
Prion Uptake in the Gut: Identification of the First Uptake and Replication Sites
After oral exposure, prions are thought to enter Peyer's patches via M cells and accumulate first upon follicular dendritic cells (FDCs) before spreading to the nervous system. How prions are actually initially acquired from the gut lumen is not known. Using high-resolution immunofluorescence and cryo-immunogold electron microscopy, we report the trafficking of the prion protein (PrP) toward Peyer's patches of wild-type and PrP-deficient mice. PrP was transiently detectable at 1 day post feeding (dpf) within large multivesicular LAMP1-positive endosomes of enterocytes in the follicle-associated epithelium (FAE) and at much lower levels within M cells. Subsequently, PrP was detected on vesicles in the late endosomal compartments of macrophages in the subepithelial dome. At 7–21 dpf, increased PrP labelling was observed on the plasma membranes of FDCs in germinal centres of Peyer's patches from wild-type mice only, identifying FDCs as the first sites of PrP conversion and replication. Detection of PrP on extracellular vesicles displaying FAE enterocyte-derived A33 protein implied transport towards FDCs in association with FAE-derived vesicles. By 21 dpf, PrP was observed on the plasma membranes of neurons within neighbouring myenteric plexi. Together, these data identify a novel potential M cell-independent mechanism for prion transport, mediated by FAE enterocytes, which acts to initiate conversion and replication upon FDCs and subsequent infection of enteric nerves.
Published in the journal:
. PLoS Pathog 7(12): e32767. doi:10.1371/journal.ppat.1002449
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002449
Summary
After oral exposure, prions are thought to enter Peyer's patches via M cells and accumulate first upon follicular dendritic cells (FDCs) before spreading to the nervous system. How prions are actually initially acquired from the gut lumen is not known. Using high-resolution immunofluorescence and cryo-immunogold electron microscopy, we report the trafficking of the prion protein (PrP) toward Peyer's patches of wild-type and PrP-deficient mice. PrP was transiently detectable at 1 day post feeding (dpf) within large multivesicular LAMP1-positive endosomes of enterocytes in the follicle-associated epithelium (FAE) and at much lower levels within M cells. Subsequently, PrP was detected on vesicles in the late endosomal compartments of macrophages in the subepithelial dome. At 7–21 dpf, increased PrP labelling was observed on the plasma membranes of FDCs in germinal centres of Peyer's patches from wild-type mice only, identifying FDCs as the first sites of PrP conversion and replication. Detection of PrP on extracellular vesicles displaying FAE enterocyte-derived A33 protein implied transport towards FDCs in association with FAE-derived vesicles. By 21 dpf, PrP was observed on the plasma membranes of neurons within neighbouring myenteric plexi. Together, these data identify a novel potential M cell-independent mechanism for prion transport, mediated by FAE enterocytes, which acts to initiate conversion and replication upon FDCs and subsequent infection of enteric nerves.
Introduction
Prions are infectious proteins composed of an abnormally folded isoform of the prion protein (PrPSc), the accumulation of which causes variant Creutzfeldt–Jakob disease, scrapie, and bovine spongiform encephalopathy, among other diseases. Prions propagate by converting endogenous, cellular prion protein (PrPC) into PrPSc containing a β-sheet core. Isolated PrPSc can be found in a wide range of aggregation states, from small oligomers to amyloid, and at least in larger aggregates the C-terminal portion of PrPSc acquires resistance to protease treatment [1]. PrPC is a ubiquitously expressed protein that is most abundant in the nervous system. The accumulation of PrPSc causes morphological changes in the central nervous system including astrocytosis, neuronal cell loss and spongiform pathology and, in some types of prion disease, amyloid plaque formation. Pathology builds up during a long incubation period that ends in a short clinical phase and death. Expression of PrPC in the host is required for successful infection, since it provides the substrate for the conversion to PrPSc [2]–[5].
Prions are highly resistant to denaturation by chemical and physical means, making disposal and disinfection difficult. This resistance may also contribute to their ability to survive passage through the digestive tract [6], allowing transmission of prion disease via prion-contaminated food. Many naturally occurring prion diseases are considered to be acquired orally, and are accompanied by accumulation of PrPSc in the lymphoreticular system long before invasion of the nervous system takes place [7]–[13]. Indeed, when specific components of the gut-associated lymphoid tissues (GALT) are absent, the transport of prions from the gut lumen to the nervous system is dramatically impaired [9], [12], [14]. The exact mechanisms by which infectious prions are transmitted from the gut lumen to the central nervous system remain elusive (for reviews see [15]–[17]).
The luminal surface of the intestine limits the access of pathogenic microorganisms to the underlying host tissues, and is protected by a single layer of epithelial cells bound by tight-junctions. Located within the villus epithelium and follicle-associated epithelium (FAE) of the Peyer's patch are microfold cells (M cells), a unique epithelial cell type specialized for the transepithelial transport of macromolecules and particles (for a review of M cells see [18]). M cells enable the host's immune system to sample the intestinal lumen and mount an appropriate immune response. However, some pathogenic microorganisms exploit M cells and use them to gain entry into mucosal tissues [18]. Using an in vitro system, M cell-like cells have been shown to actively transcytose the scrapie agent through to the basolateral side of the epithelium [19], [20], and studies in mice suggest prions might likewise be translocated across the FAE by M cells in vivo [21].
Together these data imply that M cells are plausible sites for the transepithelial transport of TSE agents across the intestinal epithelium. However, other data suggest such transport might also occur independently of M cell-mediated transcytosis via enterocytes [22], [23]. Studies in which isolated sheep gut loops were injected with scrapie brain homogenate [22] suggested that disease-specific PrP was transported across the absorptive epithelium of villi into lacteals. Other studies have shown that in response to inflammatory stimuli, mononuclear phagocytes within the lamina propria, including macrophages and classical dendritic cells (DC), can insert dendrites through the tight junctions between intestinal epithelial cells. These projections enable the cells to sample the luminal contents directly [24] implying another potential route of transepithelial TSE agent transport. Clearly, the way that orally introduced, partially proteinase-resistant prions survive the proteolytic conditions in the alimentary tract without losing all their infectivity and then cross the gut epithelium is still a matter of debate.
After passing the epithelial barrier, prions are thought to be captured by underlying migratory classical dendritic cells [25] that transmit the infectious agents by an as-of-yet unknown mechanism to the germinal centres. Within the germinal centres, prions accumulate upon follicular dendritic cells (FDCs), which are specialized stromal, mesenchymal cells of the immune system. FDCs are nonphagocytic, nonmigratory cells that trap native immune complexes on their surface through the expression of cellular complement receptors. The expression of high levels of PrPC in FDCs is regarded to be important for the accumulation and replication of prions upon their surfaces [26]–[30].
FDCs are considered to amplify the prions above the threshold level necessary to infect peripheral nerves [9], [12], [14], [31]. Prions are then thought to gain access to the central nervous system via physical interaction with peripheral nerve fibers of the enteric nervous system [32]. Retrograde axonal transport would deliver prions to their main pathological target, the brain. Due to the lack of satisfactory experimental models, some of the observations in prion transmission are based solely on in vitro studies.
