
Genomic and Proteomic Analyses of the Fungus Provide Insights into Nematode-Trap Formation
Nematode-trapping fungi are “carnivorous” and attack their hosts using specialized trapping devices. The morphological development of these traps is the key indicator of their switch from saprophytic to predacious lifestyles. Here, the genome of the nematode-trapping fungus Arthrobotrys oligospora Fres. (ATCC24927) was reported. The genome contains 40.07 Mb assembled sequence with 11,479 predicted genes. Comparative analysis showed that A. oligospora shared many more genes with pathogenic fungi than with non-pathogenic fungi. Specifically, compared to several sequenced ascomycete fungi, the A. oligospora genome has a larger number of pathogenicity-related genes in the subtilisin, cellulase, cellobiohydrolase, and pectinesterase gene families. Searching against the pathogen-host interaction gene database identified 398 homologous genes involved in pathogenicity in other fungi. The analysis of repetitive sequences provided evidence for repeat-induced point mutations in A. oligospora. Proteomic and quantitative PCR (qPCR) analyses revealed that 90 genes were significantly up-regulated at the early stage of trap-formation by nematode extracts and most of these genes were involved in translation, amino acid metabolism, carbohydrate metabolism, cell wall and membrane biogenesis. Based on the combined genomic, proteomic and qPCR data, a model for the formation of nematode trapping device in this fungus was proposed. In this model, multiple fungal signal transduction pathways are activated by its nematode prey to further regulate downstream genes associated with diverse cellular processes such as energy metabolism, biosynthesis of the cell wall and adhesive proteins, cell division, glycerol accumulation and peroxisome biogenesis. This study will facilitate the identification of pathogenicity-related genes and provide a broad foundation for understanding the molecular and evolutionary mechanisms underlying fungi-nematodes interactions.
Published in the journal:
. PLoS Pathog 7(9): e32767. doi:10.1371/journal.ppat.1002179
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002179
Summary
Nematode-trapping fungi are “carnivorous” and attack their hosts using specialized trapping devices. The morphological development of these traps is the key indicator of their switch from saprophytic to predacious lifestyles. Here, the genome of the nematode-trapping fungus Arthrobotrys oligospora Fres. (ATCC24927) was reported. The genome contains 40.07 Mb assembled sequence with 11,479 predicted genes. Comparative analysis showed that A. oligospora shared many more genes with pathogenic fungi than with non-pathogenic fungi. Specifically, compared to several sequenced ascomycete fungi, the A. oligospora genome has a larger number of pathogenicity-related genes in the subtilisin, cellulase, cellobiohydrolase, and pectinesterase gene families. Searching against the pathogen-host interaction gene database identified 398 homologous genes involved in pathogenicity in other fungi. The analysis of repetitive sequences provided evidence for repeat-induced point mutations in A. oligospora. Proteomic and quantitative PCR (qPCR) analyses revealed that 90 genes were significantly up-regulated at the early stage of trap-formation by nematode extracts and most of these genes were involved in translation, amino acid metabolism, carbohydrate metabolism, cell wall and membrane biogenesis. Based on the combined genomic, proteomic and qPCR data, a model for the formation of nematode trapping device in this fungus was proposed. In this model, multiple fungal signal transduction pathways are activated by its nematode prey to further regulate downstream genes associated with diverse cellular processes such as energy metabolism, biosynthesis of the cell wall and adhesive proteins, cell division, glycerol accumulation and peroxisome biogenesis. This study will facilitate the identification of pathogenicity-related genes and provide a broad foundation for understanding the molecular and evolutionary mechanisms underlying fungi-nematodes interactions.
Introduction
Nematode-trapping fungi are a heterogeneous group of organisms broadly distributed in terrestrial and aquatic ecosystems [1], [2]. These fungi are capable of developing specific trapping devices such as adhesive networks, adhesive knobs, and constricting rings to capture nematodes and then extract nutrients from their nematode prey [2], [3], [4]. Most nematode-trapping fungi can live as both saprophytes and parasites [1], [2]. They play important roles in maintaining nematode population density in diverse natural environments. Their broad adaptability and flexible lifestyles also make them ideal agents for controlling parasitic nematodes of plants and animals [2], [4].
Arthrobotrys oligospora (teleomorph Orbilia auricolor) is one of the best-studied nematode-trapping fungi [2], [5]. Strains of A. oligospora have been found in diverse soil environments including heavy metal-polluted soils and decaying wood [6], [7] where they live mainly as saprophytes. In the presence of nematodes, A. oligospora enters the parasitic stage by forming complex three-dimensional networks to trap nematodes (Figure 1). The trapping initiates a series of processes including adhesion, penetration, and immobilization of nematodes [2], [5]. The ability to trap nematodes makes it an attractive candidate agent for controlling parasitic nematodes of plants and animals. Indeed, two commercial biological nematicides, Royal 300 [8] and Royal 350 [9], have been developed based on two species closely related to A. oligospora: A. robusta and A. irregularis.

The formation of trapping devices by nematode-trapping fungi is an important indicator of their switch from the saprophytic to the predacious lifestyles [2], [4]. Previous studies have identified the morphological characteristics of major trapping devices, including their evolution and phylogenetic distribution [4], [10], [11], [12]. Recently, the gene expression profiles in trap cells and vegetative hyphae of the nematode-trapping fungus Monacrosporium haptotylum were analyzed using the microarray technology [13], including examining the transcriptional response of M. haptotylum to the model nematode Caenorhabditis elegans at different infection stages [14]. However, at present, no genome sequence information is available in any nematode-trapping fungi and relatively little is known about the molecular mechanism underlying trap formation or nematode-fungi interaction. Such genome sequence information could help reveal the genetic features that allow these fungi to form nematode traps and provide important information to study the molecular mechanism of infection and their lifestyle transitions.
Here the first genome sequence of a nematode-trapping fungus, A. oligospora Fres. (ATCC24927) was reported. In addition, using the proteomics approach and quantitative PCR (qPCR), proteins differentially expressed in response to nematode extract (NE) were identified and further investigated. The combined genomic, proteomic, and qPCR data led us to formulate the putative genetic and metabolic pathways involved in trap formation in A. oligospora.
Results
Genome sequencing and analysis
The genome of A. oligospora strain ATCC24927 was sequenced to 36.6-fold coverage through a Sanger/pyrosequencing hybrid shotgun approach from multiple clone types (Table S1). The 40.07 Mb genome is similar in size to that of the model ascomycete fungus Neurospora crassa [15]. The A. oligospora genome was assembled into 215 scaffolds, containing a long-range continuity as reflected by N50 scaffold size of 2037 Kb and N50 contig size of 575.8 Kb (Table S2). The assembly represents 99% of the coding regions of the genome, as assessed by mapping 50,121 assembled transcript sequences (8.9 Mb) to the genome assembly. Almost all of the transcripts (99.6%) were mapped onto the genome. A total of 11,479 protein-coding genes were predicted: 23.6% belonged to multi-gene families and 44.6% were mapped in the KOG/COG database (Figure 2A). The average gene density was one gene per 3.50 kb, with an average gene length of 1.69 kb, similar to that of N. crassa (1.67 kb) [15]. Compared to N. crassa, genes in A. oligospora have more introns (2.8 VS. 1.7) but with a shorter average intron length (90 bp VS. 134 bp) [15]. As is typical of ascomycete fungal genomes, approximately one-third (34.6%) of the predicted A. oligospora genes lacked significant homologies to known proteins from public databases (Table S2).

