
Epstein-Barr Virus Evades CD4 T Cell Responses in Lytic Cycle through BZLF1-mediated Downregulation of CD74 and the Cooperation of vBcl-2
Evasion of immune T cell responses is crucial for viruses to establish persistence in the infected host. Immune evasion mechanisms of Epstein-Barr virus (EBV) in the context of MHC-I antigen presentation have been well studied. In contrast, viral interference with MHC-II antigen presentation is less well understood, not only for EBV but also for other persistent viruses. Here we show that the EBV encoded BZLF1 can interfere with recognition by immune CD4+ effector T cells. This impaired T cell recognition occurred in the absence of a reduction in the expression of surface MHC-II, but correlated with a marked downregulation of surface CD74 on the target cells. Furthermore, impaired CD4+ T cell recognition was also observed with target cells where CD74 expression was downregulated by shRNA-mediated inhibition. BZLF1 downregulated surface CD74 via a post-transcriptional mechanism distinct from its previously reported effect on the CIITA promoter. In addition to being a chaperone for MHC-II αβ dimers, CD74 also functions as a surface receptor for macrophage Migration Inhibitory Factor and enhances cell survival through transcriptional upregulation of Bcl-2 family members. The immune-evasion function of BZLF1 therefore comes at a cost of induced toxicity. However, during EBV lytic cycle induced by BZLF1 expression, this toxicity can be overcome by expression of the vBcl-2, BHRF1, at an early stage of lytic infection. We conclude that by inhibiting apoptosis, the vBcl-2 not only maintains cell viability to allow sufficient time for synthesis and accumulation of infectious virus progeny, but also enables BZLF1 to effect its immune evasion function.
Published in the journal:
. PLoS Pathog 7(12): e32767. doi:10.1371/journal.ppat.1002455
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002455
Summary
Evasion of immune T cell responses is crucial for viruses to establish persistence in the infected host. Immune evasion mechanisms of Epstein-Barr virus (EBV) in the context of MHC-I antigen presentation have been well studied. In contrast, viral interference with MHC-II antigen presentation is less well understood, not only for EBV but also for other persistent viruses. Here we show that the EBV encoded BZLF1 can interfere with recognition by immune CD4+ effector T cells. This impaired T cell recognition occurred in the absence of a reduction in the expression of surface MHC-II, but correlated with a marked downregulation of surface CD74 on the target cells. Furthermore, impaired CD4+ T cell recognition was also observed with target cells where CD74 expression was downregulated by shRNA-mediated inhibition. BZLF1 downregulated surface CD74 via a post-transcriptional mechanism distinct from its previously reported effect on the CIITA promoter. In addition to being a chaperone for MHC-II αβ dimers, CD74 also functions as a surface receptor for macrophage Migration Inhibitory Factor and enhances cell survival through transcriptional upregulation of Bcl-2 family members. The immune-evasion function of BZLF1 therefore comes at a cost of induced toxicity. However, during EBV lytic cycle induced by BZLF1 expression, this toxicity can be overcome by expression of the vBcl-2, BHRF1, at an early stage of lytic infection. We conclude that by inhibiting apoptosis, the vBcl-2 not only maintains cell viability to allow sufficient time for synthesis and accumulation of infectious virus progeny, but also enables BZLF1 to effect its immune evasion function.
Introduction
Successful persistence of viral infection depends on the establishment of a balance between host immune responses and viral immune evasion. Epstein-Barr virus (EBV), which is carried by more than 90% of the adult human population worldwide, is a prime example of a persistent virus that is generally harmless, but which can cause serious disease including various tumours [1].
A number of immunoevasins of EBV have recently been identified as acting at different points along the MHC class I (MHC-I) presentation pathway to modulate recognition by CD8+ T cell responses [2]–[7]. However, with regards to evasion of the MHC class II (MHC-II) antigen presentation pathway, EBV is less well understood. Virus-specific CD4+ T cell responses, which include some clones with cytotoxic activity, are broadly distributed against numerous proteins encoded by the EBV genome; both latent protein antigens [8] and the larger number of lytic protein antigens [9], [10]. These observations indicate a need for EBV to also modulate MHC-II antigen presentation pathways, particularly during in EBV lytic cycle.
Mechanisms for interfering with the MHC-II antigen presentation pathway have been reported for other herpesvirus; for example, US2, US3 and pp65 of cytomegalovirus [11]–[13] and glycoprotein B (gB) of herpesimplex virus type 1 [14]. However, EBV has no homologues to these immune evasion genes of CMV, and there is no evidence that the homolog of HSV-1 gB protein encoded by the BALF4 gene of EBV targets the MHC-II pathway. An unrelated EBV glycoprotein, gp42, has however been shown to associate with MHC-II molecules and to inhibit antigen presentation to CD4+ T cells [15], [16].
More recently, the immediate-early EBV gene BZLF1, which encodes a transcription factor initiating EBV lytic cycle, was reported to be a potential modulator of MHC-II antigen presentation [17]. Ectopic expression of BZLF1 in Raji cells inhibited the expression of MHC-II molecules, apparently through repression of CIITA transcription. However, interpretation of these data is complicated by the fact that Raji is an EBV-carrying B cell line in which expression of BZLF1 can initiate virus lytic cycle and, therefore, the expression of other viral genes that may be responsible for modulating MHC-II expression. One such candidate is the early antigen, BGLF5, which has been shown to induce global mRNA degradation and thereby to reduce expression of various host proteins, including MHC-I and MHC-II [2], [3]. In addition, as the level of expression of MHC molecules does not necessarily reflect the degree of T cell recognition, it is important to assay antigen presentation using functional T cell recognition.
In this study, we demonstrated for the first time that BZLF1 does indeed impair recognition by EBV-specific CD4+ T cells. However, this was not the result of downregulation of MHC-II α/β chain molecules as expected, but through downregulation of the invariant chain (Ii, or CD74) which serves as a chaperone to ensure correct loading of antigenic peptide fragments. As CD74 also mediates cell survival, its downregulation by BZLF1 induced cell death. A pivotal role was also identified for a vBcl-2 (BHRF1) during virus lytic cycle. By abrogating the concomitant toxic properties of BZLF1, the vBcl-2 enabled the immune evasion function of BZLF1.
Results
CD4+ T cell recognition is impaired in BZLF1 expressing LCLs
To gain a complete picture of effects on the antigen presentation pathway, it is crucial to include the functional T cell recognition assays as readout. We sought to determine whether BZLF1 can impair the MHC-II antigen presentation of newly processed MHC-II/peptide complexes and also of pre-existing MHC-II/peptide complexes. Recognition of newly processed MHC-II/peptide complexes was assayed by pulsing target cells with virus particles, before co-culturing with BXLF2-specific ‘LEK’ CD4+ effector T cells recognising a virion antigen. Pre-existing MHC-II/peptide complexes were assayed with EBNA1-specific ‘SNP’ CD4+ effector T cells to detect EBNA1 antigen that is expressed in both the latent and lytic phases of infection in EBV-transformed B lymphoblastoid cell lines (LCL). Whilst antigen processing for presentation via MHC-II complexes is generally considered to occur mainly through endocytosis of exogenous antigen (e.g. virus particles), it is also possible for endogenously expressed antigen to be processed by directly accessing the MHC-II antigen processing pathway [18], [19]. EBNA1 is paradigmatic for endogenous processing of target peptides for recognition by immune CD4+ T cells [18], [20].