Here, we examined the in vivo time-course of oral prion infection in the GALT of wild-type (wt) mice that are susceptible to prion infection and of PrP-deficient (Prnp–/–) mice that do not succumb to prion disease [33]. Both wt and Prnp–/– mice were orally infected with 3 different rodent-adapted scrapie strains (ME7, RML and Sc327), and intestinal and lymphoid tissues collected at specific days post-feeding (dpf). Samples were then analysed by immunofluorescence (IF) and cryo-immunogold electron microscopy (cryo-immuno EM) in order to identify, at the ultrastructural level, the cell types and subcellular organelles that are involved in prion trafficking and early pathogenesis. Our data show that prion uptake and transfer across the follicle-associated epithelium (FAE) of the gut occurred of the follicle-associated epithelium (FAE) independently of cellular PrPC expression. We show that PrP was first transiently detectable mainly within large LAMP1-positive endosomes of FAE enterocytes and at much lower levels within M cells. Proteins of FAE enterocytes were found on vesicles in the extracellular material adjacent to FAE enterocytes, and on the surface of FDCs, and these vesicles could act to transport prions towards FDCs. Furthermore, between 7–21 dpf, increased PrP labelling was only observed on the plasma membranes of FDCs from wild-type mice, identifying these membranes as the first site of PrP conversion and replication within Peyer's patches. Together, these data identify a novel potential M cell-independent route of prion uptake and transfer from the gut lumen mediated by FAE enterocytes that may have an important influence on susceptibility to oral prion infection.
Results
Enterocytes within the FAE contain unique large apical LAMP1-positive endosomes
In orally infected animals, prions are suspected to enter the lymphoid and nervous systems from the intestine through the FAE-overlying follicles of the mucosa-associated lymphoid tissue [15]–[17]. Thus, we examined the FAE and subepithelial dome (SED) region of Peyer's patches in wild-type (wt) uninfected mice and in wt mice infected with ME7 or RML prions. For comparison we also examined normal villi in prion-infected and uninfected mice. Late endocytic compartments were identified using antibodies against LAMP1, a membrane protein specific for these compartments (for an overview, see Figure 1A). By immunofluorescence (IF) analysis, we found significantly larger, LAMP1-positive endosomes with a typical multivesicular body phenotype in the FAE as compared to those in neighbouring villi (Figure 1B, 1D and Figure S1). Large LAMP1-positive endosomes were also observed in macrophages in the SED region, but had different more pleiomorphic morphology and stained less intensively with LAMP1-antibody (Figure 2A, Figure S1). Large LAMP1 endosomes of FAE enterocytes were abundant, had a more regular round shape (appeared as ring-like structures in IF and histochemistry sections) and had predominantly an apical location between the nucleus and brush border. Occasionally, when macrophages invade into the FAE their LAMP1 positive endosomes retain their pleiomorphic morphology and are mostly located basolaterally between the enterocyte nucleus and the basal membrane (see Figure S1C). At EM level investigating the cellular contours of individual cells by following their plasma membranes it could be confirmed that the apical large LAMP1 positive endosomes were located in FAE enterocytes and not for example in FAE-invading macrophages (Figure S1). Similar observations were made in both prion-infected and uninfected wt mice (data not shown). These data suggest that FAE enterocytes could play an important role in transcytosing partially digested, gut-derived proteins to macrophages within the SED. The apical large LAMP1 positive endosomes of FAE enterocytes were so distinctive that their presence could be used as a useful landmark for detecting the FAE in IF and EM sections.


Epithelial M cells (or microfold cells) within FAE are considered as an entry point for many intestinal pathogens, both bacteria and viruses [18] and also proposed as portals of entry for prions [19]–[21]. We identified M cells in the FAE using Ulex europaeus agglutinin 1 (UEA-1) as a marker. As anticipated, UEA-1 bound strongly to the brush borders of M cells and to a lesser extent to their basolateral cell membranes and the limiting membranes of intracellular vesicles (Figure 1 C and D). The UEA-1 label was restricted to M cells in the FAE and not observed in the neighbouring villus (Figure 1E, although especially the mucus containing granules of goblet cells elsewhere in the villi were often UEA-1 positive. Occasional goblet cells could also be seen in the FAE, but were easily distinguished from the UEA-1 positive M cells by their distinct morphology. The identification of M cells in the FAE was further confirmed by using antibodies specific to the M cell markers GP-2 [34] and annexin V [35] see Figure S2. Cryo-immuno - EM revealed that UEA-1 -positive cells within the FAE had typical, short, irregular microvilli (Figure 1F and 1G, Figure S2 and S3) and often harboured lymphocytes within an intracellular pocket. Each of these observations is an established characteristic of M cells. Our analysis revealed that M cells had smaller, LAMP1-positive late endosomes and completely lacked the large late endosomes found in FAE enterocytes. Few UEA-1 positive goblet cells were seen in the FAE by EM, but were easily distinguished from M cells by their apical mucus-containing granules and pronounced ER, typical for secretory cells (see Figure S2).
Enterocytes within the FAE take up neurofilaments from orally-acquired brain homogenate
Next, groups of PrP-expressing wt mice and PrP-deficient (Prnp-/- mice) were orally exposed to prions, by feeding brain homogenates from prion-infected mice. As controls, mice were fed uninfected brain homogenate. In order to trace the fate of the brain inoculum Peyer's patches were collected at intervals after exposure, and the early cellular and subcellular localization of brain homogenate-derived neurofilaments (NF) determined by IF. Peyer's patches were also examined from age-matched mice that had not been fed brain homogenate. Both wt and Prnp-/- mice fed either prion-infected or uninfected brain homogenate showed positive immunolabeling for NF, whereas untreated control mice were negative for NF immunolabeling (data not shown). In Peyer's patches taken at 1 dpf, NFs were detected in FAE enterocytes within the large LAMP1-positive endosomes of mice exposed to prion and normal brain homogenate (Figure 2A, 3A). Both wt and Prnp–/– mice showed similar levels of NF staining in FAE enterocytes, indicating that the uptake of NF was not dependent on PrPC expression (Figure 3A). Much fewer NF-positive endosomes or other NF-containing organelles were found within M cells in the FAE of Peyer's patches from the prion-exposed or control animals (Figure 3A). By immunogold-EM the prion inoculum was locally visible in the lumen of the gut (Figure 2F and Figures S3, S4 and S5) indicating that the transcytosis process of the orally introduced PrP was still going on at 1 dpf. In addition, throughout this study, no evidence was found to indicate direct uptake from the gut lumen via macrophages or dendritic cells, and few NF-positive endosomes were seen in neighbouring villi. In samples taken at later time-points (>1 dpf), the NF signals decreased in the LAMP1-positive FAE enterocytes of all mice. Together, these data suggest that the initial uptake of the brain homogenate from the gut lumen occurred mainly via enterocytes within the FAE and that the active uptake of the inoculum was still ongoing one day after feeding.
