish peroxidase-conjugated secondary antibodies. The enhanced chemiluminescence system (ECL; Amersham/GE Healthcare) followed by exposure to chemiluminescence film was used to visualize proteins.
Measurement of ROS
Intracellular ROS were evaluated through staining cells with H2DCFDA. Cells were incubated in 10 µM H2DCFDA for 30 min at 37°C, washed, and detached. Resuspended cells were washed and immediately analyzed by flow cytometry using FACSCanto. At least 10,000 cells per sample were analyzed using CellQuest Pro acquisition and analysis software.
Mitochondrial cell-free assay
Mitochondria were isolated from 1 × 108 RAW 264.7 cells as described previously [57]. Briefly, cells were harvested by centrifugation at 600 × g and resuspended in ice-cold IB buffer (10 mM Tris-MOPS, 200 mM sucrose, 1 mM EGTA/Tris, pH 7.4). All subsequent centrifugations were performed at 4°C. The cells were then homogenized with 35 strokes in a glass potter after incubation for 10 min on ice. Cell debris was removed by centrifugation at 600 × g for 10 min, and then the supernatant was centrifuged for 10 min at 7 000 × g to precipitate mitochondria. The pellet was then resuspended in EB buffer (10 mM Tris-MOPS, 125 mM KCl, 100 µM EGTA/Tris, 1 mM KH2PO4, pH 7.4). An aliquot of the preparation was incubated with HBHA for 1 h at 37°C and centrifuged for 10 min at 7 000 × g. The pellet containing mitochondria was resuspended in the same buffer and stained with DiOC6. An average population of 50 000 mitochondria was analyzed by flow cytometry. Alternatively, the proteins contained in the supernatant were concentrated with by ultrafiltration using a 3-kDa cutoff Centricon device (Amicon, Millipore, Bellerica, MA). Immunoblot analysis for cytochrome c was performed as described above.
Statistical analysis
The data represent the mean ± standard deviation (SD) from at least three independent experiments. Statistical analyses were performed using unpaired Student's t tests with Bonferroni adjustment. A P-value of <0.05 was considered significant.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. DyeCScheeleSDolinPPathaniaVRaviglioneMC 1999 Consensus statement. Global burden of tuberculosis: estimated incidence, prevalence, and mortality by country. WHO Global Surveillance and Monitoring Project. JAMA 282 677 686
2. Hingley-WilsonSMSambandamurthyVKJacobsWRJr 2003 Survival perspectives from the world's most successful pathogen, Mycobacterium tuberculosis. Nat Immunol 4 949 955
3. MolloyALaochumroonvorapongPKaplanG 1994 Apoptosis, but not necrosis, of infected monocytes is coupled with killing of intracellular bacillus Calmette-Guerin. J Exp Med 180 1499 1509
4. TschoppJThomeMHofmannKMeinlE 1998 The fight of viruses against apoptosis. Curr Opin Genet Dev 8 82 87
5. GaoLYKwaikYA 2000 The modulation of host cell apoptosis by intracellular bacterial pathogens. Trends Microbiol 8 306 313
6. KeaneJRemoldHGKornfeldH 2000 Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J Immunol 164 2016 2020
7. RiendeauCJKornfeldH 2003 THP-1 cell apoptosis in response to Mycobacterial infection. Infect Immun 71 254 259
8. DhimanRRajeMMajumdarS 2007 Differential expression of NF-kappaB in mycobacteria infected THP-1 affects apoptosis. Biochim Biophys Acta 1770 649 658
9. ZhangJJiangRTakayamaHTanakaY 2005 Survival of virulent Mycobacterium tuberculosis involves preventing apoptosis induced by Bcl-2 upregulation and release resulting from necrosis in J774 macrophages. Microbiol Immunol 49 845 852
10. PlacidoRMancinoGAmendolaAMarianiFVendettiS 1997 Apoptosis of human monocytes/macrophages in Mycobacterium tuberculosis infection. J Pathol 181 31 38
11. KlinglerKTchou-WongKMBrandliOAstonCKimR 1997 Effects of mycobacteria on regulation of apoptosis in mononuclear phagocytes. Infect Immun 65 5272 5278
12. KeaneJBalcewicz-SablinskaMKRemoldHGChuppGLMeekBB 1997 Infection by Mycobacterium tuberculosis promotes human alveolar macrophage apoptosis. Infect Immun 65 298 304