In our initial experiments we generated LCL targets with a BZLF1-knockout recombinant EBV (BZLF1KO-LCLs), and transfected them with a doxycycline (DOX)-inducible BZLF1-expressing vector, pRTS-CD2-BZLF1. This vector expresses a CD2 surface marker from the vector backbone, allowing enrichment of viable transfected cells at 24h after transfection. The BZLF1 gene and an NGFR-IRES-GFP sequence were regulated by a bi-directional DOX-regulated promoter, which allows a further round of purification of BZLF1-expressing cells following DOX-induction. Control BZLF1KO-LCL targets were transfected with a vector containing a non-sense reverse BZLF1 sequence insert. Details of the plasmid vectors and the isolation of transfected cells are described in Figure 1A. BZLF1 - and control-vector transfected LCLs were treated with DOX for 24 h before separating the induced population according to NGFR expression. From each induced line, two populations were obtained: NGFR–/GFP– cells lacking transfected plasmid, and NGFR+/GFP+ cells in which BZLF1/GFP (or control/GFP) expression was evident (Figure 1B). The different populations were used as target cells in CD4+ T cell assays where recognition was measured by assaying the release of IFN-γ from the effector T cells.

GFP– cells from both the pRTS-CD2-control and the pRTS-CD2-BZLF1 transfectants were equally well recognised, both by BXLF2-specific CD4+ effectors following uptake and processing of virions (Figure 1C, upper histogram) and by EBNA1-specific CD4+ effectors (Figure 1D; upper histograms). In contrast, recognition of newly processed BXLF2 peptides from virus-pulsed targets (Figure 1C, lower histogram) and pre-existing peptides from EBNA1 (Figure 1D, lower histogram) was impaired in GFP+ cells expressing BZLF1 protein. The results illustrated in Figs. 1C and 1D are from one representative experiment. In three independent experiments, recognition of newly processed BXLF2 antigen by ‘LEK’ CD4+ T cells was reduced by 40–60%, and recognition of pre-existing EBNA1 peptide by ‘SNP’ CD4+ T cells was reduced by 25–40% following expression of BZLF1. These experiments demonstrate that BZLF1 expression in LCLs leads to impaired EBV-specific CD4+ effector T cell recognition.
In the BXLF2-specific CD4+ T cell recognition assays, there was very low but consistent recognition of BZLF1-expressing LCLs that had not been pulsed with EBV particles (bottom histogram, Figure 1C). This suggested that even within the short time-frame of this experiment the expression of BZLF1 can trigger lytic cycle and produce small but sufficient amounts of virus antigen for MHC-II processing. This conclusion was supported by further assays where expression of BZLF1 led to clear recognition not only by BZLF1-specific CD4+ effectors, but also by CD4+ effectors specific for the BMRF1 early antigen or the gp350 late antigen (Figure S1). Immunoblot analysis of the LCL targets showed that induction of BZLF1 led to expression of EBV lytic proteins (Figure 1E). Thus, whilst the data in Figure 1 are consistent with BZLF1 interfering with CD4+ T cell responses, further experiments with EBV-negative targets were necessary to avoid complications arising from BZLF1 initiation of the cascade of lytic cycle expression.
Effect of BZLF1 on CD4+ T cell recognition of MHC-II presented antigen
To investigate the effect of BZLF1 on MHC-II antigen presentation in EBV-negative target cells, we developed a strategy that allowed analysis of transiently transfected target cells. As intercellular antigen transfer is never detectable for EBNA1 [20], we chose this as the target antigen. To increase the sensitivity of the assay, a mutant form of EBNA1 (Cyto-EBNA1) was used as its cytosolic location renders its processing to MHC-II molecules more efficient than wild-type nuclear EBNA1 [20].
EBV-negative MJS melanoma cells expressing MHC-II DRB5*01 were transfected with cyto-EBNA1 expression plasmid together with IRES-GFP plasmid vectors for BZLF1 or other EBV lytic genes. The cultures were then assayed for antigen presentation to MHC-II DRB5*01-restricted CD4+ T cells specific for the ‘SNP’ EBNA1-derived peptide. T cell recognition was measured by the release of IFN-γ from the effector cells. The results of 5 independent experiments are summarised in Figure 2A. Mock transfected cells were not recognised, but good recognition was seen with target cells co-transfected with cyto-EBNA1 and control IRES-GFP vector. Recognition of cyto-EBNA1 was not significantly affected by co-transfection of BZLF2 or BALF4 vectors expressing the EBV lytic proteins gp42 or gp110, respectively. However, recognition of cyto-EBNA1 was substantially and reproducibly reduced by 60–80% when the target cells were co-transfected with BZLF1.

Replicate aliquots of cells analyzed by immuno-blots confirmed expression of BZLF1 in the cells transfected with the BZLF1-GFP plasmid (Figure 2B). There was no obvious change of MHC-II DR expression in any of the target cells. Whilst the levels of EBNA1 were similar in the cells co-transfected with cyto-EBNA1 and IRES-GFP, BZLF2-GFP or BALF4-GFP plasmids, there was an unexpected and marked reduction of EBNA1 expression when co-transfected with the BZLF1-GFP plasmid (Figure 2B).
Two colour flow cytometry analysis of viable cells stained for surface MHC-II DR suggested that none of the EBV lytic genes tested reduced the level of cell surface DR molecules on GFP+ cells compared to the expression on untransfected GFP– cells in the same cultures (Figure 2C). Notably, there were relatively few GFP+ cells in the cultures transfected with BZLF1-GFP; typically less than 1% compared with 15% in cultures transfected with control IRES-GFP plasmid. This indicated a toxic effect of BZLF1 in these cells, which could account for the reduced EBNA1 target antigen expression (Figure 2B) and also the reduced T cell recognition (Figure 2A).
BZLF1 downregulates anti-apoptotic Bcl-2 family members
Toxicity of BZLF1 was confirmed in other cell types, including an EBV-negative subclone of the Akata Burkitt's lymphoma, Akata-A3. Here, we used the DOX-inducible pRTS-CD2-BZLF1 as in Figure 1. Akata-A3 cultures transfected with either pRTS-CD2-BZLF1 or pRTS-CD2-control plasmids were enriched by CD2 selection to around 80% purity. Following treatment with DOX, the percentage of GFP+ cells was monitored over a period of 72 h. For the first 24 h of DOX treatment, both control - and BZLF1-transfected cultures contained indistinguishable numbers of induced GFP+ cells, but thereafter the BZLF1 transfected cultures showed a drop in the percentage of GFP+ cells such that by 72 h these BZLF1 cultures contained only around half the percentage of GFP+ cells as the control cultures (Figure 3A). These results indicate that BZLF1 is toxic to Akata-A3, but with slower kinetics than in MJS cells. The slower kinetics of toxicity in Akata-A3 allowed examination of cellular protein expression at 48 h after BZLF1 induction. Pure populations of GFP+ cells were obtained by flow cytometer sorting for GFP expression, and were analysed by immunoblotting. The first important point to note is that physiological levels of BZLF1 were obtained in these cells. Titration of the A3-BZLF1 transfectants against an LCL containing 5% BZLF1+ cells (i.e. in lytic cycle), followed by densitometry analysis of immunoblots, indicated that the level of BZLF1 in the transfected cells was around 115% of that in lytically-infected LCLs (Figure 3B). Secondly blotting for Bcl-2 and Bcl-xL expression showed a clear downregulation of these anti-apoptotic proteins in the BZLF1-expressing Akata-A3 cells (Figure 3C), which correlated with a downregulation of Bcl-2 and Bcl-xl mRNA transcripts (Figure 3D).