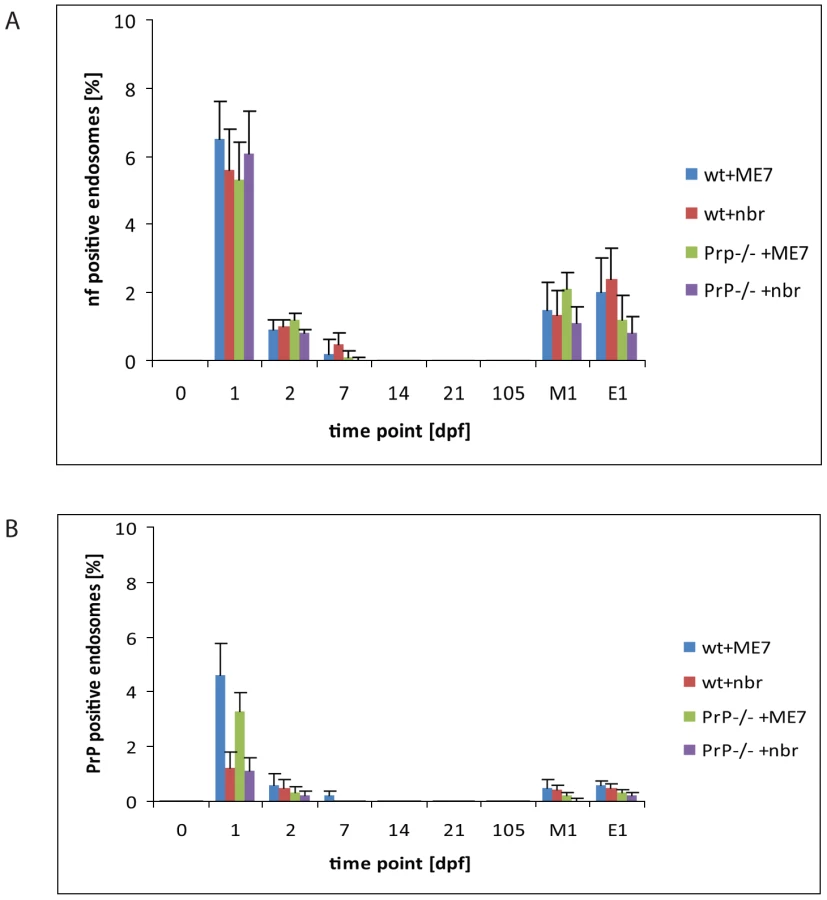
Enterocytes within the FAE take up PrPSc
To determine whether the NF immunolabeling was related to prion uptake from the gut lumen, we analysed subcellular structures of the FAE for PrP accumulation by cryo-immuno EM. No PrP-specific gold particles were found in the FAE of Peyer's patches from uninfected wt and Prnp-/-mice (day 0; Figure 3B). In tissue obtained at 1 dpf, PrP-specific immunolabelling was detected in FAE enterocytes from all mice, regardless of their PrPC expression levels. Animals fed prion-infected brain homogenate showed greater PrP labelling compared to mice fed normal brain homogenate (Figure 3B). Although the immunodetection conditions used here do not allow discrimination between cellular PrPC and pathogenic PrPSc, the PrP labelling in prion-exposed mice is likely due to PrPSc. The PrP signal was occasionally found on electron-lucent, small early endosomal vacuoles (Figure 2B and 2C), but was most abundant within large endosomal vacuoles similar to those labelled with LAMP1 (Figure 2D–F, Figure S4 and S5). As observed for the uptake of NF, PrP accumulated in FAE enterocytes. Comparatively much lower levels were found in M cells or in the enterocytes of the neighbouring villi at 1 dpf (Figure 3B). Also similar to the NF signals, the PrP signal detected within FAE enterocytes was transient: PrP labelling was decreased in samples at 2 dpf, and undetectable by 14 dpf in both wt and Prnp–/– mice (Figure 3B).
In order to discriminate the “exogenous” PrPSc within the orally administered prion inoculum from “endogenous” PrPC or PrPC within the prion inoculum an additional set of experiments was performed. Groups of wt and Prnp-/- mice were fed PK-treated brain homogenate prepared from terminally Sc327 scrapie-infected hamsters. Immunoblot analysis confirmed that treatment of the inoculum with PK destroyed any PrPC present within the brain homogenate leaving proteinase-resistant PrPSc (Figure S6). Peyer's patches were collected at 6 and 24 hours post infection, processed for cryo-immuno EM and sections were immunolabelled with mAb 3F4 directly conjugated to 10 nm gold particles. Since the mAb 3F4 recognizes only hamster PrP and does not label mouse PrP [36], the use of this mAb ensures any PrP detected is hamster-specific PrPSc from the Sc327-infected brain inoculum, and not PrPC expressed in the host mouse. Small clusters of gold labelling, indicative of PrPSc, were detected within small electron-lucent early endosomal vacuoles at 6 h postfeeding (Figure S7A and B). By 24 h after infection similar clusters of PrPSc were detected in the lumen of multivesicular bodies (Figure S7C and D). mAb 3F4-specific immunolabelling was observed predominantly in FAE enterocytes both in prion-infected wt and Prnp-/- mice, but was not detectable in control animals fed with PK-digested normal brain homogenate. Together, these data demonstrate that enterocytes within the FAE acquire PrPSc from the gut lumen. Furthermore, these data also confirm that PrP uptake from the gut lumen was independent of host PrPC expression, and also independent of the PrP sequence of the inoculum and therefore not affected by the species barrier between hamster and mouse.
Together, these data suggest that both PrPC and PrPSc cross the FAE via the endosomal system of enterocytes within the bulk flow of transcytosed material from the gut lumen. Furthermore, our data clearly show that the majority of PrP uptake from the gut lumen occurs within FAE enterocytes when compared to M cells and independently of the expression of cellular PrPC, since no substantial differences were observed between tissues from prion-exposed PrP-deficient and wt mice.
Prions are delivered to macrophages within the SED
Following transcytosis of brain material across the FAE, NFs were detected within a cluster of large LAMP1-positive organelles (Figure 2A). The SED region is known to harbour a large population of CD11c-positive cells that are often referred to as classical dendritic cells (DC), although they most likely represent a heterogeneous mixture of subpopulations of classical dendritic cells and macrophages in their different maturation forms [37], [38]. Unfortunately, CD11c-specific antibodies did not work on the aldehyde-fixed tissues used in the current study. However, most mucosal mononuclear phagocytes and all those within Peyer's patch germinal centres express CD11c indicating that this marker alone does not directly discriminate classical DC [38]. Thus, in order to discriminate between classical dendritic cells and macrophages, we used antibodies specific to major histocompability complex class II (MHC II) molecules to detect classical dendritic cells and antibodies against ferritin as a marker for macrophages (Figure 4, A and B, respectively). Macrophages express high levels of ferritin and harbour much lower levels of MHC II molecules on their surface (See Figure S8).

In 1 dpf samples, the macrophages within the SED harboured large quantities of NF (Figure 2A) which co-localized with LAMP1-positive structures. Many of these cells also appeared strongly positive for ferritin. Ferritin-positive cells that contained phagocytosed remnants of apoptotic lymphocytes, a landmark for tingible body macrophages (TBMs) were found further away from SED in the germinal centers (Figure 4C and D). In SED PrP-specific labelling was found in late endosomes of low MHC II –expressing macrophages by cryo-immuno EM (see Figure S9) indicating transcytosed prion inoculum in the SED region. Some macrophages were observed with intensely labelled intracellular LAMP1 structures, and LAMP1-positive membrane structures were also found in the surrounding extracellular space (Figure 4E). These probably secreted vesicles suggested intercellular exchange of membranous material by exocytosis and endocytosis (Figure 4E). We conclude that within the SED, PrP mostly accumulates within ferritin-positive macrophages, but not in MHC II–positive classical dendritic cells.
PrPSc accumulation upon FDCs
FDCs are typically binuclear with distinct chromatin pattern characteristics, electron-lucent cytoplasm and numerous convoluted extracellular extensions surrounding lymphocytes at different stages of differentiation. These characteristics allow FDCs to be readily identified by EM [39], [40]; typical examples of FDCs engulfing B lymphocytes are shown in Figure 5A–C.

In wt mice, oral exposure to either ME7 or RML prions resulted in a significant increase in PrP immunolabeling on the plasma membrane of FDCs beginning at 7 dpf (Figure 6A and B), which increased and became more uniform in samples taken at 21 dpf (Figure 5D–E). At 105 dpf, locally high levels of PrP label were found on FDC plasma membranes in germinal centers of PrP-infected wt (Figure S10). The increase in PrP signal (Figure 6) was observed only in prion-infected wt mice and was restricted to the plasma membrane of FDCs These observations suggest that the increase in PrP immunolobelling on FDCs of prion-infected mice was PrPSc. Initially, the increased PrP immunolabelling was often found heterogeneously on long dendritic extensions of single FDCs. Increased PrP levels were simultaneously also observed in germinal centres of mesenteric lymph nodes draining the intestines of wt mice (data not shown).

