
Rhoptry Kinase ROP16 Activates STAT3 and STAT6 Resulting in Cytokine Inhibition and Arginase-1-Dependent Growth Control
The ROP16 kinase of Toxoplasma gondii is injected into the host cell cytosol where it activates signal transducer and activator of transcription (STAT)-3 and STAT6. Here, we generated a ROP16 deletion mutant on a Type I parasite strain background, as well as a control complementation mutant with restored ROP16 expression. We investigated the biological role of the ROP16 molecule during T. gondii infection. Infection of mouse bone marrow-derived macrophages with rop16-deleted (ΔROP16) parasites resulted in increased amounts of IL-12p40 production relative to the ROP16-positive RH parental strain. High level IL-12p40 production in ΔROP16 infection was dependent on the host cell adaptor molecule MyD88, but surprisingly was independent of any previously recognized T. gondii triggered pathway linking to MyD88 (TLR2, TLR4, TLR9, TLR11, IL-1ß and IL-18). In addition, ROP16 was found to mediate the suppressive effects of Toxoplasma on LPS-induced cytokine synthesis in macrophages and on IFN-γ-induced nitric oxide production by astrocytes and microglial cells. Furthermore, ROP16 triggered synthesis of host cell arginase-1 in a STAT6-dependent manner. In fibroblasts and macrophages, failure to induce arginase-1 by ΔROP16 tachyzoites resulted in resistance to starvation conditions of limiting arginine, an essential amino acid for replication and virulence of this parasite. ΔROP16 tachyzoites that failed to induce host cell arginase-1 displayed increased replication and dissemination during in vivo infection. We conclude that encounter between Toxoplasma ROP16 and the host cell STAT signaling cascade has pleiotropic downstream effects that act in multiple and complex ways to direct the course of infection.
Published in the journal:
. PLoS Pathog 7(9): e32767. doi:10.1371/journal.ppat.1002236
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002236
Summary
The ROP16 kinase of Toxoplasma gondii is injected into the host cell cytosol where it activates signal transducer and activator of transcription (STAT)-3 and STAT6. Here, we generated a ROP16 deletion mutant on a Type I parasite strain background, as well as a control complementation mutant with restored ROP16 expression. We investigated the biological role of the ROP16 molecule during T. gondii infection. Infection of mouse bone marrow-derived macrophages with rop16-deleted (ΔROP16) parasites resulted in increased amounts of IL-12p40 production relative to the ROP16-positive RH parental strain. High level IL-12p40 production in ΔROP16 infection was dependent on the host cell adaptor molecule MyD88, but surprisingly was independent of any previously recognized T. gondii triggered pathway linking to MyD88 (TLR2, TLR4, TLR9, TLR11, IL-1ß and IL-18). In addition, ROP16 was found to mediate the suppressive effects of Toxoplasma on LPS-induced cytokine synthesis in macrophages and on IFN-γ-induced nitric oxide production by astrocytes and microglial cells. Furthermore, ROP16 triggered synthesis of host cell arginase-1 in a STAT6-dependent manner. In fibroblasts and macrophages, failure to induce arginase-1 by ΔROP16 tachyzoites resulted in resistance to starvation conditions of limiting arginine, an essential amino acid for replication and virulence of this parasite. ΔROP16 tachyzoites that failed to induce host cell arginase-1 displayed increased replication and dissemination during in vivo infection. We conclude that encounter between Toxoplasma ROP16 and the host cell STAT signaling cascade has pleiotropic downstream effects that act in multiple and complex ways to direct the course of infection.
Introduction
Pathogens living in an intracellular compartment benefit from being able to parasitize their host cell for nutrients, but they are faced with the challenge of surviving within a potentially hostile environment. This is particularly the case for microorganisms that infect cells of innate immunity such as macrophage/monocytes and dendritic cells because these cells possess potent microbial sensing and killing machinery. Many of the mechanisms that viral and bacterial pathogens use to manipulate host cell signaling pathways are well characterized, but less is known about how intracellular eukaryotic pathogens mechanistically interact with host cell signal transduction [1], [2], [3].
For one intracellular protozoan, Toxoplasma gondii, this is changing. Toxoplasma is an extremely successful microorganism as evidenced by the fact that it infects up to 50% of the human population worldwide and is also a common parasite in domestic and wild animals [4]. Within the host there is evidence that Toxoplasma preferentially targets dendritic cells, monocyte/macrophage lineage cells and neutrophils [5], [6], [7]. Infection is normally asymptomatic, and is characterized by widespread dissemination of replicative tachyzoites followed by formation of quiescent cysts in the central nervous system and skeletal muscle tissues that persist for the lifetime of the host with no overt ill effects. Yet, T. gondii is an important opportunistic pathogen insofar as infection in immunocompromised patients may have life-threatening consequences, and in utero infection can lead to major defects in the fetus [8], [9]. The parasite is normally controlled by a strong Type 1 cytokine response characterized by high-level production of cytokines such as IL-12p70, TNF-α and IFN-γ [10]. While the latter cytokines are important in resistance to infection, this proinflammatory response must be tightly controlled to avoid immunopathology that can otherwise lead to host death.
It is well established that Toxoplasma actively interferes with host cell signaling during intracellular infection of cells such as macrophages (MØ) [11]. The parasite is capable of blocking pathways leading to apoptosis and interfering with proinflammatory responses initiated by signaling through the IFN-γ receptor and Toll-like receptors (TLR). How these suppressive effects occur is less well known, although induction of suppressor of cytokine synthesis (SOCS)-1, activation of phosphatidylinositol (PI)-3 kinase signaling, inhibition of NFκB activation, and blocking chromatin remodeling have each been implicated in parasite-mediated interference with host cell signaling [12], [13], [14], [15], [16].
Recently, the signal transducer and activator of transcription (STAT) signaling pathway has emerged as a major target of exploitation by T. gondii. Infection of mouse bone marrow-derived MØ induces rapid and sustained activation of STAT3, a transcription factor through which the anti-inflammatory cytokine IL-10 exerts its function [17]. Additionally, STAT3-negative MØ are much less sensitive to parasite-mediated inhibition of TLR signaling.
Parasite strain type is a critical determinant of STAT3 activation. Of the three predominant strain types, high virulence Type I and low virulence Type III parasites are potent activators of STAT3, whereas low virulence Type II strains are incapable of sustained activation. A major advance in our understanding of how Toxoplasma interacts with the STAT signaling machinery came from the results of genetic crosses between T. gondii strain types. In these studies the rop16 locus, contained on chromosome VIIb, emerged as a determinant of activation of both STAT3 and STAT6 [18], [19].
The rop16 locus encodes rhoptry protein ROP16. This molecule, along with several other proteins, is contained within rhoptries, which are apically associated organelles that discharge their contents during invasion. While many rhoptry proteins localize to the parasitophorous vacuole membrane, some are injected into the host cell cytoplasm associated with empty (e)-vacuoles [20]. The ROP16 molecule is discharged into the cytosol during invasion, and it rapidly translocates to the host cell nucleus via a nuclear translocation sequence [19]. Although originally identified as a putative serine-threonine kinase, recent biochemical studies have shown that ROP16 from Type I parasites is capable of directly catalyzing tyrosine phosphorylation of both STAT3 and STAT6 [21], [22].
Here, we used reverse genetics to generate ROP16 negative (ΔROP16) Type 1 parasites as well as ROP16 complementation mutants (ΔROP16 : 1), and we studied the biological effects of deleting and re-inserting this gene during in vitro and in vivo infection. We report that ROP16 deletion converts Type I tachyzoites from low to high inducers of IL-12p40, and simultaneously abrogates the suppressive effect of Toxoplasma on TLR signaling. We show that ROP16 deletion eliminates the parasite's ability to block nitric oxide production mediated by signaling through the IFN-γ receptor. Yet, paradoxically, ΔROP16 tachyzoites displayed increased replication during in vivo and in vitro infection. We traced this activity to ROP16-dependent STAT6-mediated induction of arginase-1, an enzyme that degrades host cell arginine, which is required for both the production of nitric oxide by host cell inducible nitric oxide synthase, as well as being an essential amino acid nutrient the parasite requires for intracellular replication.
Results
An inhibitor of JAK2 blocks parasite-induced STAT3 phosphorylation
Signal transduction resulting in STAT3 tyrosine phosphorylation involves upstream Janus kinase (JAK) activation triggered by cytokine receptor engagement. To determine whether Toxoplasma infection stimulates JAK1 or JAK2 phosphorylation, MØ were infected with RH strain tachyzoites, and cell lysates were subjected to immunoprecipitation with anti-phosphotyrosine antibody followed by Western blotting with antibodies specific for JAK1, JAK2 and STAT3 molecules. As shown in Fig. S1A, infection triggered tyrosine phosphorylation of both JAK1 and JAK2, in addition to STAT3.