13. FayyaziAEichmeyerBSoruriASchweyerSHermsJ 2000 Apoptosis of macrophages and T cells in tuberculosis associated caseous necrosis. J Pathol 191 417 425
14. LopezMSlyLMLuuYYoungDCooperH 2003 The 19-kDa Mycobacterium tuberculosis protein induces macrophage apoptosis through Toll-like receptor-2. J Immunol 170 2409 2416
15. BasuSPathakSKBanerjeeAPathakSBhattacharyyaA 2007 Execution of macrophage apoptosis by PE_PGRS33 of Mycobacterium tuberculosis is mediated by Toll-like receptor 2-dependent release of tumor necrosis factor-alpha. J Biol Chem 282 1039 1050
16. DerrickSCMorrisSL 2007 The ESAT6 protein of Mycobacterium tuberculosis induces apoptosis of macrophages by activating caspase expression. Cell Microbiol 9 1547 1555
17. SanchezAEspinosaPEsparzaMAColonMBernalG 2009 Mycobacterium tuberculosis 38-kDa lipoprotein is apoptogenic for human monocyte-derived macrophages. Scand J Immunol 69 20 28
18. MenozziFDRouseJHAlaviMLaude-SharpMMullerJ 1996 Identification of a heparin-binding hemagglutinin present in mycobacteria. J Exp Med 184 993 1001
19. PetheKAlonsoSBietFDeloguGBrennanMJ 2001 The heparin-binding haemagglutinin of M. tuberculosis is required for extrapulmonary dissemination. Nature 412 190 194
20. ParraMPickettTDeloguGDheenadhayalanVDebrieAS 2004 The mycobacterial heparin-binding hemagglutinin is a protective antigen in the mouse aerosol challenge model of tuberculosis. Infect Immun 72 6799 6805
21. HougardyJMSchepersKPlaceSDrowartALechevinV 2007 Heparin-binding-hemagglutinin-induced IFN-gamma release as a diagnostic tool for latent tuberculosis. PLoS One 2 e926
22. ShinARLeeKSLeeJSKimSYSongCH 2006 Mycobacterium tuberculosis HBHA protein reacts strongly with the serum immunoglobulin M of tuberculosis patients. Clin Vaccine Immunol 13 869 875
23. WikerHGHarboeM 1992 The antigen 85 complex: a major secretion product of Mycobacterium tuberculosis. Microbiol Rev 56 648 661
24. LimJHParkJKJoEKSongCHMinD 1999 Purification and immunoreactivity of three components from the 30/32-kilodalton antigen 85 complex in Mycobacterium tuberculosis. Infect Immun 67 6187 6190
25. KroemerG 1999 Mitochondrial control of apoptosis: an overview. Biochem Soc Symp 66 1 15
26. GreenDRReedJC 1998 Mitochondria and apoptosis. Science 281 1309 1312
27. ElmoreS 2007 Apoptosis: a review of programmed cell death. Toxicol Pathol 35 495 516
28. CorySAdamsJM 2002 The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2 647 656
29. KroemerGZamzamiNSusinSA 1997 Mitochondrial control of apoptosis. Immunol Today 18 44 51
30. BoyaPRoquesBKroemerG 2001 New EMBO members' review: viral and bacterial proteins regulating apoptosis at the mitochondrial level. EMBO J 20 4325 4331
31. RojasMOlivierMGrosPBarreraLFGarciaLF 1999 TNF-alpha and IL-10 modulate the induction of apoptosis by virulent Mycobacterium tuberculosis in murine macrophages. J Immunol 162 6122 6131
32. SantucciMBAmicosanteMCicconiRMontesanoCCasariniM 2000 Mycobacterium tuberculosis-induced apoptosis in monocytes/macrophages: early membrane modifications and intracellular mycobacterial viability. J Infect Dis 181 1506 1509
33. Mueller-OrtizSLWangerARNorrisSJ 2001 Mycobacterial protein HbhA binds human complement component C3. Infect Immun 69 7501 7511
34. SchlesingerLSBellinger-KawaharaCGPayneNRHorwitzMA 1990 Phagocytosis of Mycobacterium tuberculosis is mediated by human monocyte complement receptors and complement component C3. J Immunol 144 2771 2780
35. MullerARudelT 2001 Modification of host cell apoptosis by viral and bacterial pathogens. Int J Med Microbiol 291 197 207
36. DuanLGanHGolanDERemoldHG 2002 Critical role of mitochondrial damage in determining outcome of macrophage infection with Mycobacterium tuberculosis. J Immunol 169 5181 5187
37. AdamsJMCoryS 1998 The Bcl-2 protein family: arbiters of cell survival. Science 281 1322 1326