Increased PrP labelling was not observed in wt mice fed normal brain homogenate, suggesting that the labelling we found on FDCs of prion-infected mice was disease-related PrPSc. No PrP immunolabelling was observed on FDC in prion-exposed Prnp–/– mice, indicating that the increased PrP signal was unlikely to be due to accumulation of PrPSc from the prion inoculum. No accumulation of PrP was observed in samples taken from wt spleens during the first 21 dpf (data not shown). Thus, coincident with the disappearance of PrP from FAE enterocytes, PrP accumulated on the plasma membrane of FDCs within Peyer's patches and mesenteric lymph nodes in a PrPC-dependent fashion. Together, these data imply the first site of PrPSc conversion and replication following oral exposure.
To characterise the PrP on FDCs, and to investigate whether protease-resistant PrPSc had formed, sections were additionally treated with trypsin before immunolabeling with R2. This antibody recognises an epitope of PrP containing a trypsin cleavage site [41], and as seen previously for sections of mouse brain infected with RML prions [42], little R2 labelling of PrPC or PrPSc remained on FDC plasma membranes after the treatment, either at 21 days (Figure 5F) or 105 dpf (Figure S10).
Enhanced tingible body macrophage activity within Peyer's patches from prion-infected mice
Further IF and cryo-immuno EM of cells in the vicinity of FDCs revealed PrP within LAMP1-positive, late endocytic compartments in cells that had all the morphological landmarks of tingible body macrophages (TBMs) (Figure 4C and D; Figure 7; Figure S10). TBMs are a subset of large mononuclear phagocytes that reside in germinal centers of secondary lymphoid tissues. TBM contain many phagocytised apoptotic cells in various states of degradation (referred as tingible bodies). The detection of PrP within these compartments implied that these cells were most probably scavenging PrP. The number of TBM cell profiles positive for PrP increased during the course of prion infection, suggesting enhanced TBM activity in the Peyer's patches of infected wt mice (Figure S11). Compared to FDCs at the same time points, a higher portion of the PrP labelling of R2 antibody in the late endosomes / lysosomes of TBMs was unaffected after trypsin treatment (Figure 7E and S10F), suggesting that the early PrP labelling on FDC plasma membranes may be protease-sensitive PrPSc, but that PrPSc targeted for degradation may have acquired a degree of protease resistance. Again, tissues from uninfected mice and prion-infected Prnp-/- mice did not show PrP in TBMs (data not shown), which argues that this increase is specific to the propagation of PrPSc.

Early detection of prion infection within the enteric nervous system
In samples from prion-infected wt mice obtained at 21 dpf, the first signs of PrP accumulation within myenteric (Auerbach's) plexi were observed between the inner circular and outer longitudinal layers of the muscularis in regions close to Peyer's patches (Figure 8A and B). In samples from prion-infected, wt mice obtained at 105 dpf, almost all the plexi closely associated with Peyer's patches had strong accumulations of PrP (data not shown). Congruent with observations above, increased PrP was only detected within the enteric nervous systems of wt mice exposed to prion-infected brain homogenate. We have previously shown that caveosomes are involved in intracellular PrPC trafficking in cultured CHO cells [43]. We found no evidence for a role of caveosomes in PrP trafficking in vivo within the stroma of Peyer's patches. However, smooth muscle cells neighbouring infected plexi appeared rich in caveosomes (Figure 8C), which often contained PrP (Figure 8D), implying a potential mechanism through which PrP may be disseminated within the muscle layer of the intestine. After trypsin treatment of cryosections of prion-infected wt animals, approximately 9% of PrP label remained on plasma membranes of neurons at myenteric plexi of sections collected at 21 dpf, and increased to 15% for sections obtained at 105 dpf, indicating the presence of protease-resistant PrPSc on enteric nerves at relatively early phases of prion infection (Figure 8E and F).

A33 links the FAE to FDCs within lymphoid follicles
The definitive epithelial marker A33 (Gpa33; [44]) is expressed at high levels on the basolateral plasma membrane of the FAE and villous enterocytes (Figure 9A). Although Gpa33 is not expressed by macrophages and classical dendritic cells (See Figure S12), cryoimmuno EM revealed the presence of A33 protein within the endosomes of SED macrophages, indicating delivery of A33-positive membranes from enterocytes to macrophages (Figure 9B). Careful analysis revealed A33-positive vesicles within the germinal centres of Peyer's patches (Figure 9C) from each mouse group. In contrast, no A33 immunostaining was detected in Peyer's patches from A33-deficient control mice, confirming the specificity of the A33-specific antibody (Figure S13).

Weak A33 immunolabeling was also observed within the germinal centrers of mesenteric lymph nodes, whereas those in the spleen were negative (data not shown). The presence of small amounts of A33 antigen within the germinal centres of mesenteric lymph nodes most likely represents membrane trafficking from the gut epithelium to the B cell follicles as described [45]. Within the germinal centres, A33 was present on the surface of FDCs and often in close proximity to TBMs. It is therefore tempting to speculate that the scavenging macrophages may be carrying epithelial membrane components (including PrPSc on A33-positive membranes) from the FAE to the germinal centre, where they are deposited on the surface of the FDCs. Our EM data (Figure 9) suggest that LAMP1-positive, late endocytic multivesicular bodies of FAE enterocytes fuse with the plasma membrane [46], releasing their intravesicular membrane contents as exosomes in the extracellular space, where they may be phagocytosed within the SED by macrophages and further spread in a “taste and spit” -manner of repetitive endo - and exocytosis events by macrophages. Consistant with this idea, exosome-like membrane structures were shown to be positive for A33, PrP, and LAMP1 (Figure 9D–F, respectively). Thus, together these data provide further evidence for the association of FAE enterocyte-derived antigens such as A33 upon the surface of FDCs within Peyer's patches and mesenteric lymph nodes.
Discussion
Over the last few years, it has been noted that several amyloid or protein-misfolding diseases are characterized by prion-like propagation mechanisms [47], [48]. The main difference that distinguishes the prion diseases from other amyloid or protein-misfolding diseases is the fact that prions can be transmitted from one animal to another as truly infectious diseases. In many cases, this transmission occurs via the oral route, emphasizing the relevance of the present study. Here, we analyzed the initial uptake of PrP from the intestinal lumen via enterocytes of the FAE. While this initial uptake proved to be independent of the expression of cellular PrPC, the subsequent propagation and disease progression required an endogenous supply of PrPC.
The precise cellular mechanism through which prions are acquired from the gut lumen and transferred to FDCs within Peyer's patches is not known, although it has been shown that the accumulation of prions upon FDCs within Peyer's patches is critical for their efficient spread to the enteric nervous system [9], [12], [14]. Many other aspects of prion invasion have remained hypothetical, especially due to the lack of physiologically relevant in-vivo studies performed at the subcellular level. Prion detection during the early stages of oral infection has also been a problem. In previous immuno EM studies, PrPSc was first detected 70 days after oral exposure and only in denatured samples [11], [22].
We were able to investigate earlier stages after oral inoculation, by using adaptations of high-resolution methods developed in previous studies [42], [49]. The GALT are filled with endogenous immunoglobulins, which limit the use of mouse antibodies for immunodetection. However, many PrP-specific monoclonal antibodies (e.g., mAb 6H4 [50] and mAb R2 [51]) were raised in Prnp–/– mice. To avoid interference of endogenous mouse immunoglobulins and to permit better penetration in the tissue analysed, we collaborated with Aurion (Wageningen, The Netherlands) to conjugate mAb 6H4 and Fab fragments mAb R2 to UltraSmall gold particles. The PrP binding sites were visualised by silver enhancement, which enabled us to detect directly by EM the binding sites of the primary PrP antibody. Unconjugated R2 Fab fragments and UltraSmall gold-labelled Fab fragments of another PrP antibody have previously been used for IF and cryo-immuno EM on normal and prion-infected mouse brain, and R2 has been shown to recognize both PrPC and PrPSc on undenatured tissue sections [42], [51]. By applying these novel PrP antibody derivates, we gained higher PrP detection levels and were able to follow PrP trafficking in nondenatured GALT as early as 1 dpf, as inoculated PrP, and 7 dpf, as replicating PrP on the surface of FDCs.