EBV vBcl-2 attenuates BZLF1 toxicity
EBV encodes a well-characterised Bcl-2 homolog, BHRF1, as an early lytic cycle protein [21]. We therefore investigated whether the toxicity of BZLF1 could be reversed by this vBcl-2. To address this possibility, we first examined the MJS line as it appears to be particularly sensitive to BZLF1. In the representative experiment shown in Figure 4A, MJS cells were transfected with IRES-GFP, BZLF1-GFP, or with BZLF1-GFP and a BHRF1 expression plasmid. Cultures were sampled at various time points post-transfection for analysis of GFP expression by flow cytometry. In the IRES-GFP control transfectants, about 10% of the viable cells were GFP+ at 8 h, rising to around 17% by 30 h. In marked contrast, MJS cells transfected with BZLF1-GFP showed only about 4% GFP+ viable cells at 8 h post-transfection, and thereafter the percentage of GFP+ cells gradually fell at later time points. However, when a BHRF1 expression vector was co-transfected with the BZLF1-GFP plasmid, the percentage of viable GFP+ cells continued to increase to about 10% by 30 h.

As in the previous set of experiments with Akata-A3 transfectants, the toxicity in MJS cells was mediated by physiological levels of BZLF1 (Figure 4B). Interestingly, the partial reversal of toxicity by co-expressed BHRF1 was achieved by levels of the vBcl-2 that were substantially lower than the levels observed in lytically-infected LCLs (Figure 4B). BHRF1-mediated protection from BZLF1-induced toxicity was also observed in other EBV-negative B cells (Figure S2). These results suggest that both the toxicity of BZLF1 and its reversal by BHRF1 are phenomena that are likely to be physiologically relevant during normal lytic cycle in EBV-infected cells.
BZLF1 causes marked downregulation of CD74 but not MHC-II DR molecules at the cell surface
The protection afforded by BHRF1 in MJS cells enabled us to re-examine the effect of BZLF1 on MHC-II antigen presentation without the confounding effect of induced cell death. MJS cells co-transfected with BHRF1 together with either IRES-GFP or BZLF1-GFP were examined for expression of cellular proteins in viable GFP+ sorted subpopulations at 48 hr post-transfection. Immuno-blot analysis of cell lysates showed that, as with the Akata-A3 cells (Figure 3C), BZLF1 also downregulated Bcl-2 and Bcl-xl protein expression in MJS cells (Figure 4C).
Interestingly, whilst the total cellular level of MHC-II DRα molecules was only slightly reduced following BZLF1 expression, the level of invariant chain, CD74, was markedly downregulated (Figure 4C). In BZLF1-transfected cultures, cell surface MHC-II DR expression was reproducible slightly elevated by around 10–20% in the GFP+ gated cells (Figure 4D, top right histogram); in contrast, there was a marked reduction in the expression of CD74 (about 50%) on the surface of GFP+ cells compared with GFP– cells (Figure 4D, lower right panel). One possible explanation for the discordance between total and cell surface MHC-II DR levels may be an altered localisation of MHC-II molecules when CD74 is downregulated. Analysis of control transfections showed that the levels of cell surface MHC-II DR (Fig 4D, top left histogram) and CD74 (bottom left histogram) were indistinguishable between GFP+ and GFP– gated cells in the same culture.
Similar experiments performed with the Akata-A3 cell line using the DOX inducible vector (Fig 5A) revealed that expression of BZLF1 in these B cells gave similar results to those seen with MJS cells.
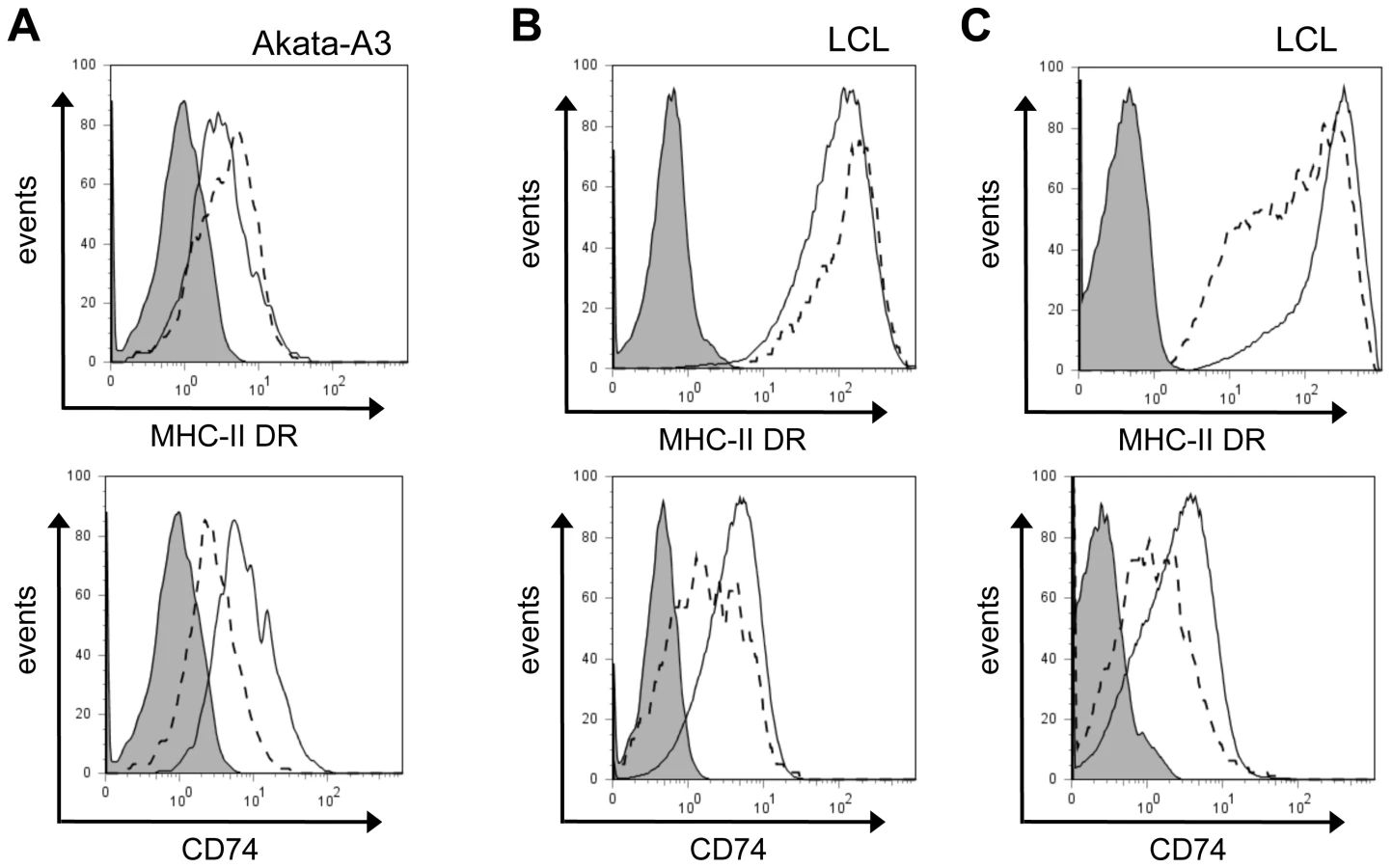
To study the surface levels of MHC-II DR and CD74 in the context of lytic cycle in normal EBV-transformed normal B cells, we examined selected LCL cultures that showed a clear subpopulation of cells spontaneously in lytic cycle. These LCLs were stained for surface DR or CD74 on viable cells, then were fixed and permeabilized for staining of intracellular BZLF1. The representative result in Figure 5B shows that the BZLF1+ cells spontaneously entering lytic cycle also showed a slight elevation of surface MHC-II DR (around 20%) and a clear reduction of surface CD74 (typically, 30–40%) compared with the BZLF1– latently infected population in the same LCL culture, i.e. mirroring the results obtained with BZLF1-transfected cells in previous experiments. Interestingly, when these same LCL cultures were co-stained for VCA to analyse the minor subpopulation of lytically-infected cells that have progressed to late lytic cycle, both MHC-II DR and CD74 were seen to be downregulated (Figure 5C).
