
Recurrent Signature Patterns in HIV-1 B Clade Envelope Glycoproteins Associated with either Early or Chronic Infections
Here we have identified HIV-1 B clade Envelope (Env) amino acid signatures from early in infection that may be favored at transmission, as well as patterns of recurrent mutation in chronic infection that may reflect common pathways of immune evasion. To accomplish this, we compared thousands of sequences derived by single genome amplification from several hundred individuals that were sampled either early in infection or were chronically infected. Samples were divided at the outset into hypothesis-forming and validation sets, and we used phylogenetically corrected statistical strategies to identify signatures, systematically scanning all of Env. Signatures included single amino acids, glycosylation motifs, and multi-site patterns based on functional or structural groupings of amino acids. We identified signatures near the CCR5 co-receptor-binding region, near the CD4 binding site, and in the signal peptide and cytoplasmic domain, which may influence Env expression and processing. Two signatures patterns associated with transmission were particularly interesting. The first was the most statistically robust signature, located in position 12 in the signal peptide. The second was the loss of an N-linked glycosylation site at positions 413–415; the presence of this site has been recently found to be associated with escape from potent and broad neutralizing antibodies, consistent with enabling a common pathway for immune escape during chronic infection. Its recurrent loss in early infection suggests it may impact fitness at the time of transmission or during early viral expansion. The signature patterns we identified implicate Env expression levels in selection at viral transmission or in early expansion, and suggest that immune evasion patterns that recur in many individuals during chronic infection when antibodies are present can be selected against when the infection is being established prior to the adaptive immune response.
Published in the journal:
. PLoS Pathog 7(9): e32767. doi:10.1371/journal.ppat.1002209
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002209
Summary
Here we have identified HIV-1 B clade Envelope (Env) amino acid signatures from early in infection that may be favored at transmission, as well as patterns of recurrent mutation in chronic infection that may reflect common pathways of immune evasion. To accomplish this, we compared thousands of sequences derived by single genome amplification from several hundred individuals that were sampled either early in infection or were chronically infected. Samples were divided at the outset into hypothesis-forming and validation sets, and we used phylogenetically corrected statistical strategies to identify signatures, systematically scanning all of Env. Signatures included single amino acids, glycosylation motifs, and multi-site patterns based on functional or structural groupings of amino acids. We identified signatures near the CCR5 co-receptor-binding region, near the CD4 binding site, and in the signal peptide and cytoplasmic domain, which may influence Env expression and processing. Two signatures patterns associated with transmission were particularly interesting. The first was the most statistically robust signature, located in position 12 in the signal peptide. The second was the loss of an N-linked glycosylation site at positions 413–415; the presence of this site has been recently found to be associated with escape from potent and broad neutralizing antibodies, consistent with enabling a common pathway for immune escape during chronic infection. Its recurrent loss in early infection suggests it may impact fitness at the time of transmission or during early viral expansion. The signature patterns we identified implicate Env expression levels in selection at viral transmission or in early expansion, and suggest that immune evasion patterns that recur in many individuals during chronic infection when antibodies are present can be selected against when the infection is being established prior to the adaptive immune response.
Introduction
It has proven to be very difficult to elicit protective immunity through an HIV vaccine [1], although a recent vaccine trial in Thailand, RV144, yielded encouraging results [2]. A protective vaccine will need to elicit immune responses that interact effectively with the spectrum of circulating viral strains, and HIV is a remarkably diverse virus [3], [4], [5]. Against this backdrop of variation, if viruses sampled early in infection exhibit a more constrained pattern of diversity at than chronic viruses, i.e. exhibit statistically enriched signature patterns related to transmission or establishing infection, then designing vaccines that incorporate such signatures may be beneficial, and such signatures may yield insight into the biology of viral transmission and disease progression.