The results of our time-course studies, employing IF and cryo-immuno EM with two distinct in-vivo mouse models, revealed the cellular and intracellular sites of prion trafficking after oral prion infection. Figure 10 represents a proposed model for prion neuroinvasion from gut lumen via Peyer's patches to enteric nervous system based on the results of the present study. In all gut samples examined, we found PrP accumulation in large, late endocytic multivesicular bodies of FAE enterocytes. Although PrP was also observed within M cells, this was at much lower levels than that within FAE enterocytes. Furthermore, our data indicated that PrP transcytosis across the FAE was independent of cellular PrPC expression. In prion-infected wt mice, we subsequently found PrP in late endosomes of SED macrophages followed by a gradual increase upon the surface of FDCs within germinal centres. Relatively large amounts of PrP could also be found in late endosomes of TBMs in germinal centres. Subsequently, beginning at 21 dpf, and following accumulation upon FDCs, increased levels of PrP were observed on the surface of neurons at submucosal and myenteric plexi. At 105 dpf (approximately one third through the incubation period for 129/Ola mice orally exposed to ME7 scrapie prions) PrP-immunolabelling on neurons had increased.
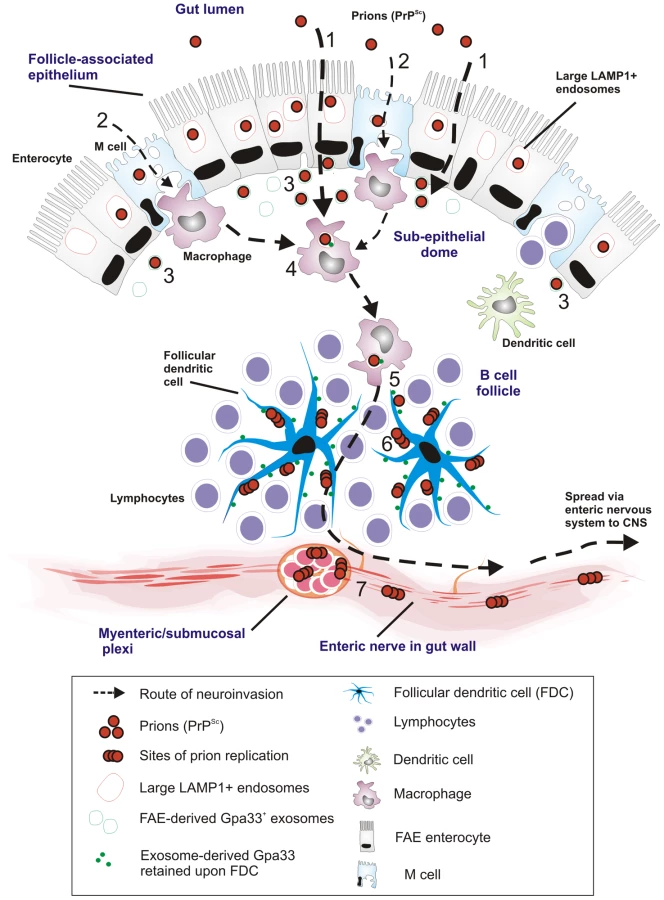
Our observations of increasing PrP on FDCs are consistent with previous reports. However, our data did not support the prevailing view concerning the initial uptake and trafficking of orally administered prions. In the gut, the FAE-overlying follicles of the mucosa-associated lymphoid tissue are key players in initiating mucosal immune responses and are strongly suspected to be the major entry site from the intestine into the lymphoid and nervous systems in animals orally infected with prions [22]. Within the FAE specifically, an in-vitro study suggested that M cells were the main target and gatekeepers for prion invasion [18], [19], [21]. In contrast, data from our in-vivo study suggest that M cells are not the primary target for prion entry through the FAE. Instead, most of the PrP was found in the endosomes of enterocytes within the FAE. These appear to be specialized enterocytes with enlarged late endosomal compartments compared to the “normal” enterocytes in the villi.
Data in the current study appear to differ significantly between those data in the study by Foster et al. [21] where PrP was mainly detected in association with Peyer's patch and cecal patch M cells. The reasons for this apparent discrepancy are uncertain. In the current study mice were fed doses of scrapie prions that require amplification in the GALT prior to neuroinvasion [9], [25]. The use of such physiologically-relevant doses is important as direct neuroinvasion can occur following exposure to higher doses. In the Foster study [21] a substantially higher dose of prions was delivered directly into the stomach of recipient mice than that used in the current study. Due to the high concentration of prions within the intestine it is plausible that other cell populations were principally involved in acquiring prions.
We found indirect evidence that the FAE enterocytes may exocytose the intravacuolar contents (including endocytosed PrP from the gut lumen) of their late endosomes into the extracellular space of the SED. These exosome-like structures (for a review, see [52]) are LAMP1 - and A33-positive and may be part of normal machinery [45] for antigen presentation to the immune system, since we found traces of the epithelial marker A33 within macrophages and upon FDCs. The concept of exosomes as membrane carriers was originally described in [53]. A33-related exosome secretion from intestinal epithelium has been described in general [54] and specifically related to prion transmission upon oral challenge [55]. However, it is difficult to find direct evidence for this highly dynamic process at the ultrastructural level in in-vivo models.
Genetic ablation of PrP abrogates susceptibility of mice to prion diseases [4]. For PrP trancytosis through the FAE, PrPC is not required, since transcytosis also occurs in PrP-deficient animals. Therefore, we conclude that PrP is likely to be transcytosed across the FAE in a non-specific manner within the bulk flow of other lumen-derived, digested, and endocytosed material. After transcytosis through the FAE, the migrating CD11c-positive cells (generally considered to be classical dendritic cells within the SED) are thought to endocytose and transfer the prions to germinal centres where prion replication may occur [25]. From there, by as-of-yet unknown cellular mechanisms, the prions are able to infect the peripheral enteric nervous system and gain further access to the main pathological target of prion diseases, the central nervous system. In macrophages of the SED, we found PrP in LAMP1-positive compartments of ferritin-positive cells and to a lesser extent in MHC II-positive cells (Figure S9). These observations suggest that PrP trafficking through the SED occurs in endosomal compartments of more macrophage-like mononuclear phagocytic cells rather than classical dendritic cells. Furthermore, in the SED, PrP was only detected in endosomal compartments of these macrophages and not on the cell surface. Transient depletion of CD11c-positive cells before oral exposure to prions has been shown to markedly delay the course of neuroinvasion [25]. When we retrospectively analysed the Peyer's patches of these CD11c-positive-cell-depleted mice (diphtheria toxin–treated, CD11c-diphtheria toxin receptor-transgenic mice), we found that the ferritin-positive macrophages within the SED were likewise transiently depleted (unpublished data). Interestingly, protease-resistant PrP has been reported to be transcytosed in a complex together with ferritin through intestinal epithelial cells [23], but the relevance of this finding for the ferritin-positive macrophages that are active in PrP endocytosis remains unclear.
The subsequent accumulation of PrP upon the surface of FDCs was only observed in Peyer's patches from orally exposed wt mice, and increased throughout the duration of the experiment. These PrPC-expressing, nonmigratory cells have a limited endosomal/phagocytic apparatus and are specialized to present native immuno complexes on their surface. These observations strongly suggest conversion and replication of PrPSc de novo upon the surfaces of FDCs and not merely the accumulation of inoculated PrPSc.