Comparative genomic analysis
The orthologous genes between A. oligospora and other 10 fungal genomes were identified based on bidirectional best hits (BBHs) using BLAST [16] (Table S3). The other 10 genomes used for comparison were divided into non-pathogen (Aspergillus nidulans, N. crassa and Saccharomyces cerevisiae) and pathogen (Fusarium graminearum, Magnaporthe oryzae, Verticillium dahliae, A. fumigatus, Coccidioides immitis, Histoplasma capsulatum and Chaetomium globosum) groups. Based on orthology analysis, the genes in A. oligospora can be classified into four categories: ao (only found in A. oligospora, 6157 genes), ao/pathogen (found in A. oligospora and pathogen genomes, 961 genes), ao/pathogen/non-pathogen (found in all genomes, 4249 genes), and ao/non-pathogen (found in A. oligospora and non-pathogen genomes, 112 genes) (Figure 3A). The results showed that A. oligospora shared many more genes with pathogenic fungi than with non-pathogenic fungi. The genes shared between A. oligospora and other pathogenic fungi may be functionally related to pathogenicity in these fungi.

Genome evolution
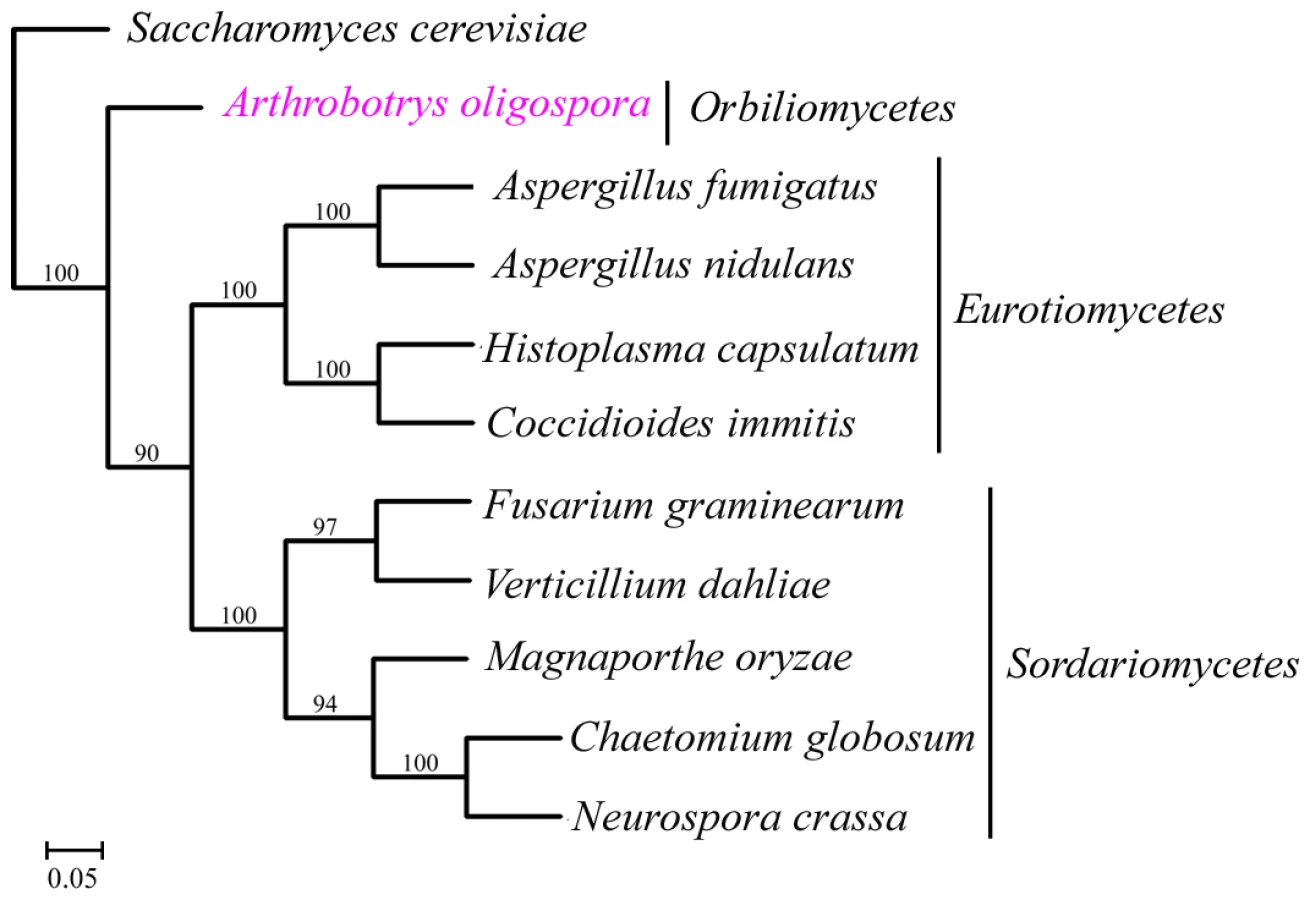
A total of 529 orthologous proteins were found in all the 11 analyzed fungal genomes. These orthologous sequences were concatenated to infer phylogenomic relationships among these fungi using Neighbor-joining, Maximum parsimony and Maximum likelihood methods. The same tree topology was found by all the three phylogenetic methods (Figure 4). From the phylogenomic trees, Eurotiomycete fungi (A. nidulans, A. fumigatus, C. immitis and H. capsulatum) were clustered into one clade, Sordariomycete fungi (F. graminearum, V. dahliae, C. globosum, N. crassa and M. grisea) were clustered into a different clade, while A. oligospora formed a separate branch. Our result was consistent with results from previous phylogenomic analyses of the Ascomycota [17], [18].
Our analysis revealed that 0.47% of the assembly consisted of repetitive sequences (Table S4), higher than that in the ascomycete wheat pathogen F. graminearum (0.10%) [19], but lower than that in N. crassa (3.15%) [15]. The relatively low percentage of repetitive sequences in N. crassa was attributed to a genome-wide defense system known as repeat-induced point mutation (RIP), a phenomenon hypothesized to be widespread among filamentous ascomycetes [15], [19]. To determine whether RIP contributed to the low percentage of repetitive sequences in A. oligospora, the RIP indices for the whole genome as well as separately for the coding regions, non-coding regions, exons, introns, multigene families, and repetitive sequences were calculated. Using the default setting (TpA/ApT≥0.89, (CpT+ApT)/(ApC+GpT) ≤1.03) [20], a positive RIP response was detected in the repetitive sequences with both RIP indices (and very high AT content) (Table S5), likely contributing to the lack of the repetitive sequences in A. oligospora. RIP could have similarly contributed to the relatively low proportion of genes in multigene families (25.11%) in A. oligospora. For protein coding genes, although no positive RIP responses were detected using average RIP indices, a large number of regions contained signatures of RIP (Table S6). Specifically, using the 200-bp windows with 100-bp shifts, about one third of the genome sequences had RIP-positive sequences.