BZLF1 downregulates CD74 post-translationally
As both MHC-II and CD74 can be transcriptionally regulated by CIITA [22], and BZLF1 was previously reported to transcriptionally repress the CIITA promoter [17], we investigated whether BZLF1 in our experiments might be modulating CD74 transcription via CIITA. Using HEK293 as a model CIITA–/MHC-II–/CD74– line, we overexpressed CIITA from a heterologous promoter, which in turn induced MHC-II and CD74 protein expression. However, when BZLF1 was co-expressed, surface expression of MHC-II DR was slightly elevated and CD74 was markedly downregulated (Figure S3), exactly as seen in our earlier experiments with MJS and B cells. This suggests that BZLF1 is modulating CD74 expression by a CIITA-independent mechanism. Furthermore, in Akata-A3 B cells, whilst MHC-II DR transcripts were slightly reduced following BZLF1 expression, no reduction in CD74 transcripts was observed (Fig 6A). Pulse-chase metabolic labeling experiments with 35S-methionine, revealed that BZLF1 has no effect on CD74 translation or protein maturation (Figure 6B). The exact mechanism of CD74 regulation was not actively pursued further, although experiments performed for other reasons (e.g. Figure S4) indicated that BZLF1 may be regulating trafficking of CD74 to or from the plasma membrane.

Inhibition of immune CD4+ T cell recognition by BZLF1 is retained when toxicity is attenuated by vBcl-2
We next revisited whether BZLF1 retains the ability to impair CD4+ T cell recognition when its toxicity is attenuated by BHRF1. MJS cells were co-transfected with cyto-EBNA1 target antigen plasmid and control-IRES-GFP or BZLF1-GFP plasmid vectors without (Figure 7A) or with (Figure 7B) BHRF1 expression plasmid. As in earlier experiments, immune CD4+ T cell recognition of the processed EBNA1 target was substantially impaired by expression of BZLF1 in the absence of BHRF1 (Figure 7A, histogram), which correlated with a clear reduction in the amount of EBNA1 antigen expression (Figure 7A, blots). However, when co-expressed with BHRF1 the BZLF1 had no significant effect on the levels of EBNA1 target antigen (Figure 7B, blots), and the immune CD4+ T cell recognition was inhibited to an even greater extent than when BHRF1 was not expressed (Figure 7A, histogram).

These data demonstrate that BZLF1 can indeed interfere with MHC-II antigen presentation to cause a substantial impairment of CD4+ effector T cell recognition.
Downregulation of CD74 is a mechanism for impaired CD4+ T cell recognition
As BZLF1 impairs T cell recognition without downregulating MHC-II DR expression, we considered it likely that the downregulation of CD74 might be responsible by qualitatively altering the MHC-II/peptide complexes available at the cell surface. To directly test this, we first generated an MJS line in which CD74 was over-expressed from a strong heterologous promoter, to see if we could maintain high levels of surface CD74 after BZLF1 expression and reverse the impaired CD4+ T cell recognition. However, despite massive over-expression of total cellular CD74, the amount of CD74 at the cell surface was barely increased; and the ability of BZLF1 to downregulate surface CD74 was unaffected (Figure S4). This is consistent with BZLF1 downregulating surface CD74 by a post-translational trafficking mechanism, but the experiment was otherwise uninformative.
We next carried out the reverse experiment, asking whether downregulation of CD74 by itself was sufficient to impair CD4+ T cell recognition. Again, we first tested this in MJS cells, using an shRNA inhibition approach. Successful knockdown of CD74 in MJS cells was associated with considerable cell death. However, using MJS-BHRF1 cells, we achieved efficient knockdown of CD74 and retained cell viability (Figure 8A and 8C). Importantly, knockdown of CD74 was associated with significant impairment of CD4+ T cell recognition of transiently expressed cyto-EBNA1 (Figure 8B, upper histogram). This was a specific effect, as pre-incubation of control and CD74-KO targets with saturating concentrations of synthetic EBNA1 target peptide resulted in both targets being equally-well recognised (Figure 8B, lower histogram). Immunoblots confirmed that the downregulation of CD74 in this experiment had no effect on the expression of the EBNA1 target antigen (Figure 8C). Finally, shRNA-mediated knockdown of CD74 in EBV-transformed LCLs (Figure 8D) resulted in a similarly impaired recognition by CD4+ effector T cells (Figure 8E, left histogram). The specificity of this effect in LCLs was demonstrated by the observation that CD74 knockdown had no effect on CD8+ effector T cell recognition (Figure 8E, right histogram).

Discussion
We have demonstrated that the EBV-encoded BZLF1 protein can interfere with recognition by immune CD4+ effector T cells, but it comes at a cost of toxicity. During EBV lytic cycle, this toxic property of BZLF1 can be overcome by expression of the vBcl-2 BHRF1 at an early stage of lytic infection. Unexpectedly, the impaired CD4+ effector T cell recognition did not correlate with levels of surface MHC-II molecules, but rather with a marked downregulation of CD74 (Ii) on the surface of target cells. CD74 not only facilitates appropriate peptide loading to MHC-II complexes in the endolysosomal vesicles [23]–[25] but, as a surface receptor for Macrophage Migration Inhibitory Factor (MIF), it also enhances cell survival through transcriptional upregulation of Bcl-2 family members [26], [27]. Therefore, downregulation of CD74 is a likely mechanism accounting for both the immune-evasion and death-inducing functions of BZLF1.