Taken together, these observations implicate two cellular compartments with distinct roles during the initial stages of orally-acquired prion disease: (1) endosomal compartments active in transient transcytosis and/or degradation/storage of PrPSc (LAMP1-positive endosomes in FAE enterocytes, M cells, villous enterocytes, SED macrophages, and germinal centre TBMs, and caveosomes in submucosal smooth muscle cells) and (2) the cell surface of FDCs and enteric neurons, where prion replication most likely occurs. Importantly, no increase of PrP surface labelling was observed on macrophages in the course of the early infection, even if they had high labelling densities in their endosomal compartments. Our data suggest uptake into the endosomal compartments occurs independently of cellular PrPC expression, whereas PrP accumulation upon FDCs and enteric neurons is critically dependent upon the expression of PrPC. Further, we were able to confirm the relevance of our results by using three different mouse infection models, with Prnp-/- mice resistant to disease as controls.
In our previous cryoimmuno EM studies, on FVB mouse hippocampus infected with RML prions, the PrPC levels were also specifically examined and were not found to increase as a result of prion disease. Increases in R2 labelling could thus be attributed to the formation of PrPSc. This labelling was also mainly on plasma membranes and on early endocytic or recycling vesicles rather than late endosomal compartments [42].
A high proportion of the R2 labelling in RML prion-infected FVB hippocampus was found to be trypsin sensitive [42], as was the case with the labelling on FDCs and enteric neurons in the present study. These results are consistent with reports that protease-sensitive forms of PrPSc predominate in some types of prion disease [56], [57]. Prions in the oral inoculum probably included protease-resistant forms that were able to withstand the general proteolytic degradation that occurs in the intestinal lumen and be processed by the endosomal machinery of FAE enterocytes without losing their infectivity.
We observed a spread of PrPSc to secondary lymphoid tissues. This may be due to several different mechanisms. First, PrPSc may spread by cell-cell contacts within the Peyer's patches and via lymphatics to the mesenteric lymph nodes. Additionally, the local concentration of the prion inoculum appears to have an impact on the speed of disease progression, which could explain why PrP accumulates first in Peyer's patches, where the enlarged endocytic capacity of FAE enterocytes sufficiently loads the locally dense population of SED macrophages.
We cannot exclude the possibility that the intestinal routing of prions after oral exposure may vary depending on the combination of prion and host strain. To address this issue in the current study, we used two different mouse-passage prion isolates that have been shown to have distinct cellular requirements for replication in lymphoid tissues [2], [27] and the hamster-passaged Sc237 scrapie strain. Our analysis showed consistent data from each of these distinct prion agents. However, there are examples of other prion strain and host combinations that do not appear to require accumulation and amplification in the GALT prior to neuroinvasion, such as BSE in cattle [58], some strains of TME [59], CWD in some cervid species [60] and atypical scrapie [61]. Of course, in each of these examples, the prions must still be transcytosed across the FAE prior to establishing infection within enteric nerves in the submucosa.
At 105 dpf (the latest time point we examined), the increased PrP labelling remained restricted to germinal centres and their neighbouring enteric plexi. For clarity, at 105 dpf, the animals infected with ME7 did not show any clinical symptoms of prion disease. The onset of clinical signs were observed at approximately 272 dpf at the dose we used in our studies. PrPSc has been reported to be found in the FAE of perorally challenged hamsters at 60 days [6] or 69 days [8] after intestinal infection with 263K scrapie, and it has been suggested that the FAE might serve as a site for prion release from the host some time after oral infection. In our study, we did not see a spread of PrPSc back to the FAE. 263K scrapie is considered to be a highly neurotropic prion strain, whereas ME7 is considered more lymphotropic. One could argue that during 263K scrapie infection, PrPSc returns to the gut epithelium via infected enteric nerves which presumably infiltrate the FAE. The same nerves may have been infected at the time of prion exposure. This does not appear to occur in mice, or if it does, it happens at later stages in the incubation period.
Together, these data suggest that uptake via large late endosomal compartments of FAE enterocytes represents a novel potential M cell-independent mechanism through which prions are acquired from the gut lumen. While these data do not exclude a role for M cells or villous enterocytes in the initial uptake of prions from the gut lumen, much lower levels of PrP were detected within them when compared to FAE enterocytes. Our data show that the transcytosis of prions to the germinal centres of Peyer's patches is PrPC-independent as it occurs also in PrPC-deficient animals. In contrast, PrPC expression is required for the observed high labelling densities on plasma membranes of FDCs and enteric neurons. Indeed, our data suggest that FDCs within Peyer's patches are the first site of prion conversion and replication after oral exposure. These findings provide insight into the subcellular localisation and trafficking of prions, which might provide suitable targets to arrest oral prion infection. Furthermore, these data identify a novel, previously unrecognised, enterocyte-dependent route of prion uptake and transfer from the gut lumen that may have an important influence on susceptibility to oral prion infection.
Materials and Methods
Mice and oral prion exposure
Prnp-/- mice were bred and maintained on a 129/Ola background [33]. Age - and sex-matched 129/Ola mice were used as wt controls. For oral infection with ME7 prions, both wt and Prnp-/- (n = 12 for each group) were fed individual food pellets doused with 50 µl of a 1% (wt/vol) brain homogenate prepared from wt mice terminally affected with ME7 scrapie prions. Food pellets doused with 50 µl of a 1% (wt/vol) brain homogenate prepared from uninfected mice (“normal brain homogenate”) were used as a control. These experiments were approved by the Roslin Institute's Protocols and Ethics Committee and carried out according to the strict regulations of the UK Home Office ‘Animals (scientific procedures) Act 1986’.
For oral infection with RML prions, wt FVB and Prnp-/- mice (n = 10 for each group) were infected by gavage with 100 µl of 1% brain homogenate from wt mice infected with mouse-adapted RML scrapie prions. An additional group of 6 wt and 6 Prnp-/-mice was infected with 1% brain homogenate from Syrian hamsters infected with hamster-adapted Sc237 scrapie prions. As respective controls, 100 µl of 1% brain homogenates prepared from uninfected wt mice or Syrian hamsters was administered by gavage as well. The brain homogenates from infected and uninfected Syrian hamsters were treated with proteinase K before being administered to the mice (See legend of Figure S6 for more details). The ME7 and RML samples were used without proteinase treatment. Use of these mice was according to the Public Health Services/National Institutes of Health Guide for the Care and Use of Laboratory animals. A33-deficient mice were created as described [62].
Tissue collection, fixation and cryo-sectioning
Mice were culled at 0, 1, 2, 7, 14, 21 and 105 dpf, and Peyer's patches, mesenteric lymph nodes, and spleens were collected and prepared for IF and cryoimmuno EM. Briefly, tissue samples were dissected from animals and immersion-fixed in a solution containing 2% paraformaldeyde and 0.2% glutaraldehyde in PHEM-buffer (25 mM HEPES, 10 mM EGTA, 60 mM PIPES, 2 mM MgCl2; pH 7.2). Fixed tissues were embedded in gelatin, infused in sucrose, and frozen in liquid nitrogen [63]. Frozen samples were cut on a cryo-ultramicrotome as semithin sections for IF (200 nm, -100°C) or as ultrathin sections for cryoimmuno EM (70 nm, -120°C). Sections were picked up with sucrose for IF or with a mixture of methylcellulose/sucrose for cryo-immuno EM.