The embryonic lethality of deleting JAK1 and JAK2 precluded us from examining the role of these molecules in parasite-triggered STAT3 activation, although experiments with Jak3−/− and Tyk2−/− MØ revealed that these kinases were not involved (Fig. S1B). However, we found that JAK inhibitor I, a relatively nonspecific reagent, partially blocked parasite-mediated STAT3 activation (Fig. S1C). Furthermore, Toxoplasma-induced STAT3 phosphorylation was almost completely blocked by a JAK2-specific inhibitor (Fig. S1D). In contrast, STAT3 phosphorylation stimulated by recombinant IL-6 was largely unaffected by the JAK2 inhibitor. This result is consistent with data suggesting that IL-6-mediated STAT3 activation involves JAK1 and JAK3 molecules [23]. While the most straightforward conclusion of these results is that parasite-triggered JAK2 activation mediates STAT3 phosphorylation, this interpretation must be treated with caution in light of recent evidence that ROP16 directly acts on STAT molecules in a manner sensitive to JAK chemical inhibition [21].
Generation of ROP16 deletion parasite (ΔROP16) and ROP16 complemented strain (ΔROP16 : 1)
To examine the biological role of ROP16 regarding STAT activation and modification of host responses, we generated Type 1 parasites lacking expression of this molecule. The ROP16 coding region was targeted and deleted in the Type I RH strain KU80 knockout background [24] (Fig. S2A). PCR assays performed on MPA-resistant clones validated efficient isolation of ROP16 knockouts (ΔROP16) as assessed by deletion of the ROP16 coding region (Fig. S2B), as well as by the correct targeted integration of the HXGPRT marker into the deleted ROP16 locus in each MPA-resistant clone (Fig. S2C, D). The ΔROP16 strain was then complemented with a single copy of a functional allele of ROP16 from type I RH by replacement of HXGPRT at the ROP16-deleted locus to generate strain ΔROP16 : 1 (Fig. S2E). The ΔROP16 : 1 complemented strain exhibited the expected genotype at the ROP16 locus (Fig. S2F, G, H).
ROP16 deletion attenuates STAT3 tyrosine phosphorylation
We examined phosphorylation kinetics in MØ infected with either wild-type or ROP16 knockout (ΔROP16) or complemented (ΔROP16 : 1) parasites (Fig. 1A). As expected, wild-type RH strain tachyzoites induced rapid and sustained STAT3 tyr705 (pY-) phosphorylation. In contrast, MØ infected with ΔROP16 parasites displayed early STAT3 tyrosine phosphorylation, but this response was not sustained and phosphorylation levels dropped to background within 1.5 h of infection. Re-introduction of the rop16 gene into ΔROP16 knockout parasites restored sustained STAT3 activation in the ΔROP16 : 1 strain (Fig. 1A). Because initial STAT3 phosphorylation is clearly independent of ROP16, but prolonged STAT3 activation requires ROP16, we conclude that activation is a biphasic process mediated initially by a host or parasite kinase independently of ROP16. Yamamoto et al. (22) did not previously report transient ROP16-independent activation of STAT3, most likely because samples were not analyzed prior to 3 hr post-infection. Phosphorylation at the ser727 residue is required for full activation of STAT3 [25]. Although this response was more difficult to detect, we found that parasite-mediated ser727 STAT3 phosphorylation did not require ROP16 expression (Fig. S3).

Because ROP16 deletion did not completely abrogate STAT3 tyrosine phosphorylation, we examined whether the active transcription factor translocated to the nucleus during infection with ROP16 knockout parasites. Nuclear extracts were prepared from infected MØ and probed for total or pY-STAT3. As demonstrated in Fig. 1B, cells infected with wild-type parasites displayed rapid nuclear accumulation of pY-STAT3. In the absence of ROP16, we also detected rapid accumulation of phosphorylated STAT3 that was greatly reduced by 1.5 hr post-infection even though total STAT3 levels remain elevated. In the complemented ΔROP16 : 1 strain, levels of phosphorylated STAT3 were maintained in a manner similar to infection with parental parasites. These data are consistent with those of others showing continuous STAT3 shuttling from cytoplasm to nucleus independent of phosphorylation status [26].
ROP16 down-modulates IL-12p40 production in infected cells
We, and others, have shown that Type II Toxoplasma strains, including PTG and ME49, induce higher levels of the IL-12p40 compared to infection with Type I strains such as RH [27], [28]. Genetic segregation analysis of crosses between parasite strains identified the rop16 locus as a determinant of IL-12 induction [19]. Therefore, we tested whether deletion of ROP16 would convert the parasite into a strong IL-12 inducer, as would be predicted from the forward genetics studies. As shown in Fig. 2, at 24 hr post-infection parental RH strain tachyzoites induced only low levels of IL-12. In contrast, deletion of ROP16 converted the parasite into a strong IL-12 inducer. Confirming a role for the rop16 gene in this response, complemented ΔROP16 : 1 parasites induced low levels of IL-12 similar to wild-type tachyzoites.

Because the common adaptor of Toll-like receptor (TLR) signaling, MyD88, has been implicated in immunity to T. gondii and in particular in IL-12 MØ responses during Type II infection [29], [30], [31], we examined whether ΔROP16-mediated IL-12p40 production was dependent upon this molecule. As shown in Fig. 3, the ability of ΔROP16 tachyzoites to induce high-level IL-12 production was strongly dependent on MyD88. Previous reports have provided evidence that TLR2 and TLR4 are involved in innate recognition of tachyzoites, suggesting that these receptors might mediate MyD88-dependent IL-12 production that occurs in the absence of ROP16 [32], [33]. However, ΔROP16 parasites induced equivalent levels of IL-12p40 in both wild-type and TLR2/4 double-knockout MØ (Fig. 3). Similarly, TLR9 and TLR11 have been implicated in innate immune recognition of Toxoplasma [34], [35], but lack of these molecules had no effect on high-level IL-12 production induced by parasites lacking ROP16 (Fig. 3). Finally, both IL-1β and IL-18 signaling involves MyD88 and it was therefore possible that autocrine production of these cytokines resulted in ΔROP16-mediated IL-12 release. However, MØ lacking caspase-1 (which is required for production of bioactive forms of these cytokines) also produced high amounts of IL-12p40 after infection with ΔROP16 parasites (Fig. 3). In these experiments, littermates served as WT controls and cells were obtained from mice at different facilities, likely contributing to differences on overall cytokine production from strain to strain. Nevertheless, we can conclude that parasite-induced IL-12 occurs independently of these TLR, or alternatively that in the absence of any single TLR, others can functionally substitute.

The MyD88 dependent IL-12 production we observed in the absence of ROP16 recalls previous observations of MyD88 dependent high-level IL-12 production in Type II parasite strain-infected cells [27]. One possibility that would explain the effects of ROP16 on down-regulating IL-12 production is that this rhoptry kinase might control IL-10 production. However, IL-10 was not detected in MyD88−/− MØ culture supernatants infected with ΔROP16 tachyzoites (data not shown). In addition, we previously demonstrated that IL-10 does not play a role in the inhibition of IL-12 production by WT RH parasites [17].
Deletion of ROP16 abrogates the ability of Toxoplasma to actively inhibit proinflammatory responses
Previously, we found that infection with Type I RH tachyzoites inhibited MØ responses to LPS and other TLR ligands, and we linked this inhibition phenotype to STAT3 activation [17]. Therefore, we examined whether ROP16 null parasites were defective in their ability to inhibit LPS-induced cytokines. In these experiments, MØ were infected followed 2 hr later by addition of LPS. Six hr after LPS stimulation (at a time prior to induction of IL-12 by parasites themselves) supernatants were collected for ELISA. MØ cultured in medium alone followed by LPS stimulation produced robust amounts of IL-12p40 and TNF-α (Fig. 4A and B, respectively). While cells infected with wild-type RH tachyzoites displayed a defective ability to respond to TLR signaling, ROP16-deleted parasites lost their ability to suppress LPS-induced cytokine production. However, re-introduction of the rop16 gene restored the parasites' ability to strongly suppress LPS-induced IL-12p40 and TNF-α production (Fig. 4A and B). Similar inhibitory effects of the RH strain on TLR-dependent stimulation occur during infection of DC [5], [36]. In contrast, ROP16 negative parasites failed to display this strong inhibitory effect during infection of DC (data not shown).