Pathogenicity
The whole genome blast analysis against the pathogen-host interaction (PHI) gene database [21] identified 398 putative PHI genes in A. oligospora, 294 of which belong to 86 multigene families. The gene number expanded to 706 if single linkage transitive closure was applied. The distribution analysis of the othologous sequences showed that the putative PHI genes included 80 ao-specific genes and 73 genes only found in pathogen genomes, more than those in the ao/non-pathogen category (Figure 3B). It should be noted that many putative PHI genes were not included in the pathogenicity-related gene families, suggesting that there are potentially more pathogenicity-related genes that remain to be experimentally confirmed.
The number of genes in several gene families related to fungal pathogenicity was found expanded in the A. oligospora genome (Figure 5). For example, subtilisins, a group of proteases essential for infection [22], [23], [24], [25], were found in a greater number in A. oligospora (24) than in several model ascomycetes such as the animal pathogens A. fumigatus (4), C. immitis (15) and H. capsulatum (3), plant pathogens F. graminearum (15), V. dahliae (16) and M. grisea (22), and non-pathogens A. nidulans (3), N. crassa (5). Besides subtilisin genes, the A. oligospora genome also contains a larger number of genes within enzyme families of cellulases (58), pectinesterases (18) and cellobiohydrolases (17) than other sequenced model fungi (Figure 5). As a comparison, the M. grisea genome contains the highest number of genes within the cytochrome P450 family (P450; 112); the A. fumigatus genome contains the highest numbers of genes within enzyme families of non-ribosomal peptide synthases (NRPS; 21), chitinases (18), and polygalacturonases (16); the V. dahliae genome contains the highest numbers of genes within enzyme families of polygalacturonases (16), pectinesterases (29), and pectate lyases (29); and the C. globosum genome contains the highest numbers of genes within enzyme families of cellulases (66) and xylanases (27).

Because of their ability to disrupt the physical and physiological integrity of the cuticles of nematode hosts during penetration and colonization, subtilisin-like serine proteases have been identified as important virulence factors in nematode-trapping fungi [23], [26], [27]. For example, previous studies demonstrated that a proteinase K-like serine protease PII in A. oligospora could efficiently degrade nematode cuticle [22], [26], [28]. The A. oligospora genome contains 24 genes encoding putative subtilases, which can be categorized into four subtilisin families (Figure S1). Among them, 20 belong to the proteinase K-like family, which can be further classified into five subfamilies (SF1-SF5, Figure S1). The following four genes are clustered into one of the five sub-families, SF4: PII (AOL_s00076g4), P186 (AOL_s00215g702), P233 (AOL_s00075g8), and P12 (AOL_s00170g103). P12 shares a high nucleotide sequence identity (77.9%) with the cuticle-degrading protease encoding gene spr1 of another nematode-trapping fungus Monacrosporium megalosporum [29].
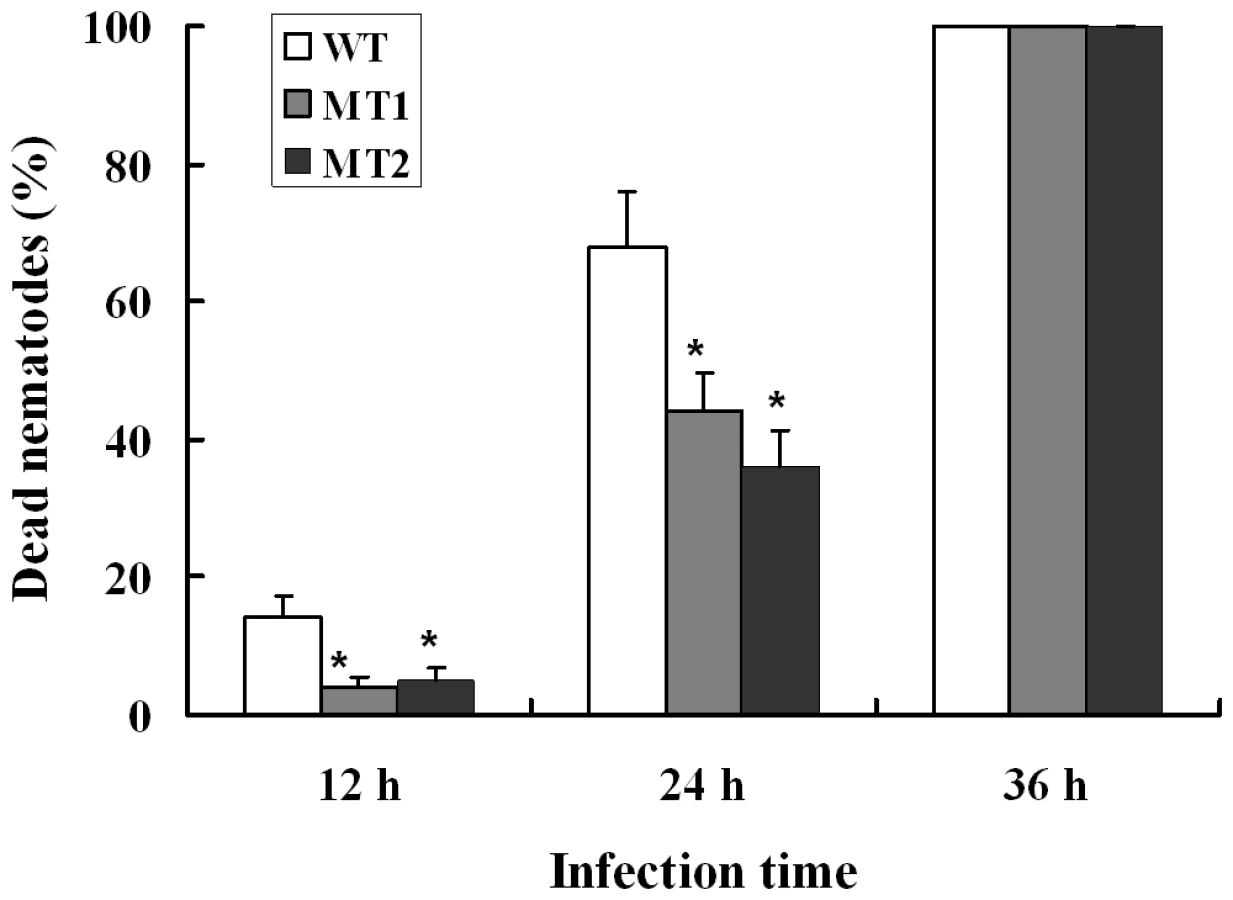
When A. oligospora was exposed to nematode extracts for 10 h, the transcription levels of P12 and P186 increased by 5.9 - and 23.4-folds (Table S7) respectively, whereas those of PII and P233 did not change significantly. In order to identify the role of P186 in infection against nematodes, the gene P186 was disrupted by homologous recombination according to the method described by Colot et al. [30]. The disruption removed 107 bp from the open reading frame of P186 and was confirmed by PCR using genomic DNA as templates with primers (P186-5F and P186-3R). The phenotypic properties and nematocidal activities of the mutants were compared with the wild strains. No obvious differences in phenotypic properties, such as growth rates, spores number, traps number and morphology, were found between the mutants and the wild strain. However, disruption of P186 greatly attenuated the pathogenicity of A. oligospora. The fatality rate of nematodes infected by P186-deletion mutants (△P186) decreased by 24–32% (Figure 6) at 24 h after infection. Our results suggested that P186 likely play major roles during nematode infection by A. oligospora. In contrast, the lack of a significant change in PII expression level upon NE induction is consistent with an earlier finding that PII gene disruption had only a limited effect on the pathogenicity of A. oligospora [22].