Reagents and antibodies
M cells were identified with biotinylated Ulex europaeus (UEA-1) lectin (L8262; Sigma). Bound lectin was detected with polyclonal rabbit anti-biotin (Rockland). The following primary antibodies were used for immunolocalisation by IF and/or cryo-immuno EM: monoclonal rat anti-LAMP1 (1D4B; BD Biosciences Pharmingen); rabbit polyclonals MHCII (JV2; generous gift from Dr. Hidde Ploegh); anti-neurofilament 200 (Sigma); anti-ferritin (F5012; Sigma); goat polyclonal anti-A33 (AF2756; R&D Systems); rabbit polyclonal anti-annexin V (Ab14196; Abcam); rat monoclonal anti-GP2/glycoprotein 2 (D277-3; MBL); monoclonal anti-PrP 6H4 [49] R2 [50] and polyclonal 1B3 [64]. To detect hamster PrP biotinylated mAb 3F4 (SIG-39640 Covance Signet Antibodies) [36] directly conjugated to Streptavidin Gold Nanoparticles (Nanocs) was used. The following secondary bridging antibodies were used for cryo-immuno EM when rat or goat primary antibodies were applied: polyclonal rabbit anti-rat and rabbit anti-goat (DAKO), respectively.
Immunofluorescence analysis
For IF microscopy, semithin cryosections on glass slides were labelled with primary antibody, followed by species-specific secondary antibodies coupled to Alexa Fluor 488 (green) or Texas red dyes (Invitrogen, Paisley, UK). Sections were mounted in fluorescent mounting medium (DakoCytomation) and examined using a Zeiss LSM5 confocal microscope (Zeiss, Welwyn Garden City, UK). Simultaneous Dapi staining was applied to visualise the nuclei and the cellular organisation of the tissue. To prevent possible false-positive signals caused by autofluorescence, sections were treated with 1% sodium borate for 5 min. A TUNEL assay was performed using the fluorescein-conjugated in situ Cell death Detection kit (Roche Applied Science) according to the manufacturer's instructions.
Cryo-immunogold EM
For cryoimmunogold EM, PrP-specific 6H4 [49] and Fab-fragments of R2 antibodies [50] were conjugated to UltraSmall gold particles (0.8 nm; Aurion, Wageningen, The Netherlands) to allow increased penetration into the cryosections and circumvent labeling artefacts caused by cross-reaction with endogenous immunoglobulins in the tissue. Labeling reactions were performed under native conditions; no antigen retrieval method was applied. The R-GENT SE-EM silver enhancement kit from Aurion was used according to the manufacturer's instructions. For other immunogold-labelling experiments, the primary antibody, or the bridging antibody, was detected via the standard protein A–gold method [63]. For double labelling, conjugates of PrP-specific antibody and UltraSmall gold were applied and briefly silver enhanced, prior to incubation with the second primary antibody that was subsequently detected by protein A–gold.
The immunolabeling of sections was done as described previously [63]. In brief, after blocking with 1% cold fish gelatin and 1% bovine serum albumin for 15 min, sections were incubated with primary antibody for 60 min, washed, and bridging rabbit antibodies were applied for 30 min when necessary. Sections were then incubated with protein A-gold (15 nm) for 20 min. The specificities of the antibodies were controlled by omission of the primary antibody. Labeled sections were viewed with a Philips CM10 electron microscope (FEI Company, Eindhoven, The Netherlands) at 80 kV.
Quantification
The quantification of the distribution of gold particles was done according to routine stereological methods in double-blind fashion. Data are presented as means ± SD. Data were analysed using a T-test and differences were considered significant when p<0.05.
Supporting Information
Zdroje
1. PrusinerSB 2007 KnipeDMHowleyPM Fields Virology Philadelphia Lippincott Williams & Wilkins 3359 3091
2. BlattlerTBrandnerSRaeberAJKleinMAVoigtlanderT 1997 PrP-expressing tissue required for transfer of scrapie infectivity from spleen to brain. Nature 389 69 73
3. BrandnerSIsenmannSRaeberAFischerMSailerA 1996 Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 379 339 43
4. BuelerHAguzziASailerAGreinerRAAutenriedP 1993 Mice devoid of PrP are resistant to scrapie. Cell 73 1339 47
5. PeretzDWilliamsonRAMatsunagaYSerbanHPinillaC 1997 A conformational transition at the N terminus of the prion protein features in formation of the scrapie isoform. J Mol Biol 273 614 22
6. KrügerDThomzigALenzGKampfKMcBrideP 2009 Faecal shedding, alimentary clearance and intestinal spread of prions in hamsters fed with scrapie. Vet Res 40 4
7. AndreolettiOBerthonPMarcDSarradinPGrosclaudeJ 2000 Early accumulation of PrP(Sc) in gut-associated lymphoid and nervous tissues of susceptible sheep from a Romanov flock with natural scrapie. J Gen Virol 81 3115 26
8. BeekesMMcBridePA 2000 Early accumulation of pathological PrP in the enteric nervous system and gut-associated lymphoid tissue of hamsters orally infected with scrapie. Neurosci Lett 278 181 4
9. GlaysherBRMabbottNA 2007 Role of the GALT in scrapie agent neuroinvasion from the intestine. J Immunol 178 3757 66
10. HeggeboRPressCMGunnesGUlvundMJTranulisMA 2003 Detection of PrPSc in lymphoid tissues of lambs experimentally exposed to the scrapie agent. J Comp Pathol 128 172 81
11. JeffreyMMcGovernGGoodsirCMBrownKLBruceME 2000 Sites of prion protein accumulation in scrapie-infected mouse spleen revealed by immuno-electron microscopy. J Pathol 191 323 32
12. PrinzMHuberGMacPhersonAJSHeppnerFLGlatzelM 2003 Oral prion infection requires normal numbers of Peyer's patches but not of enteric lymphocytes. Am J Pathol 162 1103 11
13. SigurdsonCJBarillas-MuryCMillerMWOeschBvan KeulenLJ 2002 PrP(CWD) lymphoid cell targets in early and advanced chronic wasting disease of mule deer. J Gen Virol 83 2617 28
14. MabbottNAYoungJMcConnellIBruceME 2003 Follicular dendritic cell dedifferentiation by treatment with an inhibitor of the lymphotoxin pathway dramatically reduces scrapie susceptibility. J Virol 77 6845 54
15. AguzziA 2003 Prions and the immune system: a journey through gut, spleen, and nerves. Adv Immunol 81 123 71
16. MabbottNAMacPhersonGG 2006 Prions and their lethal journey to the brain. Nat Rev Microbiol 4 201 11
17. AnoYSakudoANakayamaHOnoderaT 2009 Uptake and dynamics of infectious prion protein in the intestine. Protein Pept Lett 16 247 55
18. KraehenbuhlJPNeutraMR 2000 Epithelial M cells: differentiation and function. Annu Rev Cell Dev Biol 16 301 32
19. HeppnerFLChristADKleinMAPrinzMFriedM 2001 Transepithelial prion transport by M cells. Nat Med 7 976 7
20. MiyazawaKKanayaTTakakuraITanakaSHondoT 2010 Transcytosis of murine-adapted BSE agents in an in vitro bovine M cell model. J Virol 84 12285 12291
21. FosterNMacphersonGG 2010 Murine cecal patch M cells transport infectious prions in vivo. J Infect Dis 202 1916 1919
22. JeffreyMGonzálezLEspenesAPressCMMartinS 2006 Transportation of prion protein across the intestinal mucosa of scrapie-susceptible and scrapie-resistant sheep. J Pathol 209 4 14