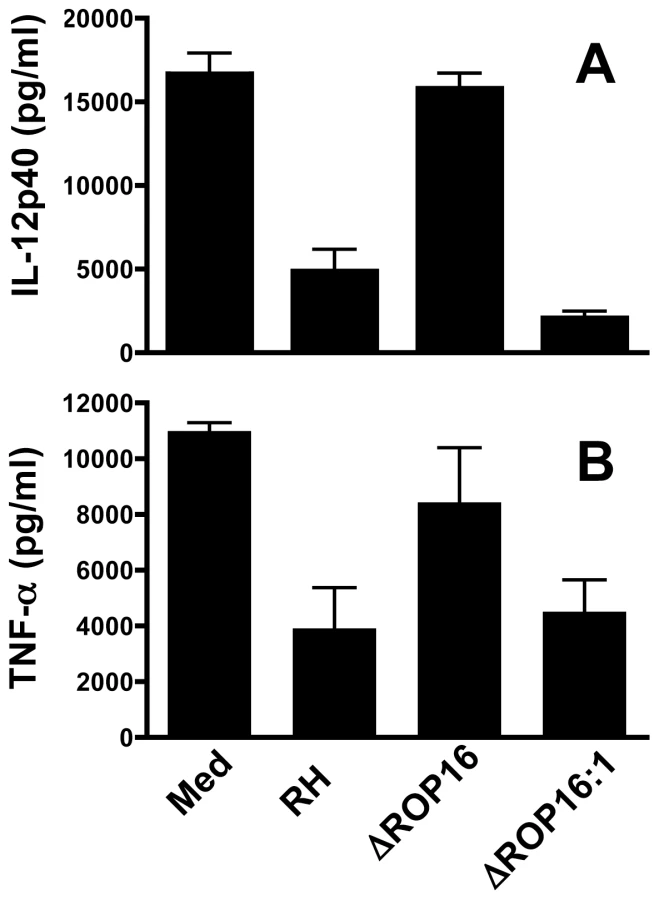
In addition to inhibition of signaling mediated through TLR pathways, Toxoplasma is capable of down-regulating IFN-γ-triggered signaling that leads to iNOS-dependent nitric oxide (NO) production [37]. Stimulation with IFN-γ alone failed to elicit significant amounts of NO in bone marrow-derived MØ, thioglycollate elicited MØ, or bone marrow-derived DC (data not shown). In addition, none of the parasite strains by themselves triggered iNOS or NO production. However, both microglial cells and astrocytes from noninfected neonatal mice produced NO after IFN-γ stimulation. When microglial cells were infected with RH tachyzoites and subjected to cytokine stimulation, NO production was severely limited (Fig. 5A). In marked contrast, infection with ROP16 null parasites failed to result in NO inhibition. IFN-γ stimulation of astrocytes resulted in lower amounts of NO, and this response was blocked during RH infection (Fig. 5B). Interestingly, when these cells were infected with ΔROP16 tachyzoites, IFN-γ-dependent NO production was even greater than that detected after stimulation with IFN-γ alone. Indeed, at priming concentrations of 4 nM and lower, IFN-γ failed to elicit detectable NO unless cells were infected with ΔROP16 parasites (Fig. 5B). Taken together, we conclude that ROP16 is a major molecule mediating inhibition of downstream targets of both TLR and IFN-γ receptor signaling pathways.

Because ΔROP16 parasites did not block and even potentiated NO production by astrocytes, and because NO is important in controlling the parasite in the brain, we examined tachyzoite survival in astrocyte-enriched cell cultures. Cells were subjected to priming with IFN-γ for 3 hr and then infected with RH or ΔROP16 parasites for 48 hr. Both parasite strains infected cells equally well and replicated in the absence of IFN-γ (data not shown). However, as shown in Fig. 5C, while RH survived and replicated within IFN-γ primed astrocytes, ΔROP16 parasites were significantly more susceptible to killing. ΔROP16 parasites displayed weak labeling with Ab to tachyzoite-specific surface antigen (SAG)-1 (p30) and the parasite form was compromised (arrows in right panel of Fig. 5C). The percent infection (Fig. 5D) and the number of tachyzoites per cell (Fig. 5E) were significantly less in ΔROP16-infected astrocytes compared to RH-infected cells.
ROP16 mediates STAT6-dependent arginase-1 induction
In addition to STAT3 activation, genetic linkage studies implicated the rop16 locus in activation of STAT6. A major downstream target of STAT6 signaling is arginase-1. This signaling cascade was of interest because arginase-1 competes with iNOS for the amino acid substrate arginine [38], [39], [40]. Therefore, we examined whether ROP16 could stimulate STAT6-dependent arginase-1 production. As shown in Fig. 6A, RH induced strong and sustained tyr641 phosphorylation of STAT6. In marked contrast, this response was completely absent in ΔROP16 mutant tachyzoites. However, in the rop16 complemented strain, STAT6 activation was restored (Fig. 6A). The absolute dependence of STAT6 phosphorylation on ROP16 differed from STAT3 activation, where only early ROP16-independent phosphorylation occurred (Fig. 1A and Fig. S4, which shows nuclear lysates from a single experiment blotted and probed for STAT3, then re-probed for STAT6). Interestingly, we detected nuclear accumulation of total STAT6, despite lack of detectable STAT6 tyrosine phosphorylation during ΔROP16 infection (Fig. S4). This may indicate phosphorylation-independent STAT nuclear accumulation as reported by others [41].

Paralleling the STAT6 phosphorylation results, wild-type and complemented parasites, but not ΔROP16 tachyzoites, induced arginase-1 synthesis, a response detectable by immunoblotting for arginase-1 protein within 6 hr of infection (Fig. 6A). Similarly, we detected induction of arginase-1 by immunofluorescence assay during RH but not ΔROP16 infection (Fig. 6B and C, respectively). Because the arginase-1 gene is controlled by STAT6 signaling, we examined whether parasite-induced induction of arginase-1 also depended upon STAT6. As shown in Fig. 6D, infection of STAT6−/− MØ with all three strains failed to induce arginase-1 protein, in contrast to infection of MØ from STAT6+/+ littermates in which we detected ROP16-dependent arginase-1 induction. We did not detect production of IL-4/IL-13 in these cultures making it very unlikely that arginase-1 induction was due to ROP16-dependent production of these cytokines (data not shown).
ROP16 modulates the availability of the essential amino acid arginine
Toxoplasma is known to be an arginine auxotroph, strictly relying on either host or exogenously supplied arginine for its replication and survival [42]. ROP16-dependent induction of arginase-1 suggested that parasites lacking this rhoptry molecule might be more resistant to conditions of arginine limitation. Therefore, we compared the ability of wild type and ΔROP16 parasites to invade and multiply within fibroblasts cultured in arginine-replete and arginine-deficient media. In normal arginine-replete medium, RH (Fig. 7A) and ΔROP16 (Fig. 7B) parasites displayed vigorous multiplication rates (quantitated in Fig. 7E). However, under arginine limiting conditions, RH replication was severely curtailed and only one cycle of replication occurred during the 38 hr growth assay (Fig. 7C and E). In striking contrast, replication of ΔROP16 parasites was markedly less affected by arginine starvation, with evidence of 4 or more cycles of division (Fig. 7D and E).

Although Toxoplasma is known as a microorganism that can infect virtually any kind of nucleated cell, the parasite is often found preferentially in macrophage/DC lineage cells during in vivo infection [7]. Therefore, we also examined the growth behavior of RH, ΔROP16 and ΔROP16 : 1 parasites under arginine high and low conditions in bone marrow-derived MØ. As shown in Fig. 7F, while all three strains replicated equivalently under arginine replete conditions, deletion of ROP16 resulted in a growth advantage to the parasites when arginine was present in limiting concentration.
Along similar lines, we collected mouse resident peritoneal cells, which are composed largely of CD11b+ resting MØ and carried out ex vivo infection with RH, ΔROP16 and ΔROP16 : 1 parasites under arginine high and low conditions. As in mouse bone marrow-derived MØ and human fibroblasts, in arginine limiting conditions deletion of ROP16 conferred a significant growth advantage on parasites as measured by overall average numbers of tachyzoites per cell (Fig. 8A). While the distribution of parasite number per cell was similar for all strains under arginine high conditions (Fig. 8B), the presence of ROP16 in RH or the complemented ΔROP16 : 1 strain reduced the number of parasites per cell under arginine limiting conditions (Fig. 8C). In contrast, the ROP16-deleted strain exhibited significantly higher numbers of parasites per cell (Fig. 8C). Representative image of cells infected with each mutant under arginine high and low conditions are shown in Fig. 8D.

The above results argued that ROP16/STAT6-dependent arginase-1 induction played a role on slowing parasite growth. To test this, we compared growth of ΔROP16 and ΔROP16 : 1 parasites in peritoneal macrophages from Stat6+/+ and Stat6−/− mice. Parasites expressing Type I ROP16 clearly displayed a growth disadvantage in WT cells that was eliminated in Stat6 gene-deleted macrophages (Fig. 8E-G). As expected ΔROP16 parasites, which do not trigger STAT6 activation, replicated equivalently in wild-type and knockout cells (Fig. 8E–G). Next, we performed the same experiment employing macrophages from Arg1−/− and Arg1+/+ littermate control mice. As shown in Fig. 8H-J, while ΔROP16 : 1 replicated less well than ΔROP16 parasites in wild-type MØ, the strains replicated equivalently in Arg1−/− cells. We conclude that ROP16 mediates STAT6-dependent arginase-1 induction, thereby modulating arginine availability and resulting in parasite sensitivity to arginine starvation conditions.