38. GuptaSYelLKimDKimCChiplunkarS 2003 Arsenic trioxide induces apoptosis in peripheral blood T lymphocyte subsets by inducing oxidative stress: a role of Bcl-2. Mol Cancer Ther 2 711 719
39. FiersWBeyaertRDeclercqWVandenabeeleP 1999 More than one way to die: apoptosis, necrosis and reactive oxygen damage. Oncogene 18 7719 7730
40. MassariPHoYWetzlerLM 2000 Neisseria meningitidis porin PorB interacts with mitochondria and protects cells from apoptosis. Proc Natl Acad Sci U S A 97 9070 9075
41. RahmaniZHuhKWLasherRSiddiquiA 2000 Hepatitis B virus X protein colocalizes to mitochondria with a human voltage-dependent anion channel, HVDAC3, and alters its transmembrane potential. J Virol 74 2840 2846
42. YangJLiuXBhallaKKimCNIbradoAM 1997 Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 275 1129 1132
43. KroemerG 1997 The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat Med 3 614 620
44. PetheKBifaniPDrobecqHSergheraertCDebrieAS 2002 Mycobacterial heparin-binding hemagglutinin and laminin-binding protein share antigenic methyllysines that confer resistance to proteolysis. Proc Natl Acad Sci U S A 99 10759 10764
45. EllerbyHMArapWEllerbyLMKainRAndrusiakR 1999 Anti-cancer activity of targeted pro-apoptotic peptides. Nat Med 5 1032 1038
46. ChenMGanHRemoldHG 2006 A mechanism of virulence: virulent Mycobacterium tuberculosis strain H37Rv, but not attenuated H37Ra, causes significant mitochondrial inner membrane disruption in macrophages leading to necrosis. J Immunol 176 3707 3716
47. DivangahiMChenMGanHDesjardinsDHickmanTT 2009 Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat Immunol 10 899 906
48. ChenMDivangahiMGanHShinDSHongS 2008 Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death. J Exp Med 205 2791 2801
49. LeeJRemoldHGIeongMHKornfeldH 2006 Macrophage apoptosis in response to high intracellular burden of Mycobacterium tuberculosis is mediated by a novel caspase-independent pathway. J Immunol 176 4267 4274
50. O'SullivanMPO'LearySKellyDMKeaneJ 2007 A caspase-independent pathway mediates macrophage cell death in response to Mycobacterium tuberculosis infection. Infect Immun 75 1984 1993
51. VelmuruganKChenBMillerJLAzogueSGursesS 2007 Mycobacterium tuberculosis nuoG is a virulence gene that inhibits apoptosis of infected host cells. PLoS Pathog 3 e110
52. HincheyJLeeSJeonBYBasarabaRJVenkataswamyMM 2007 Enhanced priming of adaptive immunity by a proapoptotic mutant of Mycobacterium tuberculosis. J Clin Invest 117 2279 2288
53. JayakumarDJacobsWRJrNarayananS 2008 Protein kinase E of Mycobacterium tuberculosis has a role in the nitric oxide stress response and apoptosis in a human macrophage model of infection. Cell Microbiol 10 365 374
54. DanelishviliLMcGarveyJLiYJBermudezLE 2003 Mycobacterium tuberculosis infection causes different levels of apoptosis and necrosis in human macrophages and alveolar epithelial cells. Cell Microbiol 5 649 660
55. ChaussabelDSemnaniRTMcDowellMASacksDSherA 2003 Unique gene expression profiles of human macrophages and dendritic cells to phylogenetically distinct parasites. Blood 102 672 681
56. WaterhouseNJGoldsteinJCvon AhsenOSchulerMNewmeyerDD 2001 Cytochrome c maintains mitochondrial transmembrane potential and ATP generation after outer mitochondrial membrane permeabilization during the apoptotic process. J Cell Biol 153 319 328
57. FrezzaCCipolatSScorranoL 2007 Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc 2 287 295
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 12
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Controlling Viral Immuno-Inflammatory Lesions by Modulating Aryl Hydrocarbon Receptor Signaling
- Fungal Virulence and Development Is Regulated by Alternative Pre-mRNA 3′End Processing in
- Cryo Electron Tomography of Herpes Simplex Virus during Axonal Transport and Secondary Envelopment in Primary Neurons
- Epstein-Barr Virus Nuclear Antigen 3C Stabilizes Gemin3 to Block p53-mediated Apoptosis