23. MishraRSBasuSGuYLuoXZouW-Q 2004 Protease-resistant human prion protein and ferritin are cotransported across Caco-2 epithelial cells: Implications for species barrier in prion uptake from the intestine. J Neurosci 24 11280 90
24. RescignoMUrbanoMValzasinaBFrancoliniMRottaG 2001 Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol 2 361 367
25. RaymondCRAucouturierPMabbottNA 2007 In vivo depletion of CD11c+ cells impairs scrapie agent neuroinvasion from the intestine. J Immunol 179 7758 66
26. MabbottNAKenneth BaillieJKobayashiADonaldsonDSOhmoriH 2011 Expression of mesenchyme-specific gene signatures by follicular dendritic cells: insights from the meta-analysis of microarray data from multiple mouse cell populations. Immunology 133 482 498
27. BrownKLStewartKRitchieDLMabbottNAWilliamsA 1999 Scrapie replication in lymphoid tissues depends on prion protein-expressing follicular dendritic cells. Nat Med 5 1308 12
28. KitamotoTMuramotoTMohriSDoh-UraKTateishiJ 1991 Abnormal isoform of prion protein accumulates in follicular dendritic cells in mice with Creutzfeldt-Jakob disease. J Virol 65 6292 5
29. KleinMAFriggRRaeberAJFlechsigEHegyiI 1998 PrP expression in B lymphocytes is not required for prion neuroinvasion. Nat Med 4 1429 33
30. ZabelMDHeikenwalderMPrinzMArrighiISchwarzP 2007 Stromal complement receptor CD21/35 facilitates lymphoid prion colonization and pathogenesis. J Immunol 179 6144 52
31. McBridePAEikelenboomPKraalGFraserHBruceME 1992 PrP protein is associated with follicular dendritic cells of spleens and lymph nodes in uninfected and scrapie-infected mice. J Pathol 168 413 8
32. BeekesMMcBridePA 2007 The spread of prions through the body in naturally acquired transmissible spongiform encephalopathies. FEBS Journal 274 588 605
33. MansonJCClarkeARHooperMLAitchisonLMcConnellI 1994 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol 8 121 7
34. HaseKKawanoKNochiTPontesGSFukudaS 2009 Uptake through glycoprotein 2 of FimH(+) bacteria by M cells initiates mucosal immune response. Nature 462 226 30
35. VerbrugghePWaelputWDieriksBWaeytensAVandesompeleJ 2006 Murine M cells express annexin V specifically. J Pathol 209 240 249
36. BoltonDCSeligmanSJBablanianGWindsorDScalaLJ 1991 Molecular location of a species-specific epitope on the hamster scrapie agent protein. J Virol 65 3667 75
37. HumeDA 2006 The mononuclear phagocyte system. Curr Opin Immunol 18 49 53
38. BradfordBMSesteraDPHumeDAMabbottNA 2011 Defining the anatomical localisation of subsets of the murine mononuclear phagocyte system using integrin alpha X (Itgax, CD11c) and colony stimulating factor 1 receptor (Csf1r, CD115) expression fails to discriminate dendritic cells from macrophages. Immunobiology 216 1228 1237
39. PetersJPRademakersLHde BoerRJvan UnnikJA 1987 Cellular composition of follicles of follicle centre cell lymphomas in relation to germinal centres of reactive lymph nodes. A morphometrical electromicroscopical study. J Pathol 153 233 44
40. DenzerKvan EijkMKleijmeerMJJakobsonEde GrootC 2000 Follicular dendritic cells carry MHC class II-expressing microvesicles at their surface. J Immunol 165 1259 65
41. PastranaMASajnaniGOniskoBCastillaJMoralesR 2006 Isolation and Characterization of a Proteinase K-Sensitive PrP(Sc) Fraction. Biochemistry 45 15710 15717
42. GodsaveSFWilleHKujalaPLatawiecDDeArmondSJ 2008 Cryo-immunogold electron microscopy for prions: toward identification of a conversion site. J Neurosci 28 12489 99
43. PetersPJMironovAJrPeretzDvan DonselaarELeclercE 2003 Trafficking of prion proteins through a caveolae-mediated endosomal pathway. J Cell Biol 162 703 17
44. JohnstoneCNWhiteSJTebbuttNCClayFJErnstM 2002 Analysis of the regulation of the A33 antigen gene reveals intestine-specific mechanisms of gene expression. J Biol Chem 277 34531 9
45. LeeJWEpardaudMSunJBeckerJEChengAC 2007 Peripheral antigen display by lymph node stroma promotes T cell tolerance to intestinal self. Nat Immunol 8 181 90
46. PetersPJBorstJOorschotVFukudaMKrähenbühlO 1991 Cytotoxic T lymphocyte granules are secretory lysosomes, containing both perforin and granzymes. J Exp Med 173 1099 109
47. AguzziARajendranL 2009 The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 64 783 90
48. FrostBDiamondMI 2010 Prion-like mechanisms in neurodegenerative diseases. Nat Rev Neurosci 11 155 9
49. MironovAJrLatawiecDWilleHBouzamondo-BernsteinELegnameG 2003 Cytosolic prion protein in neurons. J Neurosci 23 7183 93
50. KorthCStierliBStreitPMoserMSchallerO 1997 Prion (PrPSc)-specific epitope defined by a monoclonal antibody. Nature 390 74 7
51. WilliamsonRAPeretzDPinillaCBallHBastidasRB 1998 Mapping the prion protein using recombinant antibodies. J Virol 72 9413 8
52. SimonsMRaposoG 2009 Exosomes – vesicular carriers for intracellular communication. Current Opin in Cell Biol 21 575 581
53. PetersPJGeuzeHJvan der DonkHABorstJ 1990 A new model for lethal hit delivery by cytotoxic T lymphocytes. Immunol Today 11 28 32
54. van NielGMallegolJBevilacquaCCandalhCBrugiereS 2003 Intestinal epithelial exosomes carry MHC class II/peptides able to inform the immune system in mice. Gut 52 1690 7
55. FevrierBViletteDArcherFLoewDFaigleW 2004 Cells release prions in association with exosomes. Proc Natl Acad Sci U S A 101 9683 8
56. HoffmannCZieglerUBuschmannAWeberAKupferL 2007 Prions spread via the autonomic nervous system from the gut to the central nervous system in cattle incubating bovine spongiform encephalopathy. J Gen Virol 88 1048 55
57. BartzJCDejoiaCTuckerTKincaidAEBessenRA 2005 Extraneural prion neuroinvasion without lymphoreticular system infection. J Virol 79 11858 11863
58. SigurdsonCJ 2008 A prion disease of cervids: chronic wasting disease. Vet Res 39 41
59. BenestadSLArsacJNGoldmannWNöremarkM 2008 Atypical/Nor98 scrapie: properties of the agent, genetics, and epidemiology. Vet Res 39 19
60. SafarJGDeArmondSJKociubaKDeeringCDidorenkoS 2005 Prion clearance in bigenic mice. J Gen Virol 86 2913 2923
61. SafarJGGeschwindMDDeeringCDidorenkoSSattavatM 2005 Diagnosis of human prion disease. Proc Natl Acad Sci U S A 102 3501 3506
62. Pereira-FantiniPMJuddLMKalantzisAPetersonAErnstM 2009 A33 Antigen-deficient mice have defective colonic mucosal repair. Inflamm Bowel Dis 16 604 12
63. PetersPJBosEGriekspoorA 2006 Cryo-immunogold electron microscopy. Curr Protoc Cell Biol; Chapter 4: Unit 4.7
64. FarquharCFSomervilleRARitchieLA 1989 Post-mortem immunodiagnosis of scrapie and bovine spongiform encephalopathy. J Virol Methods 24 215 222
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 12
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Controlling Viral Immuno-Inflammatory Lesions by Modulating Aryl Hydrocarbon Receptor Signaling
- Fungal Virulence and Development Is Regulated by Alternative Pre-mRNA 3′End Processing in
- Cryo Electron Tomography of Herpes Simplex Virus during Axonal Transport and Secondary Envelopment in Primary Neurons
- Epstein-Barr Virus Nuclear Antigen 3C Stabilizes Gemin3 to Block p53-mediated Apoptosis