Influence of ROP16 on IL-12 production, parasite replication, and dissemination during in vivo infection
To investigate the effect of ROP16 deletion during in vivo infection, mice were i. p. inoculated with wild-type, or ΔROP16 parasites. Peritoneal exudate cells (PEC) were collected 2, 3 and 4 days post-infection and analyzed by Western blot for STAT3 and STAT6 phosphorylation. In agreement with our in vitro results, both of these signaling intermediates were phosphorylated in a ROP16-dependent manner (Fig. 9A). In Fig. S5, we i. p. infected mice with RH, ΔROP16 and ΔROP16 : 1 and collected PEC 5 days later. The results confirm that deletion and re-introduction of ROP16 controls STAT3 and STAT6 activation in vivo. PEC collected 24 hr post-infection were analyzed for ex vivo IL-12 production. As shown in Fig. 9B, cells from ΔROP16-infected mice produced significantly higher levels of IL-12p40 relative to cells from mice infected with wild-type parasites. Both wild-type and ΔROP16-infected mice succumbed with the same kinetics after i. p. infection, despite evidence for increased IL-12 production during ΔROP16 infection (Fig. S6).
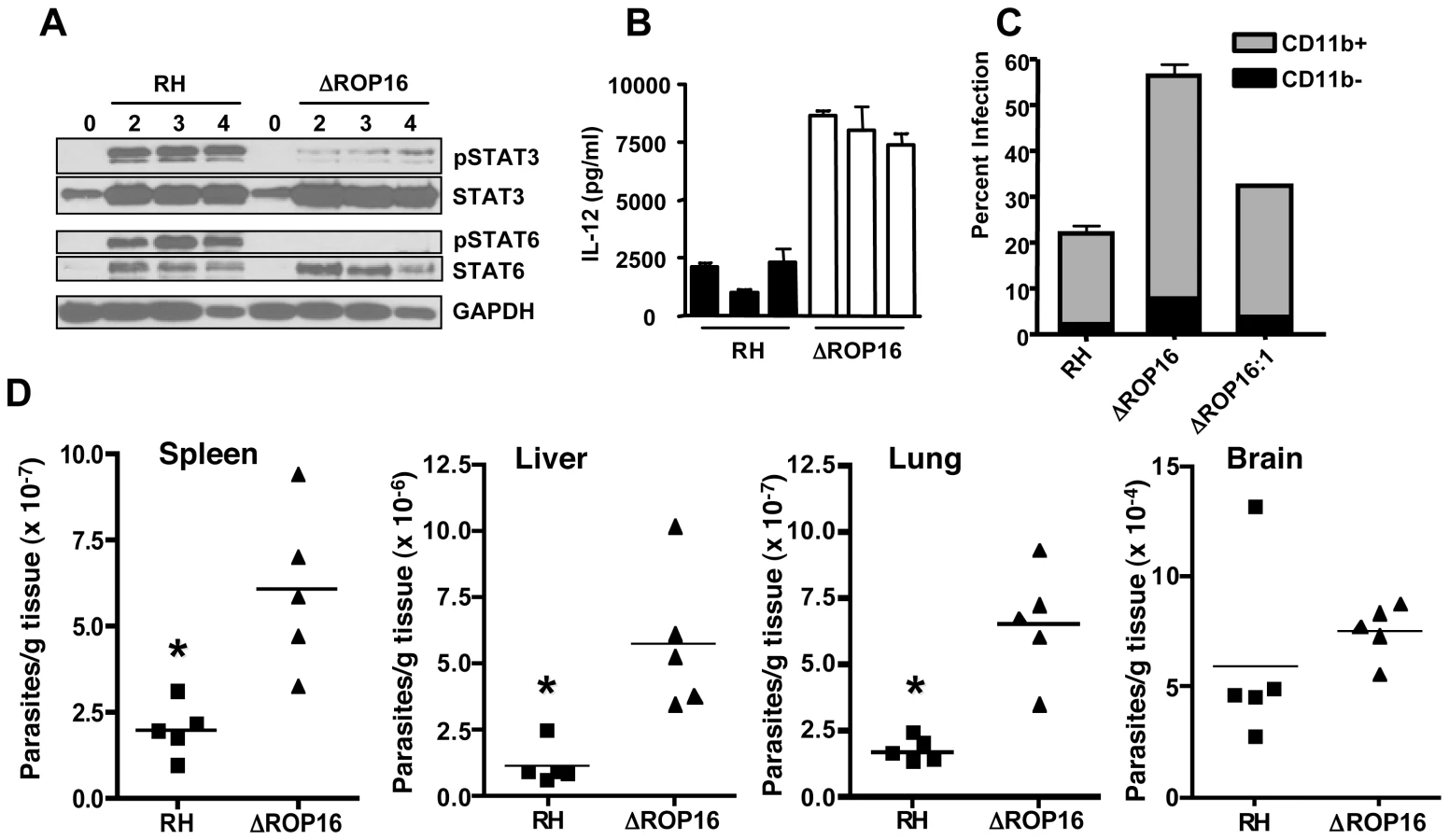
We examined the course of acute infection in the peritoneal cavity following parasite inoculation with 106 tachyzoites. At 6 hr post infection (Fig. S7), similar levels of CD11b+ cells were observed in the RH and ΔROP16 infections, and the percent infection was equivalent. Flow cytometric analysis revealed the CD11b+ cells to be inflammatory monocytes and approximately 10% neutrophils (data not shown). By 24 hr post-infection, the percentage of infected cells was approximately 2-fold higher in mice infected with ΔROP16 parasites. After 72 hr of infection, 80% of PECs were infected with ROP16 knockout parasites compared to 54% with the wild-type parasite strain. We also infected mice with a 10-fold lower dose of parasites and collected PEC 72 hr later for parasite quantitation by qPCR. The results (Fig. S7B) show increased numbers of parasites per host cell during infection with ΔROP16 relative to WT parasites.
We performed similar experiments employing ΔROP16 : 1 tachyzoites in parallel with RH and ΔROP16 parasites. As shown in Fig. 9C, while deletion of rop16 promoted parasite infection in the peritoneal cavity, re-introduction of the gene into the deletion mutant decreased tachyzoite-positive cells to levels similar to RH infection. Increased parasite level resulting from ROP16 deletion was also found in various tissues (liver, lung, spleen) harvested from 3-day infected mice. Levels of the parasite B1 gene were significantly higher after infection with ΔROP16 parasites relative to the parental RH strain in all tissues except for the brain (Fig. 9D). In Fig. 10, we assessed downstream IFN-γ production and parasite numbers in spleen following infection. In the spleen IFN-γ levels displayed a ROP16-dependent increase (Fig. 10A). There was a concomitant increase in parasites per host cell in the spleen that depended upon the presence of Type I ROP16 (Fig. 10B).

Evidence that ROP16-dependent arginase-1 induction in infected MØ limits in vivo parasite replication and dissemination
We next examined whether ROP16 was involved in arginase-1 induction during in vivo infection. To test this, we infected mice by i. p. injection with 106 parasites of either strain and measured Arg-1 expression levels by qPCR. Within 4 days of infection, we detected strong Arg-1 up regulation during RH infection. Notably, RH induced approximately two-fold more Arg-1 than ΔROP16 (Fig. 11A). Likewise, when mesenteric lymph node lysates were subjected to Western blotting for arginase-1 (Fig. 11B), we found that the protein was expressed at higher level during RH infection.

We tested the hypothesis that ROP16-mediated arginase-1 expression in infected cells limited the replication and dissemination of the parasite during in vivo infection. Straightforward infection of arginase-1 knockout or MØ-specific arginase-1 knockout mice was problematic because of issues of lethality for arginase-1 knockout mice. In addition, cytokines such as IL-4 produced during infection could mediate arginase-1 induction in infected and noninfected cells, and this could also complicate the results. Therefore, we adopted the approach outlined in Fig. 11C. Wild-type and Arg1−/− CD45.1+ MØ were infected with RH or with ΔROP16 parasites. After removing extracellular tachyzoites, infected MØ were transferred into the peritoneal cavities of wild-type CD45.2+ congenic mice. Two days after transfer, PEC were collected and host cell infection levels were analyzed by flow cytometry. We reasoned that if ROP16-mediated arginase-1 induction limited in vivo growth, emergence of ΔROP16 parasites and subsequent infection of CD45.2 host cells would occur more rapidly than during RH infection. The results are plotted as the ratio of percent host infection in recipients of infected Arg1−/− MØ relative to recipients of Arg1+/+ MØ. Indeed, there was large increase in infection rate when wild-type parasites entered the host in Arg1−/− MØ compared to Arg1+/+ cells (Fig. 11D). In contrast, the MØ genotype made no difference in dissemination to the host during ΔROP16 infection.
Discussion
Toxoplasma displays an unusual population structure in that 3 clonal lineages predominate in Europe and North America [43]. Type I strains are highly virulent in mice, whereas strain types II and III are less virulent and can establish latent infection. Importantly, there is evidence that Type I strains also cause more serious disease in humans [44]. Studies in mice suggest that the immune response is an important determinant of virulence, although its exact role is unclear. For example, infection with Type I strains is associated with overproduction of proinflammatory cytokines, whereas the response during Type II infection is more restrained [45], [46]. Paradoxically, Type II tachyzoites stimulate higher levels of MØ IL-12 production compared to Type I parasites during in vitro infection [27], [28].