CD74 and MHC-II genes can both be regulated by the transcription factor, CIITA [22], and in many instances CD74 and MHC-II genes appear to be co-ordinately expressed. As CIITA expression is negatively regulated by BZLF1 through the suppression of its promoter [17], we might have expected that BZLF1 would downregulate both CD74 and MHC-II. However, from the present work, it is clear that BZLF1 selectively downregulates CD74. Transcription of CD74 is not solely regulated by CIITA [28], which could potentially explain discordant regulation of CD74 and MHC-II genes. Nevertheless, we found that the dominant mechanism by which BZLF1 downregulates CD74 was in fact post-translational.
Although BZLF1 can inhibit CIITA transcription [17], we consistently found cell surface MHC-II DR to be slightly elevated in BZLF1 expressing EBV-negative cells (Figure 4D, 5A). This mirrors what is observed following synchronous induction of lytic cycle in EBV-positive B cells, where cell surface MHC-II DR is initially elevated although it then falls between 12 to 24 h post-induction to a level that is around 40% of that in latently infected cells [29]. In the present study, we also observed that the MHC-II DR is slightly elevated in BZLF1+ cells in spontaneously lytic LCLs (Figure 5B), but that the level of surface DR was reduced in the minor subpopulation of cells expressing late viral capsid antigen (Figure 5C). Together, these results suggest that the initial rise in surface MHC-II DR expression in lytically infected cells is likely to be due to BZLF1 expression, while the later reduction in MHC-II DR in lytic cycle may be due to BGLF5, which acts as a host shutoff protein and contributes to immune evasion [2], [3], and/or a delayed effect of BZLF1-mediated inhibition of CIITA.
CD74 is a polypeptide involved in the transport and peptide loading of MHC-II molecules [23]–[25], [30]. Newly synthesized MHC-II α and β chains complex with CD74 (invariant chain) in the endoplasmic reticulum. A cytosolic di-leucine-targeting motif of CD74 directs MHC-II complexes to the endocytic pathway, either directly from the trans-Golgi network or via rapid internalization from the cell surface. The majority of CD74 at the cell surface is physically associated with MHC-II molecules [31] and most, if not all, of immature MHC-II molecules (complex of α chain, β chain and invariant chain) reach the cell surface before entering the peptide-loading compartment [32]. CD74 is rapidly turned over at the cell surface. Downregulation of surface CD74 by BZLF1 may therefore indicate a reduction of available immature MHC-II complexes for processing and uptake of antigenic peptides in the endosomes, and would account for the marked effect of BZLF1 expression on the MHC-II antigen processing pathway. Indeed, when we targeted expression of CD74 through shRNA, the knockdown of CD74 itself was sufficient to inhibit CD4+ T cell recognition (Figure 8).
In addition to serving as a chaperone for MHC-II, CD74 has been reported to play an essential role in B cell maturation [33], which involves activation of transcription mediated by p65 member of the NF-κB family [34]. These two functions of CD74 are genetically separable and map to different regions of the protein [35]. More recently CD74 has been identified as a receptor for MIF, and to promote cell survival and proliferation [26]. Binding of MIF to CD74 triggers activation of the p65 member of the NF-κB family, which in turn trans-activates Bcl-2 family genes, thereby providing the cells with increased survival capacity [27]. Furthermore, antibodies that block MIF/CD74 interaction cause growth inhibition and induction of apoptosis in B-cell lines [36]. BZLF1 is known to inhibit NF-κB p65 activity [37] and is toxic for all CD74+ cell lines used in our experiments, a phenomenon that correlated with downregulation of the Bcl-2 and Bcl-xl anti-apoptotic proteins (Figure 3C, 4C). It is notable that we observed no toxicity of BZLF1 in the epithelial cell line, HEK-293, which lacks expression of MHC-II and CD74.
The kinetics of the toxicity of BZLF1 in the EBV-negative Akata-A3 B cell line is such that BZLF1-transfected cells survive only 2 days (Figure 3A). This contrasts with what is observed during the normal physiological process of lytic cycle in the EBV-positive parental Akata line, where cell viability is maintained for at least 4 days after expression of BZLF1 [29]. EBV encodes two vBcl-2 homologs, both of which are expressed early following initiation of lytic cycle by EBV. The best characterized vBcl-2 is BHRF1, a potent anti-apoptotic protein that clearly enhances survival of B lymphocytes [21] and whose molecular mechanisms are beginning to be elucidated [38]. In contrast, it is unclear whether the second vBcl-2, BALF1, actually functions to modulate apoptosis [39]. In the present study, we showed that BHRF1 alone is able to moderate BZLF1 toxicity to an extent that is consistent with the enhanced survival period of cells entering lytic cycle.
In this study we have shown for the first time that the MHC-II antigen presentation is impaired during lytic infection of normal B cells (Figure 1). About 80 antigens are expressed in the EBV lytic cycle, representing a large pool of potential target antigens as reflected in the broad repertoire of EBV-specific CD4+ T responses identified, including some clones with cytotoxic activity to these EBV antigens [9], [10]. Therefore, impairment of MHC-II antigen presentation is likely to be crucial for the lytic cycle cells to survive long enough to generate infectious virus progeny. In addition to T cell responses to newly-synthesized early and late antigens in lytic cycle, there are also responses to pre-existing MHC-II/peptide complexes from latent antigens expressed at the time of initiation of lytic cycle. In this context, it is interesting to note that recognition of EBNA1, which is expressed during both latent and lytic infection, by specific CD4+ T cells is also impaired following expression of BZLF1 and induction of lytic cycle (Figure 1D).
As BZLF1 is the first EBV antigen to be expressed during lytic cycle, a process that can be sustained for several days before cell death occurs [29], its immune-evasion functions may be paramount in EBV's strategy for attenuating anti-viral responses. In this context, the impairment of the MHC-II antigen presentation pathway by BZLF1 adds to other previously reported immune-modulating properties of BZLF1, notably; inhibition the IFN-gamma signaling pathway [40] and TNF-alpha activation [41], [42] by down-regulation of IFN-γ receptor and TNF-R1. However, with regards to modulation of MHC-II antigen presentation it is likely that multiple EBV genes will cooperate to evade immune CD4+ T cell responses, as is seen with MHC-I antigen presentation to CD8+ T cells [43]. The exonuclease/host shut-off protein, BGLF5, expressed in early lytic cycle may contribute by degrading MHC-II mRNA transcripts [2], [3] and the late BZLF2 glycoprotein, gp42, may contribute by binding to MHC-II molecules and sterically inhibiting recognition by the T cell receptor of immune CD4+ T cells [16].