Several aspects of the biology of sexual transmission of HIV motivated this systematic search for early versus chronic infection signatures. First was the genetic bottleneck at transmission. It has long been apparent that HIV-1 undergoes extensive diversification during the course of an infection [6], [7], [8], [9], [10], and that viruses sampled from early in infection are less diverse than chronic samples [11], [12], [13], [14], [15]. Improved sampling, modeling strategies, and experimental methods have added greater clarity to this, and recent studies indicate new infections are established by a single virus in approximately 80% of HIV-1 heterosexual transmission cases [16], [17], [18], [19], [20]. By an infection being established by a single virus, we mean that only one lineage is apparent in the viral population sampled early in infection, and that the sampled data is fully consistent with a single founder virus that was transmitted and that expanded in accord with a model of early viral diversification using established parameters for HIV mutation rates and generation time [17], [21], [22]. In addition, the estimated time of infection in homogeneous infections based on experimentally defined Fiebig staging is consistent with estimated times to the most recent common ancestor based on viral diversity [17], [18]. In these cases, the virus that established the infection and was presumably transmitted can be modeled and reconstructed from sequences sampled in early infection, and synthesized for further study [23]. The appropriateness of these models has been confirmed experimentally in macaques where the inoculum, infecting strains and time of infection were known [24], [25]. The rates of multi-variant transmission in men who have sex with men (MSM) [26] and in individuals with inflammatory genital infection [20] are higher, indicating that barriers to transmission may be reduced in these circumstances. The high mutation and replication rates of the virus in a newly infected host provides the baseline for acquisition of genetic diversity, enabling escape from host cytotoxic T lymphocyte (CTL) [27], [28], [29], [30], [31] and antibody [30], [32], [33] responses, and adaptation in a rapidly changing landscape of in vivo selection pressures.
Our second motivation for this study was that a sequence pattern associated with early viruses had already been defined, so a systematic extended search for more patterns seemed likely to yield results. The known pattern was that hypervariable loops of HIV-1 Env tend to be shorter and to carry fewer potential N-linked glycosylation sites (PNLGs) than their chronic counterparts [34], [35]. One hypothesis to explain this is that while larger loops may mask epitopes recognized by neutralizing antibodies, and so may be acquired during the course of infection under immune pressure, these same variable loop insertions may reduce CD4 receptor or CCR5 co-receptor access, and be disfavored at transmission [6], [36]. Our third motivation was the evidence for phenotypic trait selection at transmission: Viruses isolated during acute infection almost exclusively use the CCR5 co-receptor, while during progression HIV-1 can utilize different co-receptors, most commonly CXCR4 [17], [23], [26], [37]. In addition, cloned early viruses replicate efficiently in activated human CD4+T cells, but not in monocyte-derived macrophages [23], [26].
Here we performed a search for amino acids in Env sequences to discern patterns in amino acid substitutions (signatures) that were statistically associated either with transmission or with frequent recurrence across individuals during viral diversification in the chronic phase of the infection. We based our analyses on thousands of sequences from several hundred subjects (summarized in Table 1, with subjects individually described in Table S1). The analyses involved a series of strategies to identify signatures in single sites or sets of functionally related sites. By putting the signatures in a structural, functional, and immunological context, we then discuss what is known about the sites and the protein regions they are embedded in, to raise hypothesis regarding their possible modes of action.

Results
Sequence data
All sequences were derived using single genome amplification (SGA) methods [38] from individuals with sexually transmitted subtype B infections. We assembled as many well-characterized samples as we could that met these criteria, with contributions from many groups, with the goal of making this study as well powered as possible. Most samples were collected within the United States, with a subset from Trinidad. The demographic and clinical information relating to the subjects and samples are described in Supplement Table S1. Sequences were separated into two data sets: the ‘original’ hypothesis-raising set, and the ‘holdout’ hypothesis-validating set. Data sets were matched as described in the methods. In a second series of hypothesis-forming analyses, to increase our sample size and statistical power, we also generated a third set of sequences from acute/early infections, from infected plasma donors, and added additional sequences reported to be sampled during chronic infection from the Los Alamos database, and combined them with the original set.