Toxoplasma strain type is an important determinant of activation of STAT signaling pathways during intracellular infection. Forward genetic analysis identified ROP16 as a polymorphic parasite kinase controlling strain-specific activation of STAT3 and STAT6, as well as modulating production of IL-12p40 [19]. Here, we employed a reverse genetic approach to generate ROP16 deletion mutants as well as control complementation mutants to gain insight into the biological role of this rhoptry molecule during infection.
Previously, it was found that Type I strain parasites induces rapid and sustained STAT3 activation associated with low-level IL-12 production in mouse MØ [17], [27], [28]. In contrast, Type II Toxoplasma fails to sustain STAT3 activation, and this was genetically linked to high level IL-12 synthesis [19]. We found Type I parasites lacking ROP16 were severely defective in STAT3 tyrosine phosphorylation. Nevertheless, ΔROP16 parasites maintained an ability to trigger early STAT3 activation. This result is essentially identical to previous data from genetic crosses indicating that Type I ROP16 is required for sustained STAT3 activation rather than the initial response [19]. Nevertheless, recent in vitro kinase studies have shown that both STAT3 and STAT6 serve as direct substrates for ROP16 tyrosine kinase activity [21], [22]. Therefore, we propose that there are two STAT3 activation phases. The first occurs rapidly and independently of ROP16, and possibly involves activation of JAK molecules. The second wave is necessary for sustained STAT3 activation in Toxoplasma-infected cells and is likely dependent upon direct tyrosine kinase activity of Type I ROP16. Interestingly, STAT6 tyrosine phosphorylation differed from STAT3 tyrosine phosphorylation insofar as deletion of ROP16 completely eliminated the parasite's ability to activate this signal transducing molecule. Therefore, STAT6 activation during infection is entirely dependent upon the parasite kinase.
Deletion of ROP16 converts Type I parasites from low to high inducers of IL-12. Here, we show that this response, like that induced by the Type II Toxoplasma ME49 strain, is highly dependent upon the common adaptor MyD88. This molecule is involved in signal transduction through most TLR, as well as signaling through receptors for IL-1β and IL-18 [47]. We assessed whether TLR2, 4, 9 and 11, MyD88-dependent TLR implicated in the response to Toxoplasma [48], were responsible for high level IL-12 production induced by the ΔROP16 strain. However, using TLR knockout MØ, we found no evidence for involvement of these TLR, and we formally ruled out autocrine IL-1β and IL-18 activity using MØ from caspase-1 knockout mice. The ROP16-dependent and MyD88-dependent IL-12 production that we observe may result from redundant functions of multiple TLR, or it is possible that Toxoplasma itself may be capable of bypassing TLR and use a novel mechanism to directly trigger MyD88-dependent signaling.
Toxoplasma is known to inhibit signaling through TLR ligands such as LPS, and we previously found that this activity was dependent upon STAT3 [17]. Here, we show that ROP16 controls the ability to suppress TLR4-triggered IL-12p40 and TNF-α production. We reported recently that T. gondii interferes with LPS-induced chromatin remodeling at the TNF promoter by blocking phosphorylation and acetylation of histone H3, suggesting one mechanism for the suppressive effects of the parasite [49]. However, ROP16 does not appear to be involved in this activity, because ΔROP16 parasites maintain the ability to inhibit TLR4-mediated histone H3 modification (data not shown). The biological significance of the down-regulatory effects of Toxoplasma on TLR signaling is not yet clear. Since Toxoplasma infection is naturally acquired via oral ingestion of parasites, one possibility is that inhibition is a way to evade the activating effects of bacterial TLR ligands that the host is exposed to during T. gondii infection in the intestine [50], [51]. Alternatively, it is possible that down-regulating TLR signaling is a way for the parasite to avoid the activating effects of its own TLR ligands.
Toxoplasma also interferes with signaling mediated by IFN-γ [37], [52], [53]. In order to assess this response, we first examined bone marrow-derived and thioglycollate-elicited MØ NO production. In our hands these cells produced undetectable amounts of NO in response to IFN-γ (data not shown). However, both astrocytes and microglial cells are known to produce NO [54], [55], and indeed we found that IFN-γ stimulation of these cells resulted in NO release. While wild-type parasites were able to suppress the response, the ΔROP16 strain was defective in inhibitory activity. Insight into the functional significance of this response may come from the observation that ability to express inducible NO, in particular by microglial cells, is involved in controlling chronic infection in the mouse brain, whereas animals survive acute infection without the iNOS enzyme [56], [57], [58]. Therefore, expression of ROP16 may be a parasite mechanism to increase transmission potential by escaping the microbicidal effects of NO in the central nervous system.
Despite the finding that absence of ROP16 results in enhanced IL-12 production and defective ability to inhibit production of proinflammatory mediators, replication and dissemination of tachyzoites was enhanced by deletion of ROP16. We obtained evidence that the reduced replication and dissemination of parental RH parasites compared to ΔROP16 parasites was due to arginine starvation resulting from ROP16-dependent STAT6-mediated induction of arginase-1. The dependence of Toxoplasma-induced arginase-1 expression on STAT6 activation we observed is in contrast to previously published results by El Kasmi et al. [40]. In that study, the authors observed STAT6 activation by Type II ROP16, a result not seen by us or others [19], [21]. Likewise, the authors reported STAT6-independent arginase-1 induction during infection with Type II ROP16-expressing parasites, a result that also stands in contrast to our findings. At present we do not understand the reason for these discrepant results, but because El Kasmi et al. used ME49 without manipulation of ROP16 a certain degree of caution regarding STAT6-dependent arginase-1 induction may be warranted.
Regardless, our data are consistent with a view that ROP16-mediated induction of arginase-1 functions to limit parasite replication, and that this is a strategy to facilitate host survival and establishment of latent infection to increase transmission potential. An alternative view comes from the consideration that arginase-1 and iNOS compete for the same substrate - namely, arginine. Thus, ROP16-mediated arginase-1 induction, and consequent arginine depletion in infected cells, may represent a mechanism used by Type I strain parasites to evade the potentially lethal effects of high-level NO production. Consistent with this concept, we previously reported that Type II ME49 infection, in contrast to Type I RH infection, resulted in NO production during in vivo infection in the spleen even though overall cytokine synthesis was greater during Type I infection [45], [46].
More evidence for an in vivo role of arginase-1 during infection comes from recent findings with Leishmania major and Schistosoma mansoni [59], [60]. In those studies, arginase-1 induction in myeloid cells promoted infection by localized depletion of arginine, in turn leading to suppressed T cell responses. This could possibly have relevance to in vivo responses during Toxoplasma infection, insofar as several older studies suggested that acute infection is associated with nonspecific T cell suppression [61], [62], [63]. We are currently re-examining this issue.
Our study demonstrates ROP16 manipulates host cell signaling pathways that determine availability of arginine for parasite replication and dissemination, and host production of NO. By manipulating arginase-1 levels and consequently arginine availability to both host and parasite, ROP16 may act as a central regulator of parasite replication and transmission potential. In addition to ROP16-mediated control of host arginase-1, it seems possible, and even likely, that ROP16 has additional functions during intracellular infection. The ROP16 molecule is involved in activation of STAT3 and STAT6, transcription factors that each possess their own unique targets. Microarray analysis of host cell responses also suggests that ROP16 has multiple downstream targets [21]. Thus, ROP16 is emerging as a molecule at the nexus of the host-parasite interaction, and as such may function as one of the key determinants of strain-specific virulence and transmissibility.
While this manuscript was under review similar findings were reported by an independent group (Jensen et al. Cell Host and Microbe 2011. 9 : 472).
Materials and Methods
Ethics statement
The experiments in this study were performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocols were approved by the Institutional Animal Care and Use Committee at Cornell University (permit number 1995-0057). All efforts were made to minimize animal suffering during the course of these studies.
Mice
Female C57BL/6 mice, 6–8 wks of age, were purchased from Taconic Farms (Germantown, NY). STAT6−/− mice (provided by M. Bynoe), TLR2−/−/TLR4−/− mice (provided by D. Russell) and MyD88−/− animals, originally supplied by S. Akira (Osaka University), were bred in-house at Cornell University. Caspase-1−/− and JAK3−/− mice were obtained from The Jackson Laboratory (Bar Harbor, ME). TLR9−/− mice were bred in house at UT-Southwestern Medical School and Arg-1−/− mice were bred at the University of Cincinnati College of Medicine. Animals were housed under specific pathogen-free conditions in the Cornell University College of Veterinary Medicine animal facility, which is accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International.