Some fungi can produce secondary metabolites, including small molecular toxins that kill host cells before infection, generate a necrotrophic stage during infection, or disable host cellular functions after infection [31]. So far, 179 nematicidal compounds belonging to diverse chemical groups have been identified from nematophagous fungi, three (oligosporon, 4′,5′-dihydro-oligosporon and linoleic acid) of which were from A. oligospora [32]. Several enzyme families are commonly involved in the synthesis of these secondary metabolites in fungi, such as PKSs, NRPSs, and P450s. Genomic analysis identified five putative PKS genes and seven NRPS genes in the A. oligospora genome (Figure 5). Among the PKS genes, AOL_s00215g283 is likely involved in the production of 6-methyl salicylic acid (Figure S2). However, the functions of the seven NRPS genes could not be predicted based on the phylogenetic analysis (Figure S3). Of the P450 genes, our search against the fungal cytochrome P450 database (FCPD) [33] found that the 36 P450 (Table S8) genes in the A. oligospora genome could be classified into four classes (Group I P450, Group IV P450, pisatin demethylase-like P450, and CYP52 P450). P450 genes are known to be associated with the biosynthesis of diverse classes of secondary metabolites in other fungi [34].
Proteomic and qPCR analyses of trap formation
Although the above-mentioned genes may play important roles in pathogenicity, the formation of trap likely involves other genes and is a prerequisite for infection, and serving as the indicator for lifestyle switch from saprophytic to predacious stages in nematode-trapping fungi [1], [2], [3], [4]. At present, little is known about the molecular mechanism of trap formation. To identify the proteins involved in trap formation, a proteomic study was performed and the profiles of intracellular proteins from A. oligospora cells at two developmental stages representing the early nematode extract (NE) induction stage (10 h after treatment with NE) and the late stage of trap formation (48 h after treatment with NE) (Figures S4 and 2B) were compared. As a negative control, mycelia incubated on medium without NE but with the solvent of NE (sterile deionized water) were analyzed. The expressions of 90 and 25 proteins were found up-regulated (P<0.05), while 16 and 94 were down-regulated (P<0.05) at 10 h and 48 h, respectively. Most of the proteins up-regulated at 10 h were involved in translation, posttranslational modification, amino acid metabolism, carbohydrate metabolism, energy conversion, cell wall and membrane biogenesis (Table S7). The results suggest very active growth and metabolism during the transition from vegetative hyphae to trap cells. In contrast, compared to those at the saprophytic stage, the expressions of most proteins up-regulated at 10 h were found either down-regulated or unchanged at 48 h when the traps were already formed, consistent with the hypothesis that the proteins up-regulated at 10 h were likely related to trap formation in A. oligospora.
Similar to other organisms, A. oligospora uses signaling cascades to alter its gene expression patterns in response to environmental changes. Genes encoding the components of common fungal signal transduction pathways are all found in the A. oligospora genome. Specifically, glycosylphosphatidylinositol-specific phospholipase C (AOL_s00109g54), mitogen-activated protein kinase (MAPK, AOL_s00173g235), serine/threonine protein phosphatase 2A (regulatory subunit, AOL_s00007g146), calcyclin binding protein (AOL_s00054g214), and Ca2+/calmodulin-dependent protein kinase (AOL_s00078g95) were all up-regulated (Table S7) during the formation of traps. This result is consistent with the importance of signal sensing and transduction in the shift from saprophytic to carnivorous lifestyles in A. oligospora.
Both hyphal growth and trap formation require energy. In A. oligospora, the tricarboxylic acid (TCA) cycle was up-regulated in response to NE, as indicated by the enhanced expression of genes in the TCA cycle, such as citrate synthase (AOL_s00079g361), aconitase (AOL_s00110g24), isocitrate dehydrogenase (AOL_s00075g141), succinyl-CoA synthetase (AOL_s00043g45), and malate dehydrogenase (AOL_s00210g140 and AOL_s00215g565) (Table S7). In addition, malate synthase (AOL_s00112g112) and isocitrate lyase (AOL_s00075g130), two key enzymes in the glyoxylate cycle, were up-regulated at 10 h (Table S7), indicating that this pathway is also important for trap formation. It has been reported that the glyoxylate cycle is associated with fungal virulence [35]. Taken together, our results suggest that the TCA cycle and the glyoxylate cycle are actively involved in A. oligospora trap formation, possibly by providing energy and substrates for macromolecule biosynthesis.
Cell division and cell cycle controls also play important roles during A. oligospora trap formation. In this study, the expression levels of Cdc37 (a molecular chaperone, AOL_s00043g594) and Mih1 (a phosphatases, AOL_s00176g31) (Table S7) were up-regulated during the formation of traps (10 h). In S. cerevisiae and Schizosaccharomyces pombe, Cdc37 is required for maintaining the protein level of a cyclin-dependent kinase Cdc28, a key enzyme for regulating the G1-S and the G2-M phase transitions [36]. Similarly, Mih1 promotes cell entry into mitosis by removing the inhibitory phosphorylation placed on Cdk1 by Wee1 in S. cerevisiae [37]. In addition, several cytoskeleton proteins such as two actin-binding proteins (AOL_s00097g552 and AOL_s00079g186), an actin-related protein (AOL_s00007g186), and a microtubule-binding protein (AOL_s00097g636) (Table S7) were significantly up-regulated at 10 h after exposure to NE. In summary, these proteins facilitate trap formation by increasing mitosis and cell proliferation.
The fungal cell wall is a complex structure composed of chitin, glucan and other polymers [38]. Traps are derived from specialized vegetative hyphae that have thicker and more robust cell walls than typical vegetative hyphae [39]. A large number of genes related to cell wall biosynthesis were identified in the A. oligospora genome. Proteomics analysis revealed that the expression levels of several proteins involved in cell wall synthesis, e.g. glycosyltransferase (AOL_s00097g268), glycosidase (AOL_s00083g375), phosphoglucomutase (AOL_s00054g87) (Table S7), were significantly up-regulated during trap formation (10 h). In addition, increased expressions at the mRNA level for 1,3-beta-glucan synthase (AOL_s00054g491), chitin synthases (AOL_s00210g37, AOL_s00075g119 and AOL_s00078g76) and glucosamine 6-phosphate synthetase (AOL_s00076g99) were also detected using qPCR (Table S7). Increased expression of those enzymes required for the biosynthesis of glucan, chitin and glycan undoubtedly facilitate new cell wall formation during trap formation.