In summary, this work provides a new paradigm for viral immune evasion of MHC-II presented antigen. Targeting CD74 expression is sufficient to substantially impair MHC-II presentation of antigenic peptides even when levels of MHC-II DR molecules are barely affected. However, as CD74 also serves as an important regulator of cell survival, fresh insight is provided as to the role of vBcl-2 during lytic cycle in B cells. It is widely accepted that BHRF1 prolongs cell survival during lytic cycle to allow sufficient time for production and accumulation of new infectious virions. Now, we suggest that BHRF1 also plays a pivotal role in enabling BZLF1 to attenuate recognition by CD4+ T cell responses.
Materials and Methods
Plasmids and transfection
A derivative of the DOX-dependent expression vector pRTS-1 [44] was kindly provided by Dr J Mautner, Munich; BZLF1 and a reverse BZLF1 sequence as control were introduced into the vector by standard DNA cloning procedures to create vectors pRTS-CD2-BZLF1 and pRTS-CD2-control. The EBV lytic genes BZLF1, BZLF2, BALF4 were also subcloned into the EcoRI/NotI sites of pCDNA3-IRES-nls-GFP vector. All plasmids were verified by restriction digest and sequence analysis. The pCDNA3-cyto-EBNA1 plasmid was described previously [20]. Transient transfection of MJS cells with plasmid DNA was routinely performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Targets for the T cell recognition assay clone were generated by co-transfection of HLA-DRB5*01 MJS cells with a cyto-EBNA1 expression plasmid and IRES-GFP, BZLF2-GFP, BALF4-GFP or BZLF1-GFP expression plasmids.
Stable transfection and establishment of pBZLF1-tet cell lines
LCLs were established using the reference B95.8-based recombinant lacking the BZLF1 gene (BZLF1KO) [45]. A doxycycline (DOX)-inducible BZLF1 expression vector, pRTS-CD2-BZLF1, or control vector with the reverse BZLF1 sequence (pRTS-CD2-control) were introduced into LCLs or Akata-A3 by electroporation of 10 µg plasmid DNA into 107 cells in OptiMem medium (Invitrogen) at 280 V and 960 µF using a Biorad electroporation apparatus. Transfected cells were cultured in RPMI-1640 supplemented with 10% fetal calf serum (FCS). After 24 h, the transfected cell population was enriched by staining with OX34 antibody to rat CD2, and positively selected by magnetic cell sorting with anti-mouse IgG2a/b Microbeads and LS columns (Miltenyi Biotech) according to the manufacturer's guidelines. Cells were thereafter expanded and maintained in RPMI 1640 medium supplemented with 10% FCS. BZLF1 expression was induced by addition of 200 ng/ml DOX for 24 h, and the induced cells were positively selected by magnetic cell sorting with anti-NRFR Microbeads and LS columns (Miltenyi Biotech). The purity of the sorted cells was checked with Beckman Coulter XL flow cytometer.
MJS-DRB5*01 and MJS-BHRF1 cells
The MJS (Mel JuSol) melanoma-derived cell line [46] was maintained in RPMI 1640 medium (Gibco BRL) supplemented with 10% FCS. HLA-DRB5*01 expressing MJS cells were generated by transduction with a DRB5*01 retrovirus vector; a HLA-DRB5*01 β chain gene was cloned into retroviral expression plasmid pQCXIN (Clontech) by standard methods. Vesicular stomatitis virus-pseudotyped retrovirus particles were produced in GP2-293 cells co-transfected with the pVSV-G envelope vector. Virus in the culture supernatant at 72 h was concentrated by ultracentrifugation and used to infect 5×105 target cells overnight. Infected cells were selected with G418 (Invitrogen).
BHRF1 retroviral constructs were engineered by cloning the cDNA encoding BHRF1 into the pLZRS retroviral vector. Immediately downstream from the inserted BHRF1 gene lies an IRES sequence and the marker gene, a truncated nerve growth factor (ΔNGFR). Vesicular stomatitis virus-pseudotyped retrovirus particles were produced as above and used to transduce MJS cells. Transduced cells were magnetically sorted using MACS NGFR-specific beads as directed by the manufacturer (Miltenyi Biotech).
CD74 knockdown by shRNA lentivirus
The lentivirus plasmids containing a sequence of CD74-specific shRNA or a sequence of scrambled shRNA were purchased from Santa Cruz Biotechnology. Vesicular stomatitis virus-pseudotyped lentivirus particles were produced in FT-293 cells co-transfected with the pVSV-G and Gag-Pol expressing vectors. Virus in the culture supernatant at 72 h was concentrated by ultracentrifugation and used to infect 5×105 target cells overnight. Infected cells were selected with puromycin (Sigma).
Metabolic labelling and immunoprecipitation
Cells were starved by culturing 107 in 15 ml methionine-free RPMI medium supplemented with 10% dialysed FCS for 1 h at 37°C, then labeled for 15 min with 200 µCi of 35S protein labeling mix (PerkinElmer) in a final volume of 1 ml. After two washes with chase medium (normal RPMI medium supplemented with 10% FCS), the cells were resuspended at 2×106 cells/ml and chased at 37°C for the times indicated. Samples containing 2×106 cells were lysed in 400 µl of NP-40 buffer (0.5% Nonidet P-40, 5 mM MgCl2 and 50 mM Tris-HCl, pH 7.5) with protease inhibitor cocktail (Sigma) at 4°C for 45 min. Nuclei and insoluble debris were removed by centrifugation, and the supernatants were precleared; first with 1.2 µl normal mouse serum and 20 µl Dynabeads Protein A (Invitrogen) for 2 h at 4°C, and then with 20 µl Dynabeads Protein A and 20 µl Dynabeads protein G at 4°C overnight. The precleared lysates were immunoprecipitated for 2 h with 1 µg of mouse anti-CD74 and 20 µl Dynabeads Protein A plus 20 µl Dynabeads protein G, before washing the beads four times with NET buffer (0.5% NP-40, 150 mM NaCl2, 5 mM EDTA and 50 mM Tris-HCl, pH 7.5) and eluting by boiling in reducing gel sample buffer for 5 min. Finally, the samples were separated by SDS-PAGE on 12% Bis-Tris NuPage mini-gels with MOPS buffer (Invitrogen). After the gels were fixed and dried, they were exposed to autoradiographic film.
Antibodies
Goat antibodies to calregulin (sc6467), mouse anti-Bcl-xl, the mouse anti-CD74 and the mouse anti-DRα were purchased from Santa Cruz Biotechnology. Clone 124 mouse antibody to Bcl-2 [47] was a kind gift from the late David Mason. Antibodies to EBV antigens are described elsewhere [48]. For flow cytometry experiments, phycoerythrin (RPE)-conjugated goat anti-mouse IgG antibodies were purchased from AbD Serotec.
Flow cytometry analysis of cell surface MHC molecules
Cell surface expression of MHC-II DR or CD74 on viable cells was determined by staining with PE-conjugated anti-DR (AbD Serotec) or anti-CD74 primary antibodies followed with RPE-conjugated goat anti-mouse IgG2a (AbD Serotec). Intracellular staining to detect cells expressing nuclear BZLF1 was performed on 106 cells that were fixed using 100 µl of Ebiosciences Intracellular (IC) Fixative for 1 h on ice, followed by permeabilisation through the addition of Triton X-100 to a final concentration 0.2% for a further 30 min incubation on ice. After extensive washing with PBS, the cells were stained with 1 µg/ml of either MAb BZ.1 (anti-BZLF1) or with an IgG1 isotype control MAb for 1 hr at 37°C, followed by a 1∶50-dilution of RPE-conjugated goat anti-mouse IgG1 antibody (AbD Serotec) for 1 hr at 37°C. Stained cells were analyzed on Beckman Coulter XL flow cytometer, and the data processed using Flowjo software (Tree Star).