Parasites
Tachyzoites were maintained by twice weekly passage on human foreskin fibroblast monolayers in DMEM (Life Technologies, Gaithersburg, MD) supplemented with 1% heat-inactivated bovine growth serum (HyClone, Logan, UT), 100 U/ml penicillin (Life Technologies), and 0.1 mg/ml streptomycin (Life Technologies). Parasite cultures were tested every 4–6 weeks for Mycoplasma contamination using a commercial PCR-ELISA based kit (Roche Applied Systems, Mannheim, Germany).
Antibody sources
Cell Signaling Technology (Danvers, MA) was the source of antibodies to total and phospho-STAT3, JAK1, JAK2, ERK1/2 PARP and GAPDH. Anti-CD11b and anti-phospho-STAT6 were purchased from BD Biosciences (San Jose, CA). FITC-conjugated anti-p30 was purchased from Argene, Inc. Dr. Sidney Morris (University of Pittsburgh) generously provided the arginase-1 antibody.
Gene knockout targeting plasmid constructs
The Type I ROP16 gene locus is defined by TGGT1_063760 in the current T. gondii genome database www.Toxodb.org (version 6.0). Gene knockout targeting plasmid pΔROP16 was constructed by fusing through yeast recombinational cloning [64] a ∼1.2 kb 5′ ROP16 target flank amplified from RH genomic DNA, the HXGPRT minigene cassette [24] and a ∼1.2 kb 3′ ROP16 target flank amplified from RH genomic DNA in correct order into the yeast-shuttle plasmid pRS416. The deletion was engineered to remove a small portion of the 5′ UTR and essentially all of the coding region with the exception of a few codons near the predicted translation stop of ROP16, to create a ∼2.2 kb deletion in the ΔROP16 knockout strain. Complementation plasmid pΔROP16 : 1 was generated by fusing a c-terminal HA-tagged functional allele of Type I RH ROP16 with 5′ and 3′ ROP16 target flanks. The oligonucleotide primers used in gene knockout and complementation plasmid construction are shown in Table S1. Targeting plasmids were validated by restriction digest, and by DNA sequencing to verify 100% homology in gene targeting flanks.
Gene replacement at the rop16 locus
Approximately 10 µg of PmeI linearized pΔROP16 targeting plasmid was individually transfected into T. gondii strain RHΔku80Δhxgprt that exhibits highly enhanced homologous recombination and knockouts were then selected in mycophenolic acid (MPA) using previously described methods [24]. Individual MPA resistant clones were isolated and the genotype of the clones was evaluated by PCR as previously described [24]. Genotype validation primers are shown in Table S1. PCR 1 (rop16 deletion) used primers ΔF and ΔR. PCR 2 used primers EXF & CXR. PCR 3 (5′ integration) used primers CXF and pminiHXR. PCR 4 (3′ integration) used primers CXR and pminiHXF [24]. The ΔROP16 : 1 complemented strain was generated by retargeting the ROP16-deleted locus with plasmid pΔROP16 : 1 to delete HXGPRT and reinsert a c-terminal HA-tagged functional allele of Type I RH ROP16. Following transfection of the ΔROP16 knockout strain with 10 µg of PmeI linearized pΔROP16 : 1 targeting plasmid, parasites were selected in 6-thioxanthine (6TX) for removal of the HXGPRT selectable marker [24]. The complemented ΔROP16 : 1 strain was validated in PCR by showing correct 5′ and 3′ integration of the Type I ROP16 allele at the ROP16 locus. PCR 5 (5′ integration) used primers CXF and cvR. PCR 6 (3′ integration) used primers cvF and CXR.
Virulence assay
Adult 8-week-old female CF1 mice were obtained from Charles River Laboratories and maintained in Techniplast Seal Safe mouse cages on vent racks at the Dartmouth-Hitchcock Medical Center (Lebanon, NH) mouse facility. All mice were cared for and handled according to the Animal Care and Use Program of Dartmouth College using National Institutes of Health-approved institutional animal care and use committee guidelines. Groups of four mice were injected intraperitoneally with 0.2 ml (100 tachyzoites) and monitored daily for degree of illness and survival.
Bone marrow-derived MØ and DC preparation
MØ were derived from bone marrow by 5-day culture in L929-containing supernatants as previously described [65]. DC were prepared as described previously [66].
Generation of astrocytes and microglia
Astrocytes and microglia were generated from the brains of 3-4 day old mice as previously described [67] with minor modifications. Briefly, brains were homogenized through 40-micron cell strainers and single cell suspensions were subjected to discontinuous Percoll gradient centrifugation (70∶30∶0). Astrocytes were collected from the 30∶0 interface and microglial cells from the 70∶30 interface. Both cell types were cultured in DMEM/F12 medium supplemented with 10% bovine growth serum up to three weeks. Microglial cells were additionally supplemented with 130 ng/ml recombinant GM-CSF (PeproTech, Inc. Rocky Hill NJ). Medium was replaced every 3 days.
Cell culture
Infection was accomplished by addition of tachyzoites to cell cultures (3∶1 ratio of parasites to MØ). Plates were briefly centrifuged (3 min, 200 x g) to synchronize contact between tachyzoites and cells. For endotoxin triggering studies, LPS (100 ng/ml; S. minnesota, ultrapure, List Biological Laboratories, Campbell, CA) was added 60 min after infection, and at varying times cells were collected for biochemical assays. For measurement of cytokine release, cells were infected with various parasite ratios with and without subsequent LPS stimulation. Supernatants were collected at times indicated in text. Total cell lysates and nuclear extracts were prepared for Western blot analysis with the Nuclear Extract Kit (Active Motif, Carlsbad, CA). In some experiments, JAK inhibitor I (EMD Biosciences, La Jolla, CA) and JAK2 inhibitor III (EMD Biosciences) were added to cells 60 min prior to infection. Arginine-free medium (Sigma-Aldrich) was supplemented with dialyzed bovine growth serum, leucine, lysine and glucose. In addition, arginine-high medium was supplemented to 84 mg/L L-arginine. Arginine-low medium was not supplemented with the amino acid.
Immunoblot analysis
Cells (106/sample) were lysed in reducing SDS sample buffer, and DNA was sheared by forcing samples three times through a 27-gauge needle. After boiling for 3 min, cellular lysates were separated by 10% SDS-PAGE, and proteins subsequently electrotransferred onto nitrocellulose membranes (Schleicher & Schuell, Keene, NH). Membranes were blocked in 5% nonfat dry milk containing 0.1% Tween 20 (Sigma-Aldrich) in Tris-buffered saline, pH 7.6 (TBST), for 1 hr at room temperature, followed by incubation with primary antibodies according to the manufacturer's protocol (Cell Signaling Technology). After washing blots in TBST, antibody binding was detected with an HRP-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) in TBST containing 5% nonfat dry milk for 1 hr at room temperature. Tyrosine phosphorylated STAT6 was detected using a mouse antibody from BD Biosciences followed by anti-mouse biotin-conjugated antibody (Thermo Scientific, Rockford IL) and an anti-biotin horseradish peroxidase-conjugated antibody (Cell Signaling Technology). After washing blots in TBST, bands were visualized using an ECL system (Lumi-GLO; Cell Signaling Technology).
Pull-down assay
Tachyzoites were added to bone marrow-derived MØ (5×107) at a ratio of 8 : 1, cells were pelleted (2000 rpm, 3 min), and incubated for 20 min at 37°C. In some experiments, recombinant IL-6 (eBioscience; San Diego, CA; 100 ng/ml) was added. Cells were washed in PBS and resuspended in Cell Lysis Buffer containing a cocktail of protease and phosphates inhibitors (Cell Signaling Technology), and the suspension was subjected to brief sonication. After centrifugation (14, 000 rpm, 10 min, 4°C), the resulting pellet was resuspended in 100 µl anti-phosphotyrosine-agarose (Cell Signaling Technology) and the slurry was rotated overnight at 4°C. The agarose beads were subsequently washed in Cell Lysis Buffer, resuspended in SDS sample reducing buffer and incubated at 100°C for 3 min. After pelleting the beads, samples were subject to SDS-PAGE and immunoblot analysis.
Immunofluorescence microscopy
Coverslips bearing infected MØ or fibroblast monolayers were fixed with 4% formaldehyde in PBS, rinsed with PBS and incubated (1 h, room temperature) with indicated primary antibodies and/or FITC-conjugated anti-P30 antibody diluted in PBS containing 1% BSA and 10% donkey serum. Coverslips were washed 3 times with PBS and then incubated (1 h, room temperature) with secondary antibody in the same diluent. After washing 3 times with PBS, coverslips were mounted with Pro-Long Gold antifade (Invitrogen, Carlsbad, CA). Actin staining was accomplished with Alexa-fluor 594-conjugated phalloidin according to manufacturer's instructions (Invitrogen).