Trap cells in nematode-trapping fungi contain numerous dense bodies that are related to peroxisomes [2], [5], [13]. A number of genes encoding peroxisomal biogenesis factors and related proteins were found (Table S9) in the A. oligospora genome. While no significant change in protein level was found in cells at 10 h after exposure to NE, the qPCR analyses of 10 selected peroxisomal genes revealed that four peroxisomal genes were significantly up-regulated during trap formation (Table S7). A previous study demonstrated that in M. haptotylum, the transcription level of a peroxisomal membrane protein (Peroxin-11) was up-regulated by 72% in nematode trapping knobs when compared to saprotrophic mycelia [13]. These data suggest that up-regulation of peroxisomal proteins is likely involved in the formation of dense bodies in trap cells in A. oligospora.
Glycogen is present in most fungi as a carbon storage molecule. The expression levels of glycogen phosphorylase (AOL_s00109g17) and hexokinase (AOL_s00112g89) were significantly up-regulated during A. oligospora trap formation (10 h) (Table S7), indicating accelerated glycogen degradation and glycolysis. In M. haptotylum, the glycogen phosphorylase (gph1) gene was also reported to be up-regulated during knob formation [13]. Moreover, the expression level of glycerol 3-phosphate dehydrogenase (AOL_s00054g748), a key enzyme in the synthesis of glycerol from dihydroxyacetone phosphate, increased by 5.7-fold during the formation of traps (Table S7). Accumulation of glycerol in response to NE was also confirmed experimentally (Figure S5). In contrast, degradation of fatty acids that generate other substrates for glycerol synthesis was down-regulated as indicated by the decreased expression of several beta-oxidation enzymes such as thiolase (AOL_s00210g122) and 3-hydroxyacyl-CoA dehydrogenase (AOL_s00110g113) (Table S7). Based on the proteomic and qPCR analyses, glycerol production during trap formation in A. oligospora likely occurred via the glycogen degradation and glycolysis pathways. In appressorium-forming fungi, such as M. grisea, glycerol plays a key role in generating hydrostatic turgor pressure to breach the host cuticles by a mechanical force [40]. The results here are consistent with the hypothesis that glycerol accumulation in A. oligospora likely function similarly to that of M. grisea during the penetration of nematode cuticle.
A previous study proposed that the capture of nematodes by A. oligospora was mediated by lectins located on the fungal cell surface [41]. An A. oligospora lectin (AOL) was subsequently identified [42]. However, AOL-deletion mutants showed similar saprophytic growth and nematode pathogenicity as their wild-type progenitor strain [42]. Our study revealed that seven genes encoding lectins with different sugar specificities were present in the A. oligospora genome (Table S10). Interestingly, none of the seven lectin genes, including AOL (AOL_s00080g288), showed any significant change in expression at either the mRNA or the protein level in response to NE.
The adhesive proteins comprising extracellular fibrils on trap cell surface can aid in nematode capturing [2], [5]. A total of 17 adhesion-associated protein-encoding genes (Table S10) were found in the A. oligospora genome. Our transcriptional analyses via qPCR revealed that the expressions of five adhesin encoding genes were up-regulated at 10 h (Table S7). One up-regulated adhesion gene, AOL_s00076g567, is a homolog of MAD1, an adhesin in the entomopathogenic fungus Metarhizium anisopliae that mediates its attachment to insects [43]. The other four adhesion proteins (AOL_s00043g50, AOL_s00007g5, AOL_s00210g231 and AOL_s00076g207) were up-regulated by 5.4-, 4.2-, 21.7 - and 1.7-fold, respectively, during the formation of traps (10 h) (Table S7). These data support previous findings that adhesions on trap surfaces are important for capturing nematodes.
The combined genomic, proteomic, and qPCR data suggest a model of nematode trap formation in A. oligospora (Figure 7). In this model, A. oligospora recognizes nematode signals and activates a diversity of downstream cellular processes. These processes include (i) increased cell proliferation and septum formation with enhanced cell wall biosynthesis and cytoskeleton assembling; (ii) enhanced glycerol synthesis and accumulation leading to increased intracellular turgor pressure in preparation for host colonization and penetration within the nascent trapping cells; (iii) formation of dense bodies possibly involving the peroxisome biogenesis; and (iv) synthesis of adhesive proteins on the surface of the trapping cells to enhance nematode capture. These complex and coordinated processes are fueled by energy and building blocks supplied from the glyoxylate and TCA cycles.

Discussion
Nematode trapping device is not only an important indicator of the switch from the saprophytic to the predacious lifestyle, but also a pivotal tool for capturing nematodes for nematode-trapping fungi [2], [4]. However, due to the lack of genome data of this group of fungi, there is currently limited information about the molecular mechanism of trap formation. Previous studies were mainly focused on the morphology and morphogenesis of traps. In this study, the major biological pathways, and the proteins and genes likely involved in trap formation in A. oligospora were first identified by proteomic and qPCR analyses. And a model on the formation of nematode trapping device in A. oligospora was proposed based on the combined genomic, proteomic, and qPCR data. However, the detailed spatial and temporal dynamics among the cellular processes, and their interactions during trap formation, remain to be elucidated.
A. oligospora is the first Orbiliomycete fungus to have its whole genome sequenced. Compared with other pathogenic and non-pathogenic fungi, A. oligospora contained abundant orphan genes not found in other sequenced fungi (53.64%) (Figure 3A), a result consistent with the phylogenomic analysis that A. oligospora is phylogenetically very distant from other sequenced Ascomycota (Figure 4). The A. oligospora specific genes may be related to its complex life-styles. Specifically, A. oligospora is not only a saprophyte, but also a nematode pathogen, a pathogen of other fungi, and a colonizer of plant roots [44]. The genes shared between A. oligospora and 10 other pathogenic and non-pathogenic ascomycete fungi (37.02%, Figure 3A) likely represent housekeeping genes for this group of organisms. Interestingly, A. oligospora share many more genes with pathogenic fungi than with non-pathogenic fungi. Those shared between A. oligospora and pathogens likely contribute to fungal pathogencity in general.
The distribution patterns of the pathogenicity-related gene families and putative PHI genes in different genome categories suggested that the ao-specific genes and the genes in the ao/pathogen category were more abundant than those in the ao/non-pathogen category. In fact, some pathogenicity-related gene families were completely absent in the ao/non-pathogen category. The genes specific to the ao/non-pathogen category are likely not involved in pathogenicity while the ao-specific genes may be very important for the pathogenicity of A. oligospora. Further analysis of PHI genes showed that carbohydrate-degrading enzymes (for example, polygalacturonase, xylanase and pectate lyase) represent a big proportion of ao-specific genes. Similarly, MFS transporter (11%), pectate lyase (6.8%) and short-chain dehydrogenase (6.8%) were the common genes specific to the ao/pathogens category.
Subtilisins and other pathogenicity-related genes play very important roles during infections [23], [34], [45]. As is typical for pathogens and saprobes of animals, the A. oligospora contains abundant pathogenicity genes encoding subtilisins and chitinases. Increasing evidences showed that increasing the copy number of virulence genes can improve the pathogenicity of pathogenic fungi. This strategy has been successfully used in A. oligospora [22] and several fungi used for biocontrol, such as M. anisopliae [46], Beauveria bassiana [47] and Trichoderma harzianum [48]. The qPCR analysis suggests that the proteases P12 and P186 likely play more important roles than PII during the infection process, and disruption of the gene P186 further confirmed that P186 is an important pathogenicity factor in A. oligospora. Therefore, increasing the copy numbers of P12 and P186 through genetic engineering may effectively improve the pathogencity of A. oligospora and other fungi. Moreover, no obvious phenotypic differences were found between the mutants and the wild strains, suggesting that the P186 may specifically work in pathogenicity without interfering with other metabolic and reproductive processes.