Western blotting
Total cell lysates were denatured in reducing sample buffer (final concentration: 2% SDS, 72.5 mM Tris-HCl pH 6.8, 10% glycerol, 0.2 M sodium 2-mercaptoethane-sulfonate, 0.002% bromophenol blue), then sonicated and heated to 100°C for 5 min. Solubilized proteins equivalent to 105 cells/20 µl sample were separated by sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) on 4–12% acrylamide gradient Bis-Tris NuPage mini-gels with MOPS running buffer (Invitrogen). Following electroblotting to polyvinylidene difluoride membranes, immunoblotting with specific primary antibodies followed by detection with appropriate alkaline phosphatase-conjugated secondary antibodies and a CDP-Star™ chemiluminescence detection kit (Tropix, Applied Biosystems) was performed as previously described [5].
T cell function assays
T cells were grown in 10% FCS in RPMI-1640 medium supplemented with 30% supernatant from the IL-2-producing MLA 144 cell line and 50 U/ml recombinant IL-2. The effector CD4+ T cell clones ‘LEK’ (specific for amino acids 126–140 of BXLF2 and restricted through DRB5*01) and ‘SNP’ (specific for amino acids 474–493 of EBNA1 and restricted through DRB5*01) were generated as described elsewhere [10], [20]. The effector CD8+ T cell clone ‘HPV’ (specific for aa 407–417 of EBNA1 and restricted through MHC-I B35.01) was described elsewhere [49]. The capacity of CD4+ and CD8+ T cell clones to recognize target LCLs or MJS cells was measured by IFNγ ELISA (Endogen). Briefly, 104 effector T cells were incubated for 18 h at 37°C in V-bottom microtest plate wells with 105 target cells, before assaying the supernatants for IFN-γ release by ELISA (Endogen) in accordance with the manufacturer's recommended protocol.
QRT-PCR assay
Total RNA was isolated from cultured cell lines using QIAGEN RNeasy kit and treated with DNase I (Turbo DNA-free kit; Ambion). Quantitative reverse-transcription polymerase chain reaction (QRT-PCR) assays for DRA, CD74, Bcl-2 and BCl-xl were performed with TaqMan® Gene Expression Assays (applied biosystem), duplexed with GAPDH assays for normalization.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. RickinsonABKieffE 2007 Epstein-Barr virus. KnipeDMHowleyPM Fields Virology Philadelphia Walters Kluwer/Lippincott, Williams & Wilkins 2655 2700
2. RoweMGlaunsingerBvan LeeuwenDZuoJSweetmanD 2007 Host shutoff during productive Epstein-Barr virus infection is mediated by BGLF5 and may contribute to immune evasion. Proc Natl Acad Sci U S A 104 3366 3371
3. ZuoJThomasWvan LeeuwenDMiddeldorpJMWiertzEJ 2008 The DNase of gammaherpesviruses impairs recognition by virus-specific CD8+ T cells through an additional host shutoff function. J Virol 82 2385 2393
4. HislopADRessingMEvan LeeuwenDPudneyVAHorstD 2007 A CD8+ T cell immune evasion protein specific to Epstein-Barr virus and its close relatives in Old World primates. J Exp Med 204 1863 1873
5. ZuoJCurrinAGriffinBDShannon-LoweCThomasWA 2009 The Epstein-Barr virus G-protein-coupled receptor contributes to immune evasion by targeting MHC class I molecules for degradation. PLoS Pathog 5 e1000255
6. ZuoJQuinnLLTamblynJThomasWAFeederleR 2011 The Epstein-Barr Virus-Encoded BILF1 Protein Modulates Immune Recognition of Endogenously Processed Antigen by Targeting Major Histocompatibility Complex Class I Molecules Trafficking on both the Exocytic and Endocytic Pathways. J Virol 85 1604 1614
7. ZeidlerREissnerGMeissnerPUebelSTampeR 1997 Downregulation of TAP1 in B Lymphocytes by Cellular and Epstein-Barr Virus-Encoded Interleukin-10. Blood 90 2390 2397
8. LongHMHaighTAGudgeonNHLeenAMTsangC-W 2005 CD4+ T-Cell Responses to Epstein-Barr Virus (EBV) Latent-Cycle Antigens and the Recognition of EBV-Transformed Lymphoblastoid Cell Lines. J Virol 79 4896 4907
9. AdhikaryDBehrendsUMoosmannAWitterKBornkammGW 2006 Control of Epstein-Barr virus infection in vitro by T helper cells specific for virion glycoproteins. J Exp Med 203 995 1006
10. LongHMLeeseAMChagouryOLConnertySRQuarcoopomeJ 2011 Cytotoxic CD4+ T Cell Responses to EBV Contrast with CD8 Responses in Breadth of Lytic Cycle Antigen Choice and in Lytic Cycle Recognition. J Immunol 187 92 101
11. TomazinRBonameJHegdeNRLewinsohnDMAltschulerY 1999 Cytomegalovirus US2 destroys two components of the MHC class II pathway, preventing recognition by CD4+ T cells. Nat Med 5 1039 1043
12. HegdeNRTomazinRAWisnerTWDunnCBonameJM 2002 Inhibition of HLA-DR Assembly, Transport, and Loading by Human Cytomegalovirus Glycoprotein US3: a Novel Mechanism for Evading Major Histocompatibility Complex Class II Antigen Presentation. J Virol 76 10929 10941
13. OdebergJPlachterBBrandenLSoderberg-NauclerC 2003 Human cytomegalovirus protein pp65 mediates accumulation of HLA-DR in lysosomes and destruction of the HLA-DR α-chain. Blood 101 4870 4877
14. TemmeSEis-HübingerAMMcLellanADKochN 2010 The Herpes Simplex Virus-1 Encoded Glycoprotein B Diverts HLA-DR into the Exosome Pathway. J Immunol 184 236 243
15. RessingMEvan LeeuwenDVerreckFAWGomezRHeemskerkB 2003 Interference with T cell receptor-HLA-DR interactions by Epstein-Barr virus gp42 results in reduced T helper cell recognition. Proc Natl Acad Sci U S A 100 11583 11588
16. RessingMEvan LeeuwenDVerreckFAWKeatingSGomezR 2005 Epstein-Barr Virus gp42 Is Posttranslationally Modified To Produce Soluble gp42 That Mediates HLA Class II Immune Evasion. J Virol 79 841 852
17. LiDQianLChenCShiMYuM 2009 Down-Regulation of MHC Class II Expression through Inhibition of CIITA Transcription by Lytic Transactivator Zta during Epstein-Barr Virus Reactivation. J Immunol 182 1799 1809
18. PaludanCSchmidDLandthalerMVockerodtMKubeD 2005 Endogenous MHC Class II Processing of a Viral Nuclear Antigen After Autophagy. Science 307 593 596
19. ZhouDLiPLinYLottJMHislopAD 2005 Lamp-2a Facilitates MHC Class II Presentation of Cytoplasmic Antigens. Immunity 22 571 581