PCR detection of Toxoplasma gondii
DNA extraction from tissue samples was carried using DNeasy Blood and Tissue Kit (Qiagen Inc. Valencia, CA) following the manufacturer's instructions. Real-time PCR was performed targeting the highly conserved 35-fold-repetitive B1 gene in T. gondii [68]. A 25 µl-reaction mixture was prepared using 2X Power SYBR Green PCR Master Mix (Applied Biosystems, Carlsbad, CA) with 0.3 µM forward primer 5′-GGA-GGA-CTG-GCA-ACC-TGG-TGT-CG-3′ and reverse primer 5′-TTG-TTT-CAC-CCG-GAC-CGT-TTA-GCA-G-3′ [69]. Reactions were carried out in an Applied Biosystems 7500 Fast Real Time PCR System with the following thermal cycling conditions: 50°C for 2 min, 95°C for 10 min, followed by 40 cycles at 95°C for 15 sec and 60°C for 1 min. All PCR amplifications were subjected to dissociation analysis to confirm the specificity of the reaction. Quantification of parasites was accomplished by using a standard curve constructed with a series of known quantity of the T. gondii RH strain over a range of 6 logs corresponding to 105 to 1 parasite per reaction. Uninfected tissues were used as negative controls and distilled sterile water used as zero template controls.
Parasites per cell were determined by qPCR for detection of the parasite B1 gene and host argininosuccinate lyase (NM_133768), a gene that is absent from the Toxoplasma genome. Aliquots of 106 cells were centrifuged to pellet cells and extracellular parasites. DNA was extracted with the E.Z.N.A tissue DNA kit (Omega Bio-Tek). Using the Syber Green PerfeCTa low ROX kit (Quanta Biosciences) B1 primers (listed above) were used to determine parasite number and argininosuccinate lyase forward (5′TCT-TCG-TTA-GCT-GGC-AAC-TCA-CCT-3′) and reverse (5′ATG-ACC-CAG-CAG-CTA-AGC-AGA-TCA-3′) primers were used to determine mouse cell number in replicate samples. Standard curves were constructed as described above for both species.
Quantitative reverse transcriptase PCR of Arg-1 gene
Total RNA was prepared from brain tissue by Trizol purification and 1 µg was converted to cDNA using a commercially available method (Quanta Biosciences, Gaithersburg, MD). Quantitative PCR was performed on the Arg1 gene and normalized to the expression of the housekeeping gene GAPDH utilizing the SYBR green method (Quanta biosciences, Gaithersburg, MD) and ABI 7500 fast machine (Life Technologies Corporation, Carlsbad, CA). Expression relative to noninfected control samples was calculated utilizing the ΔΔCt method. The primer sequences employed were: 5′-AAG-AAT-GGA-AGA-GTC-AGT-GTG-G-3′ (Arg1 forward); 5′-GGG-AGT-GTT-GAT-GTC-AGT-GTG-3′ (Arg1 reverse); 5′-AAT-GGT-GAA-GGT-CGG-TGT-G-3′ (GAPDH forward); 5′-GTG-GAG-TCA-TAC-TGG-AAC-ATG-TAG-3′ (GAPDH reverse).
Cytokine ELISA and NO assay
IL-12(p40) was measured by ELISA as described [15], and TNF-α and IFN-γ was measured using a commercial kit according to the manufacturer's instructions (eBiosciences). Production of nitric oxide was detected by measurement of NO2/NO3 by the Griess reaction as described [70].
Statistical analysis
Student's t-test was used to analyze statistical differences between groups. Values for P<0.05 were considered significant. All experiments were repeated a minimum of two times.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. BhavsarAPGuttmanJAFinlayBB 2007 Manipulation of host-cell pathways by bacterial pathogens. Nature 449 827 834
2. RoyCRMocarskiES 2007 Pathogen subversion of cell-intrinsic innate immunity. Nat Immunol 8 1179 1187
3. SacksDSherA 2002 Evasion of innate immunity by parasitic protozoa. Nature Immunol 3 1041 1047
4. DubeyJP 2007 The history and life-cycle of Toxoplasma gondii. WeissLMKimK Toxoplasma gondii The model apicomplexan: Perspective and methods San Diego Academic Press 1 17
5. BierlyALShufeskyWJSukhumavasiWMorelliADenkersEY 2008 Dendritic cells expressing plasmacytoid marker PDCA-1 are Trojan horses during Toxoplasma gondii infection. J Immunol 181 8445 8491
6. ChtanovaTSchaefferMHanSJvan DoorenGGNollmannM 2008 Dynamics of Neutrophil Migration in Lymph Nodes during Infection. Immunity 29 487 496
7. CourretNDarcheSSonigoPMilonGBuzoni-GatelD 2006 CD11c and CD11b expressing mouse leukocytes transport single Toxoplasma gondii tachyzoites to the brain. Blood 107 309 316
8. PetersonELiesenfeldO 2007 Clinical disease and diagnostics. WeissLMKimK Toxoplasma gondii The model apicomplexan: Perspectives and methods Amsterdam Academic Press 81 100
9. PfaffAWLiesenfeldOCandolfiE 2007 Congenital toxoplasmosis. AjiokaJWSoldatiD Toxoplasma molecular and cellular biology Norfolk Horizon Bioscience 93 110
10. DenkersEYGazzinelliRT 1998 Regulation and function of T cell-mediated immunity during Toxoplasma gondii infection. Clin Microbiol Rev 11 569 588
11. LengJButcherBADenkersEY 2009 Dysregulation of macrophage signal transduction by Toxoplasma gondii: Past progress and recent advances. Parasite Immunol 31 717 728
12. KimLDenkersEY 2006 Toxoplasma gondii triggers Gi-dependent phosphatidylinositol 3-kinase signaling required for inhibition of host cell apoptosis. J Cell Sci 119 2119 2126
13. LengJButcherBAEganCEAbdallahDSDenkersEY 2009 Toxoplasma gondii prevents chromatin remodeling initiated by TLR-triggered macrophage activation. J Immunol 182 489 497
14. ZimmermannSMurrayPJHeegKDalpkeAH 2006 Induction of Suppressor of Cytokine Signaling-1 by Toxoplasma gondii Contributes to Immune Evasion in Macrophages by Blocking IFN-{gamma} Signaling. J Immunol 176 1840 1847
15. ButcherBAKimLJohnsonPFDenkersEY 2001 Toxoplasma gondii tachyzoites inhibit proinflammatory cytokine induction in infected macrophages by preventing nuclear translocation of the transcription factor NFkB. J Immunol 167 2193 2201
16. ShapiraSHarbOMargaritJMatrajtMHanJ 2005 Initiation and termination of NFkB signaling by the intracellular protozoan parasite Toxoplasma gondii. J Cell Sci 118 3501 3508
17. ButcherBAKimLPanopoulosAWatowichSSMurrayPJ 2005 Cutting Edge: IL-10-independent STAT3 activation by Toxoplasma gondii mediates suppression of IL-12 and TNF-a in host macrophages. J Immunol 174 3148 3152
18. SaeijJPBoyleJPCollerSTaylorSSibleyLD 2006 Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Science 314 1780 1783
19. SaeijJPCollerSBoyleJPJeromeMEWhiteMW 2007 Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature 445 324 327
20. HakanssonSCharronAJSibleyLD 2001 Toxoplasma evacuoles: a two-step process of secretion and fusion forms the parasitophorous vacuole. EMBO J 20 3132 3144
21. OngYCReeseMLBoothroydJC 2010 Toxoplasma rhoptry protein 16 (ROP16) subverts host function by direct tyrosine phosphorylation of STAT6. J Biol Chem 285 28731 28740
22. YamamotoMStandleyDMTakashimaSSaigaHOkuyamaM 2009 A single polymorphic amino acid on Toxoplasma gondii kinase ROP16 determines the direct and strain-specific activation of Stat3. J Exp Med 206 2747 2760
23. MurrayPJ 2007 The JAK-STAT signaling pathway: input and output integration. J Immunol 178 2623 2629
24. FoxBARistucciaJGGigleyJPBzikDJ 2009 Efficient gene replacements in Toxoplasma gondii strains deficient for nonhomologous end joining. Eukaryot Cell 8 520 529
25. ShenYSchlessingerKZhuXMeffreEQuimbyF 2004 Essential role of STAT3 in postnatal survival and growth revealed by mice lacking STAT3 serine 727 phosphorylation. Mol Cell Biol 24 407 419
26. ReichNCLiuL 2006 Tracking STAT nuclear traffic. Nat Rev Immunol 6 602 612
27. KimLButcherBALeeCWUematsuSAkiraS 2006 Toxoplasma gondii genotype determines MyD88-dependent signaling in infected macrophages. J Immunol 177 2584 2591
28. RobbenPMMordueDGTruscottSMTakedaKAkiraS 2004 Production of IL-12 by macrophages infected with Toxoplasma gondii depends on the parasite genotype. J Immunol 172 3686 3694
29. HitzigerNDellacasaIAlbigerBBarraganA 2005 Dissemination of Toxoplasma gondii to immunoprivileged organs and role of Toll/interleukin-1 receptor signalling for host resistance assessed by in vivo bioluminescence imaging. Cell Micro 6 837 848