The nematode-trapping fungi can kill nematodes by producing secondary metabolites [32]. Several classes of secondary metabolite producing genes were found in the A. oligospora genome, suggesting that the potential repertoire of compounds is quite diverse as predicted. Some of these genes were not found in other closely related species (Figures S3 and S4) and could potentially contribute to producing novel metabolites. Further work may identify new nematicides from these secondary metabolite-producing genes in A. oligospora.
Attraction and recognition are the early steps during A. oligospora – nematodes interaction [49]. Nematodes are likely attracted by compounds released from the mycelia of nematode-trapping fungi [50]. However, the chemical attractant(s) involved in nematode-fungal recognition remain largely unknown. Recently, it was found that the nematoxic bacterium Pseudomonas aeruginosa could produce acylated homoserine lactones (AHL), the signal molecules in quorum sensing, as attractants for nematodes [51]. Interestingly, a homologous gene involved in biosynthesis of AHL was found in A. oligospora genome. Moreover, in our recent study, a “Trojan horse” mechanism of bacterial pathogenesis against nematodes was reported [52], in which the bacterium Bacillus nematocida B16 lures nematodes by emitting potent volatile organic compounds (VOCs), and seven VOCs are confirmed to lure nematodes, while the related genes had not been identified. These studies should help us identify the potential compounds involved in attraction and uncover the mechanism of recognition between the nematode-trapping fungi and nematodes.
Previous studies showed that lipid droplets accumulate in the trophic hyphae of A. oligospora at later stages of infection, and new vegetative mycelium developed from the trap that had originally captured the nematode when the lipid droplets disappeared [12]. Our results showed that several peroxisomal proteins as well as the key enzymes of the glyoxylate cycle were up-regulated during trap formation, which suggested that part of the nutrients released from the nematodes might first be converted to lipids by the fungus. The lipids were then degraded via the beta-oxidation pathway located on the peroxisome and further metabolized through the glyoxylate cycle to support growth of new vegetative hyphae. However, some of the enzymes involved in the beta-oxidation pathway were down-regulated during the early stage of trap formation. Further studies focusing on individual gene knockouts will be necessary to determine the roles of these enzymes in lipid metabolism during trap formation.
Fungi that are pathogenic to invertebrates, especially those targeting nematodes and insects, are of great importance for maintaining ecological balance in natural environments and for improving agricultural production [2], [53]. Here the first genome of this group of fungi was reported and the key features related to their pathogenicity were described. The information presented here on a nematophagous fungus should facilitate our understanding of these fascinating carnivorous fungi, and provide a basis to analyze the similarities and differences between nematophagous and other pathogenic fungi. These data also have important ramification for the practical development of improved biological control agents. Our proposed model for trap formation based on the comprehensive genomic, qPCR and proteomics analysis could serve as a roadmap for further investigating the molecule mechanism underlying the transition between saprophytic and predatory lifestyles in fungi.
Methods
Full methods and any associated references are available in the Supporting information file (Text S1). The major methods described in the file were listed as follows. 1) Strain and growth conditions. 2) Genome sequencing, assembly, prediction and analysis. 3) Comparative genome, evolution and gene family analyses. 4) RIP analysis. 5) QPCR analysis. 6) Trap formation and fungus-nematode interaction. 7) Two-dimensional gel electrophoresis (2-DE) analysis. 8) Matrix assisted laser desorption ionization/time of flight (MALDI-TOF) analysis. 9) Disruption of the gene p186.
Fungal strain
A. oligospora Fres. (ATCC24927) was purchased from the American Type Culture Collection (ATCC) and maintained on cornmeal agar (CMA). This fungus was originally isolated from soil in Sweden and provided to ATCC by Nordbring-Hertz B.
Genome sequencing, assembly and analysis
The A. oligospora was sequenced by the ABI 3730 Sanger sequencing platform to a depth of 2 ×. An additional 34.6 × sequencing data coverage was provided by the Roche 454 Genome Sequencer Titanium/FLX platforms. GS De Novo Assembler developed by Roche was used to assemble the sequencing data. Repetitive sequences in the genome assembly were identified by searching the Repbase database [54] using RepeatMasker and by de novo repetitive sequence search using RepeatModeler (http://www.repeatmasker.org/RepeatModeler.html).
Gene prediction and annotation
Ab initio gene prediction was performed on the genome assembly by Augustus, GlimmerHMM, and SNAP trained with transcript sequences of A. oligospora from this study, and by GeneMark-ES formulated for fungal genomes. A final set of gene models was selected by EvidenceModeler [55], combining ab initio gene predictions with supports by transcript alignments from A. oligospora and other fungal species. Predicted genes were annotated by BLAST searches against protein databases, and by InterProScan searches against protein domain databases.
Non-coding RNAs analysis
tRNAs were predicted by tRNAscan-SE [56]. Ribosomal RNAs were identified by a BLAST search with known rRNA modules of other fungal genomes. Other non-coding RNAs, including snRNAs and miRNAs, were predicted by searching the Rfam database [57] using Infernal (http://infernal.janelia.org).
Orthology and phylogenomic analysis
Predicted proteins in A. oligospora were compared with the predicted proteins of 10 sequenced fungal genomes. All proteins were searched against all other proteins in these genomes using BLASTP. The matches with E≤1e−5 and at least 30% sequence identity [58] over 60% of both protein lengths [15] were taken as homologous sequences. Bidirectional best hits (BBHs) [16] from the homologous sequences were taken as orthologous sequences. A total of 529 orthologous proteins were obtained and concatenated to infer the phylogenomic relationships among these taxa with PHYLIP [59] using different methods, including Neighbor-joining (NJ), Maximum parsimony (MP) and Maximum likelihood (ML).
Multiple gene family analysis
Multigene families were constructed from the homologous sequences based on single linkage transitive closure [15]. A total of 2882 of the 11479 genes were clustering into 789 multigene families. Several gene families related to fungal pathogenicity were manually selected based on previous knowledge and gene annotations. The members of the pathogenicity-related gene families were expanded via homology search followed by clustering based on single linkage transitive closure. In addition, pathogenicity-related genes were also predicted by a whole genome blast analysis against the PHI gene database [21].
Evolution analysis of pathogenicity-related gene families
The pathogenicity-related gene families of subtilisin, NRPS and PKS were selected to perform phylogenetic analysis. NJ tree was obtained using MEGA 4.1 [60] using bootstrap analysis with 1,000 replicates. MP tree was constructed using PAUP*4.0b8 [61] with a heuristic search (initial trees were obtained by 100 replicates with random addition and branch swapping with the TBR algorithm). Non-parameter bootstrap (1,000 replicates) was performed to assess the support level for each node on the MP trees. ML tree was obtained using PHYML version 3.0 [62]. Bootstrap analysis with 100 replicates was applied.