20. LeungCSHaighTAMackayLKRickinsonABTaylorGS 2010 Nuclear location of an endogenously expressed antigen, EBNA1, restricts access to macroautophagy and the range of CD4 epitope display. Proc Natl Acad Sci U S A 107 2165 2170
21. HendersonSHuenDRoweMDawsonCJohnsonG 1993 Epstein-Barr virus-coded BHRF1 protein, a viral homologue of Bcl-2, protects human B cells from programmed cell death. Proc Natl Acad Sci U S A 90 8479 8483
22. ChangCHFlavellRA 1995 Class II transactivator regulates the expression of multiple genes involved in antigen presentation. J Exp Med 181 765 767
23. RochaNNeefjesJ 2008 MHC class II molecules on the move for successful antigen presentation. EMBO J 27 1 5
24. van den HoornTPaulPJongsmaMLMNeefjesJ 2011 Routes to manipulate MHC class II antigen presentation. Curr Opin Immunol 23 88 95
25. RochePATeletskiCLStangEBakkeOLongEO 1993 Cell surface HLA-DR-invariant chain complexes are targeted to endosomes by rapid internalization. Proc Natl Acad Sci U S A 90 8581 8585
26. StarletsDGoreYBinskyIHaranMHarpazN 2006 Cell-surface CD74 initiates a signaling cascade leading to cell proliferation and survival. Blood 107 4807 4816
27. LantnerFStarletsDGoreYFlaishonLYamit-HeziA 2007 CD74 induces TAp63 expression leading to B-cell survival. Blood 110 4303 4311
28. ZhuLJonesPP 1990 Transcriptional control of the invariant chain gene involves promoter and enhancer elements common to and distinct from major histocompatibility complex class II genes. Mol Cell Biol 10 3906 3916
29. RessingMEKeatingSEvan LeeuwenDKoppers-LalicDPappworthIY 2005 Impaired Transporter Associated with Antigen Processing-Dependent Peptide Transport during Productive EBV Infection. J Immunol 174 6829 6838
30. RomagnoliPGermainRN 1994 The CLIP region of invariant chain plays a critical role in regulating major histocompatibility complex class II folding, transport, and peptide occupancy. J Exp Med 180 1107 1113
31. MoldenhauerHenneKarhausenMÖLler 1999 Surface-expressed invariant chain (CD74) is required for internalization of human leucocyte antigen-DR molecules to early endosomal compartments. Immunology 96 473 484
32. OngGoldenbergHansenMattes 1999 Cell surface expression and metabolism of major histocompatibility complex class II invariant chain (CD74) by diverse cell lines. Immunology 98 296 302
33. ShacharIFlavellRA 1996 Requirement for Invariant Chain in B Cell Maturation and Function. Science 274 106 108
34. MatzaDWolsteinODiksteinRShacharI 2001 Invariant Chain Induces B Cell Maturation by Activating a TAFII105-NF-kB-dependent Transcription Program. J Biol Chem 276 27203 27206
35. MatzaDLantnerFBogochYFlaishonLHershkovizR 2002 Invariant chain induces B cell maturation in a process that is independent of its chaperonic activity. Proc Natl Acad Sci U S A 99 3018 3023
36. SteinRQuZCardilloTMChenSRosarioA 2004 Antiproliferative activity of a humanized anti-CD74 monoclonal antibody, hLL1, on B-cell malignancies. Blood 104 3705 3711
37. MorrisonTEKenneySC 2004 BZLF1, an Epstein-Barr virus immediate-early protein, induces p65 nuclear translocation while inhibiting p65 transcriptional function. Virology 328 219 232
38. KvansakulMWeiAHFletcherJIWillisSNChenL 2010 Structural Basis for Apoptosis Inhibition by Epstein-Barr Virus BHRF1. PLoS Pathog 6 e1001236
39. MarshallWLYimCGustafsonEGrafTSageDR 1999 Epstein-Barr Virus Encodes a Novel Homolog of the bcl-2 Oncogene That Inhibits Apoptosis and Associates with Bax and Bak. J Virol 73 5181 5185
40. MorrisonTEMauserAWongATingJPKenneySC 2001 Inhibition of IFN-gamma signaling by an Epstein-Barr virus immediate-early protein. Immunity 15 787 799
41. MorrisonTEMauserAKlingelhutzAKenneySC 2004 Epstein-Barr virus immediate-early protein BZLF1 inhibits tumor necrosis factor alpha-induced signaling and apoptosis by downregulating tumor necrosis factor receptor 1. J Virol 78 544 549
42. BristolJARobinsonARBarlowEAKenneySC 2010 The Epstein-Barr virus BZLF1 protein inhibits tumor necrosis factor receptor 1 expression through effects on cellular C/EBP proteins. J Virol 84 12362 12374
43. RoweMZuoJ 2010 Immune responses to Epstein-Barr virus: molecular interactions in the virus evasion of CD8+ T cell immunity. Microbes Infect 12 173 181
44. KellyGLLongHMStylianouJThomasWALeeseA 2009 An Epstein-Barr Virus Anti-Apoptotic Protein Constitutively Expressed in Transformed Cells and Implicated in Burkitt Lymphomagenesis: The Wp/BHRF1 Link. PLoS Pathog 5 e1000341
45. FeederleRKostMBaumannMJanzADrouetE 2000 The Epstein-Barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J 19 3080 3089
46. JohnsonJPDemmer-DieckmannMMeoTHadamMRRiethmüllerG 1981 Surface antigens of human melanoma cells defined by monoclonal antibodies. I. Biochemical characterization of two antigens found on cell lines and fresh tumors of diverse tissue origin. Eur J Immunol 11 825 831
47. PezzellaFTseAGCordellJLPulfordKAGatterKC 1990 Expression of the bcl-2 oncogene protein is not specific for the 14;18 chromosomal translocation. Am J Pathol 137 8
48. CroftNPShannon-LoweCBellAIHorstDlKremmerE 2009 Stage-Specific Inhibition of MHC Class I Presentation by the Epstein-Barr Virus BNLF2a Protein during Virus Lytic Cycle. PLoS Pathog 5 e1000490
49. BlakeNLeeSRedchenkoIThomasWStevenN 1997 Human CD8+ T cell responses to EBV EBNA1: HLA class I presentation of the (Gly-Ala)-containing protein requires exogenous processing. Immunity 7 791 802
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 12
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Controlling Viral Immuno-Inflammatory Lesions by Modulating Aryl Hydrocarbon Receptor Signaling
- Fungal Virulence and Development Is Regulated by Alternative Pre-mRNA 3′End Processing in
- Cryo Electron Tomography of Herpes Simplex Virus during Axonal Transport and Secondary Envelopment in Primary Neurons
- Epstein-Barr Virus Nuclear Antigen 3C Stabilizes Gemin3 to Block p53-mediated Apoptosis