30. ScangaCAAlibertiJJankovicDTilloyFBennounaS 2002 Cutting edge: MyD88 is required for resistance to Toxoplasma gondii infection and regulates parasite-induced IL-12 production by dendritic cells. J Immunol 168 5997 6001
31. SukhumavasiWEganCEWarrenALTaylorGAFoxBA 2008 TLR adaptor MyD88 is essential for pathogen control during oral toxoplasma gondii infection but not adaptive immunity induced by a vaccine strain of the parasite. J Immunol 181 3464 3473
32. Debierre-GrockiegoFCamposMAAzzouzNSchmidtJBiekerU 2007 Activation of TLR2 and TLR4 by glycosylphosphatidylinositols derived from Toxoplasma gondii. J Immunol 179 1129 1137
33. Del RioLButcherBABennounaSHienySSherA 2004 Toxoplasma gondii triggers MyD88-dependent and CCL2(MCP-1) responses using distinct parasite molecules and host receptors. J Immunol 172 6954 6960
34. FoureauDMMielcarzDWMenardLCSchulthessJWertsC 2010 TLR9-dependent induction of intestinal alpha-defensins by Toxoplasma gondii. J Immunol 184 7022 7029
35. YarovinskyFZhangDAndersonJFBannenbergGLSerhanCN 2005 TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science 308 1626 1629
36. McKeeASDzierszinskiFBoesMRoosDSPearceEJ 2004 Functional inactivation of immature dendritic cells by the intracellular parasite Toxoplasma gondii. J immunol 173 2632 2640
37. LuderCGKAlgnerMLangCBleicherNGrossU 2003 Reduced expression of the inducible nitric oxide synthase after infection with Toxoplasma gondii facilitates parasite replication in activated murine macrophages. Internat J Parasitol 33 833 844
38. ModolellMCorralizaIMLinkFSolerGEichmannK 1995 Reciprocal regulation of the nitric oxide synthase/arginase balance in mouse bone marrow-derived macrophages by TH1 and TH2 cytokines. Eur J Immunol 25 1101 1104
39. RutschmanRLangRHesseMIhleJNWynnTA 2001 Stat6-dependent substrate depletion regulates nitric oxide production. J Immunol 166 2173 2177
40. El KasmiKCQuallsJEPesceJTSmithAMThompsonRW 2008 Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat Immunol 9 1399 1406
41. SehgalPB 2008 Paradigm shifts in the cell biology of STAT signaling. Semin Cell Dev Biol 19 329 340
42. FoxBAGigleyJPBzikDJ 2004 Toxoplasma gondii lacks the enzymes required for de novo arginine biosynthesis and arginine starvation triggers cyst formation. Int J Parasitol 34 323 331
43. SibleyLDAjiokaJW 2008 Population structure of Toxoplasma gondii: clonal expansion driven by infrequent recombination and selective sweeps. Annu Rev Microbiol 62 329 351
44. BoothroydJCGriggME 2002 Population biology of Toxoplasma gondii and its relevance to human infection: do different strains cause different disease? Curr Opin Micro 5 438 442
45. GavrilescuLCDenkersEY 2001 IFN-g overproduction and high level apoptosis are associated with high but not low virulence Toxoplasma gondii infection. J Immunol 167 902 909
46. MordueDGMonroyFLa ReginaMDinarelloCASibleyLD 2001 Acute toxoplasmosis leads to lethal overproduction of Th1 cytokines. J Immunol 167 4574 4584
47. KawaiTAkiraS 2006 TLR signaling. Cell Death Differentiat 13 816 825
48. DenkersEY 2010 Toll-like receptor initiated host defense against Toxoplasma gondii. J Biomed Biotechnol 2010 737125 737125
49. LengJButcherBAEganCEAbi AbdallahDSDenkersEY 2009 Toxoplasma gondii prevents chromatin remodeling initiated by Toll-like receptor-triggered macrophages activation. J Immunol 182 489 497
50. HeimesaatMMBereswillSFischerAFuchsDStruckD 2006 Gram-Negative Bacteria Aggravate Murine Small Intestinal Th1-Type Immunopathology following Oral Infection with Toxoplasma gondii. J Immunol 177 8785 8795
51. HeimesaatMMFischerAJahnHKNiebergallJFreudenbergM 2007 Exacerbation of Murine Ileitis By Toll-Like Receptor 4 Meditated Sensing of Lipopolysaccharide From Commensal Escherichia coli. Gut 56 941 948
52. KimSKFoutsAEBoothroydJC 2007 Toxoplasma gondii dysregulates IFN-gamma-inducible gene expression in human fibroblasts: insights from a genome-wide transcriptional profiling. J Immunol 178 5154 5165
53. LangCAlgnerMBeinertNGrossULuderCG 2006 Diverse mechanisms employed by Toxoplasma gondii to inhibit IFN-gamma-induced major histocompatibility complex class II gene expression. Microbes Infect 8 1994 2005
54. BorysiewiczEFilDKonatGW 2009 Rho proteins are negative regulators of TLR2, TLR3, and TLR4 signaling in astrocytes. J Neurosci Res 87 1565 1572
55. WilmsHSieversJRickertURostami-YazdiMMrowietzU 2010 Dimethylfumarate inhibits microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, IL-1beta, TNF-alpha and IL-6 in an in-vitro model of brain inflammation. J Neuroinflammation 7 30
56. Deckert-SchluterMBluethmannHKaeferNRangASchluterD 1999 Interferon-gamma receptor-mediated but not tumor necrosis factor receptor type 1 - or type 2-mediated signaling is crucial for the activation of cerebral blood vessel endothelial cells and microglia in murine Toxoplasma encephalitis. Am J Pathol 154 1549 1561
57. Scharton-KerstenTYapGMagramJSherA 1997 Inducible nitric oxide is essential for host control of persistent but not acute infection with the intracellular pathogen Toxoplasma gondii. J Exp Med 185 1 13
58. SchluterDDeckert-SchluterMLorenzEMeyerTRollinghoffM 1999 Inhibition of inducible nitric oxide synthase exacerbates chronic cerebral toxoplasmosis in Toxoplasma gondii-susceptible C57BL/6 mice but does not reactivate the latent disease in T. gondii-resistant BALB/c mice. J Immunol 162 3512 3518
59. ModolellMChoiBSRyanROHancockMTitusRG 2009 Local suppression of T cell responses by arginase-induced L-arginine depletion in nonhealing leishmaniasis. PLoS Negl Trop Dis 3 e480
60. PesceJTRamalingamTRMentink-KaneMMWilsonMSEl KasmiKC 2009 Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS Pathog 5 e1000371
61. CandolfiEHunterCARemingtonJS 1994 Mitogen - and antigen-specific proliferation of T cells in murine toxoplasmosis is inhibited by reactive nitrogen intermediates. Infect Immun 62 1995 2001
62. HaqueSKhanIHaqueAKasperL 1994 Impairment of the cellular immune response in acute murine toxoplasmosis: Regulation of IL-2 production and macrophage-mediated inhibitory effects. Infect Immun 62 2908 2916
63. KhanIAMatsuuraTKasperLH 1996 Activation-mediated CD4+ T cell unresponsiveness during acute Toxoplasma gondii infection in mice. International Immunol 8 887 896
64. OldenburgKRVoKTMichaelisSPaddonC 1997 Recombination-mediated PCR-directed plasmid construction in vivo in yeast. Nucleic Acids Res 25 451 452
65. KimLButcherBADenkersEY 2004 Toxoplasma gondii interferes with lipopolysaccharide-induced mitogen-activated protein kinase activation by mechanisms distinct from endotoxin tolerance. J Immunol 172 3003 3010
66. BennounaSBlissSKCurielTJDenkersEY 2003 Cross-talk in the innate immune system: neutrophils instruct early recruitment and activation of dendritic cells during microbial infection. J Immunol 171 6052 6058
67. ButchiNBDuMPetersonKE 2010 Interactions between TLR7 and TLR9 agonists and receptors regulate innate immune responses by astrocytes and microglia. Glia 58 650 664
68. BurgJLGroverCMPoulettyPBoothroydJC 1989 Direct and sensitive detection of a pathogenic protozoan, Toxoplasma gondii, by polymerase chain reaction. J Clin Microbiol 27 1787 1792
69. CassaingSBessieresMHBerryABerrebiAFabreR 2006 Comparison between two amplification sets for molecular diagnosis of toxoplasmosis by real-time PCR. J Clin Microbiol 44 720 724
70. HibbsJBTaintorRVavrinZRachlinE 1988 Nitric oxide: a cytotoxic activated macrophage effector molecule. Biochem Biophys Res Com 157 87 92
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 9
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- HTLV-1 Propels Thymic Human T Cell Development in “Human Immune System” Rag2 gamma c Mice
- Hostile Takeover by : Reorganization of Parasite and Host Cell Membranes during Liver Stage Egress
- Exploiting and Subverting Tor Signaling in the Pathogenesis of Fungi, Parasites, and Viruses
- A Viral Ubiquitin Ligase Has Substrate Preferential SUMO Targeted Ubiquitin Ligase Activity that Counteracts Intrinsic Antiviral Defence