Repeat-induced point mutation (RIP) analysis
The RIP indices, TpA/ApT and (CpA+TpG)/(ApC+GpT), were determined to detect RIP relics [20], [63], [64]. The AT content and RIP indices in all sequences were calculated. Windows of 500-bp and 200-bp with 100-bp shifts were performed separately, for the whole genome as well as individually for the coding regions, non-coding regions, exons, introns, multigene families, and repetitive sequences. RIP regions were detected in the 200-bp windows with 100-bp shifts with TpA/ApT ≥0.89 and (CpA+TpG)/(ApC+GpT) ≤1.03.
Quantitative PCR
Quantitative PCR was conducted with 2 µl reverse transcribed product in a 7300 Real-Time PCR system (Applied Biosystems, California, USA) using Power SYBR Green PCR Master Mix (Applied Biosystems). 18S rDNA gene was used as the internal control. Fold changes were calculated using the formula 2−(ΔΔCt), where ΔΔCt is ΔCt (treatment)-ΔCt (control), ΔCt is Ct (target gene) - Ct (18S), and Ct is the threshold cycle (User's Manual for ABI 7300 Real-Time PCR System).
Proteomic analysis
Total proteins were extracted by following the method by Fernández-Acero et al. [65]. Approximately 1 mg of protein samples was brought to a final volume of 170 µl for 2-D analysis. Protein spots were manually excised from Coomassie Brilliant Blue G-250 (BBI) stained gels. Proteins were digested by trypsin for 16–20 h at 37°C. Peptides were analyzed by MALDI-TOF using a 4700 series Proteomics Analyzer (Applied Biosystems). Proteins were identified by searching PMF generated against a local database using Mascot algorithm of the GPS software.
GenBank accession numbers
The Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession ADOT00000000. Other genes, accession nos. PII, X94121/AOL_s00076g4; spr1, AB120125; Cdc37, Z47813/AOL_s00043g594; Mih1, JO4846/AOL_s00176g31; Cdc28/Cdk1, Z36029; Wee1, X73966; Peroxin-11 (PEX11), EF419889; gph1, AY635194; AOL, X97093; Mad1, DQ438337/AOL_s00076g567.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. PramerD 1964 Nematode-trapping fungi. Science 144 382 388
2. Nordbring-HertzBJanssonHBTunlidA 2006 Nematophagous fungi. In Encyclopedia of life sciences. Chichester John Wiley & Sons, Ltd
3. SchmidtARDörfeltHPerrichotV 2007 Carnivorous fungi from Cretaceous Amber. Science 318 1743
4. YangYYangECAnZQLiuXZ 2007 Evolution of nematode-trapping cells of predatory fungi of the Orbiliaceae based on evidence from rRNA-encoding DNA and multiprotein sequences. Proc Natl Acad Sci U S A 104 8379 8384
5. Nordbring-HertzB 2004 Morphogenesis in the nematode-trapping fungus Arthrobotrys oligospora-an extensive plasticity of infection structures. Mycologist 18 125 133
6. MoMHChenWMSuHYZhangKQDuanCQ 2005 Heavy metal tolerance of nematode-trapping fungi in Lead-polluted soils. Appl Soil Ecol 31 11 19
7. PfisterDHLiftikME 1995 Two Arthrobotrys anamorphs from Orbilia auricolor. Mycologia 87 684 688
8. CayrolJCFrankowskiJPLanieceAd'HardemareGTalonJP 1978 Contre les nématodes en champignonniere. Mise an point d'une méthode de lutte biologique a l'aide d'un hyphomycete predateur: Arthrobotrys robustus souche antipolis' (Royal 300). Rev Hortie 184 23 30.y
9. CayrolJC 1983 Lutte biologique contre les Meloidogyne au moyen d'Arthrobotrys irregularis (in French). Revue Nèmatol 6 265 273
10. LiYanKevinDHJeewonRCaiLVijaykrishnaD 2005 Phylogenetics and evolution of nematode-trapping fungi (Orbiliales) estimated from nuclear and protein coding genes. Mycologia 97 1034 1046
11. VeenhuisMNordbring-HertzBHarderW 1985 An electron-microscopical analysis of capture and initial stages of penetration of nematodes by Arthrobotrys oligospora. Antonie van Leeuwenhoek 51 385 398
12. VeenhuisMVan WijkCWyssUNordbring-HertzBHarderW 1989 Significance of electron dense microbodies in trap cells of the nematophagous fungus Arthrobotrys oligospora. Antonie Van Leeuwenhoek 56 251 261
13. AhrénDTholanderMFeketeCRajashekarBFrimanE 2005 Comparison of gene expression in trap cells and vegetative hyphae of the nematophagous fungus Monacrosporium haptotylum. Microbiology 151 789 803
14. FeketeCTholanderMRajashekarBAhrénDFrimanE 2008 Paralysis of nematodes shifts in the transcriptome of the nematode-trapping fungus Monacrosporium haptotylum during infection of Caenorhabditis elegans. Environ Microbiol 10 364 375
15. GalaganJECalvoSEBorkovichKASelkerEUReadND 2003 The genome sequence of the filamentous fungus Neurospora crassa. Nature 422 859 868
16. OverbeekRFonsteinMD'SouzaMPuschGDMaltsevN 1999 The use of gene clusters to infer functional coupling. Proc Natl Acad Sci U S A 96 2896 2901
17. FitzpatrickDALogueMEStajichJEButlerG 2006 A fungal phylogeny based on 42 complete genomes derived from supertree and combined gene analysis. BMC Evol Biol 6 99
18. RobbertseBReevesJBSchochCLSpataforaJW 2006 A phylogenomic analysis of the Ascomycota. Fungal Genet Biol 43 715 725
19. CuomoCAGüldenerUXuJRTrailFTurgeonBG 2007 The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science 317 1400 1402
20. MargolinBSGarrett-EngelePWStevensJNFritzDYGarrett-EngeleC 1998 A methylated Neurospora 5S rRNA pseudogene contains a transposable element inactivated by repeat-induced point mutation. Genetics 149 787 797
21. WinnenburgRBaldwinTKUrbanMRawlingsCKohlerJ 2006 PHI-base: a new database for pathogen host interactions. Nucleic Acids Res 34 D459 D464
22. ÅhmanJJohansonTOlssonMPuntPJvan den HondelCAMJJ 2002 Improving the pathogenicity of a nematode-trapping fungus by genetic engineering of a subtilisin with nematotoxic activity. Appl Environ Microbiol 68 3408 3415
23. YangJKTianBYLiangLMZhangKQ 2007 Extracellular enzymes and the pathogenesis of nematophagous fungi. Appl Microbiol Biotechnol 75 21 31
24. LiJYuLYangJKDongLQTianBY 2010 New insights into the evolution of subtilisin-like serine protease genes in Pezizomycotina. BMC Evol Biol 10 68
25. LiangLMMengZHYeFPYangJKLiuSQ 2010 The crystal structures of two cuticle-degrading proteases from nematophagous fungi and their contribution to infection against nematodes. FASEB J 24 1391 1400
26. TunlidARosenSEkBRaskL 1994 Purification and characterization of an extracellular serine protease from the nematode-trapping fungus Arthrobotrys oligospora. Microbiology 140 1687 1695
27. ZouCGTuHHLiuXYTaoNZhangKQ 2010 PacC in the nematophagous fungus Clonostachys rosea controls virulence to nematodes. Environ Microbiol 12 1868 1877
28. ÅhmanJEkBRaskLTunlidA 1996 Sequence analysis and regulation of a gene encoding a cuticle-degrading serine protease from the nematophagous fungus Arthrobotrys oligospora. Microbiology 142 1605 1616
29. KandaSAimiTKanoSIshiharaSKitamotoY 2008 Ambient pH signaling regulates expression of the serine protease gene (spr1) in pine wilt nematode-trapping fungus, Monacrosporium megalosporum. Microbiol Res 163 63 72
30. ColotHVParkGTurnerGERingelbergCCrewCM 2006 A high-throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc Natl Acad Sci U S A 103 10352 10357
31. SoanesDMRichardsTATalbotNJ 2007 Insights from sequencing fungal and oomycete genomes: what can we learn about plant disease and the evolution of pathogenicity? Plant Cell 19 3318 3326
32. LiGHZhangKQXuJPDongJYLiuYJ 2007 Nematicidal substances from fungi. Recent Pat Biotechnol 1 212 233
33. ParkJLeeSChoiJAhnKParkB 2008 Fungal cytochrome P450 database. BMC Genomics 9 402
34. IdnurmAHowlettBJ 2001 Pathogenicity genes of phytopathogenic fungi. Mol Plant Pathol 2 241 255
35. LorenzMCFinkGR 2001 The glyoxylate cycle is required for fungal virulence. Nature 412 83 86
36. LiangJFantesP 2007 The Schizosaccharomyces pombe Cdc7 protein kinase required for septum formation is a client protein of Cdc37. Eukaryot cell 6 1089 1096
37. PalGParazMTZKelloggDR 2008 Regulation of Mih1/Cdc25 by protein phosphatase 2A and casein kinase 1. J Cell Biol 180 931 945
38. AdamsDJ 2004 Fungal cell wall chitinases and glucanases. Microbiology 150 2029 2035
39. HigginsMLPramerD 1967 Fungal morphogenesis: Ring formation and closure by Arthrobotrys dactyloides. Science 155 345 346
40. ThinesEWeberRWTalbotNJ 2000 MAP kinase and protein kinase A-dependent mobilization of triacylglycerol and glycogen during appressorium turgor generation by Magnaporthe grisea. Plant Cell 12 1703 1718
41. Nordbring-HertzBMattiassonB 1979 Action of a nematode-trapping fungus shows lectin-mediated host-microorganism interaction. Nature 281 477 479
42. BaloghJTunlidARosénS 2003 Deletion of a lectin gene does not affect the phenotype of the nematode-trapping fungus Arthrobotrys oligospora. Fungal Genet Biol 39 128 135
43. WangCSSt LegerRJ 2007 The MAD1 adhesin of Metarhizium anisopliae links adhesion with blastospore production and virulence to insects, and the MAD2 adhesin enables attachment to plants. Eukaryot Cell 6 808 816
44. BordalloJJLopez-LlorcaLVJanssonHBSalinasJPersmarkL 2002 Colonization of plant roots by egg-parasitic and nematode-trapping fungi. New Phytol 154 491 499
45. YangJKGanZWLouZYTaoNMiQL 2010 Crystal structure and mutagenesis analysis of a chitinase CrChi1 from the nematophagous fungus Clonostachys rosea in complex with the inhibitor caffeine. Microbiology 156 3566 3574
46. St LegerRJJoshiLBidochkaMJRobertsDW 1996 Construction of an improved mycoinsecticide over-expressing a toxic protease. Proc Natl Acad Sci U S A 93 6349 6354
47. FangWGLengBXiaoYHJinKMaJC 2005 Cloning of Beauveria bassiana chitinase gene Bbchit1 and its application to improve fungal strain virulence. Appl Environ Microbiol 71 363 370
48. FloresAChetIHerrera-EstrellaA 1997 Improved biocontrol activity of Trichoderma harzianum by over-expression of the proteinase-encoding gene prb1. Curr Genet 31 30 37
49. SaxenaGDayalRMukerjiKG 1987 Interaction of nematodes with nematophagus fungi: induction of trap formation, attraction and detection of attractants. FEMS Microbiol Lett 45 319 327
50. BalanJKrižkováLNemecPVollekV 1974 Production of nematode-attracting and nematicidal substances by predacious fungi. Folia Microbiol 19 512 519
51. BealeELiGTanMWRumbaughKP 2006 Caenorhabditis elegans senses bacterial autoinducers. Appl Environ Microbiol 72 5135 5137
52. NiuQHHuangXWZhangLXuJPYangDM 2010 A Trojan horse mechanism of bacterial pathogenesis against nematodes. Proc Natl Acad Sci U S A 107 16631 16636
53. WangCSt LegerRJ 2007 A scorpion neurotoxin increases the potency of a fungal insecticide. Nat Biotechnol 25 1455 1456
54. JurkaJKapitonovVVPavlicekAKlonowskiPKohanyO 2005 Repbase Update, a database of eukaryotic repetitive elements. Cytogent Genome Res 110 462 467
55. HaasBJSalzbergSLZhuWPerteaMAllenJE 2008 Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol 9 R7
56. LoweTMEddySR 1997 tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25 955 964
57. Griffiths-JonesSMoxonSMarshallMKhannaAEddySR 2005 Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res 33 D121 D124
58. RostB 1999 Twilight zone of protein sequence alignments. Protein Eng 12 85 94
59. FelsensteinJ 1989 PHYLIP-Phylogeny inference package (Version 3.2). Cladistics 5 164 166
60. TamuraKDudleyJNeiMKumarS 2007 MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol 24 1596 1599
61. SwoffordD 2002 PAUP*: phylogenetic analysis using parsimony (*and other methods). Sunderland, MA, USA Sinauer Associates Inc
62. GuindonSGascuelO 2003 A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52 696 704
63. HaneJKOliverRP 2008 RIPCAL: a tool for alignment-based analysis of repeat-induced point mutations in fungal genomic sequences. BMC Bioinformatics 9 478
64. WattersMKRandallTAMargolinBSSelkerEUStadlerDR 1999 Action of repeat-induced point mutation on both strands of a duplex and on tandem duplications of various sizes in Neurospora. Genetics 153 705 714
65. Fernández-AceroFJJorgeICalvoEVallejoICarbúM 2007 Proteomic analysis of phytopathogenic fungus Botrytis cinerea as a potential tool for identifying pathogenicity factors, therapeutic targets and for basic research. Arch Microbiol 187 207 215
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 9
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- HTLV-1 Propels Thymic Human T Cell Development in “Human Immune System” Rag2 gamma c Mice
- Hostile Takeover by : Reorganization of Parasite and Host Cell Membranes during Liver Stage Egress
- Exploiting and Subverting Tor Signaling in the Pathogenesis of Fungi, Parasites, and Viruses
- A Viral Ubiquitin Ligase Has Substrate Preferential SUMO Targeted Ubiquitin Ligase Activity that Counteracts Intrinsic Antiviral Defence