Analyses strategies
We performed a series of exploratory tests to identify signatures that were significantly associated with Env protein sequences from either viruses sampled in early infection or viruses collected during chronic infection. We used an approach that accounts for the non-independence of the sequences due to phylogenetic relationships and adjusts for multiple tests (see the results and methods sections for more details) [39]. By “signature” we mean a mutational pattern that compared to expectations from unselected inheritance either (i) is enriched among the early virus, or (ii) recurs in chronic infection and yet is rare among the early variants,. We began with a search for statistically significant enrichment of single amino acids found at each position in the Env alignment. We next grouped small sets of alignment positions based on their contribution to a potential N-linked glycosylation site (PNLG) motif, membership in an inferred functional domain (functional groups), or spatial proximity defined using structural models (contact sets). We then systematically looked for signatures based on combinations of amino acid changes within these three groups, enabling us to identify additional patterns that were significantly different between early and chronic sequences.
The first approach we used tested for correlations between early versus chronic status and the amino acids found in the consensus sequences derived from individual patients, using the same methods as we have used previously [39], [40]. A consensus sequence represents the most common amino acid found at each alignment position within an individual. Consensus sequences from homogeneous early infection cases generally correspond to the modeled transmitted virus [17], [18]. The second approach we used included all sequences from each subject, modifying our earlier published methods to enable inclusion of multiple sequences per subject, as illustrated in Supplement Fig. S1. Fig. S1 shows the phylogenetic tree based on all of the available data, highlights characteristic phylogenetic patterns from examples of early and chronic infection, and illustrates the strategy we used to incorporate all sequences from every subject into the signature analysis. We initially required associations both be statistically supported in the “test” data set with a q-value of <0.2, and that they show a consistent association in a separate analysis in the “holdout” data test set. A q-value is a false discovery rate [41] that adjusts for multiple tests, critical in this study as thousands of tests were conducted. We chose a relatively high q-value cut off in our initial analysis; thus we expect approximately 20% of our sites from our first round of analysis to be by chance. We then used then conservative strategy of requiring validation in a completely separate holdout set to minimize false positives (Type I errors). This was very stringent, and we only found a small number of signatures. Therefore, we subsequently did an analysis combining data from all subjects, test and holdout and plasma donors, using a cross-validation strategy to test the statistical robustness of the observed signature sites. This provided an alternate view of the data that minimizes false negatives (Type II errors).
Identification of a signature at position 12 in the Envelope signal peptide
Using just the consensus sequences from each subject, only one signature amino acid at position 12 in Env was identified through an analysis of all amino acids found at each single alignment position in Env in both the test and holdout sets. Mutating away from His at position 12 (expressed here as !H12) was statistically enriched in chronic viruses, while a stable His was enriched in early viruses (p = 0.001, for details see Table 2). The distribution of amino acids at position 12 for each subject is shown in Supplement Fig. S2A. H12 is the most common amino acid among both early and chronic viruses, but it was enriched among early sequences. This was true for the within-subject consensus sequences (74% in early versus 57% in chronics were His), as well as all of the natural sequences (3114/4181, 74%, of early sequences were His, as compared to 1150/2122, 54%, of chronic sequences). Thus H12 is enriched among early infection relative to chronic sequences (odds ratio = 2.5, 95% CI 2.2–2.8, Fisher's p<2×10−16). However, as demonstrated in Bhattacharya et al. [39], a simple analysis testing for enrichment can be profoundly biased by lineage effects, as sequences are not independent but related by shared phylogenetic history. Thus without a phylogenetic correction even such apparently strong associations should be viewed with caution. In the case of the !H12 chronic signature we have such support (Table 2), and in all of the other signature identification strategies employed here (Tables 3–5) we have used a phylogenetic correction.
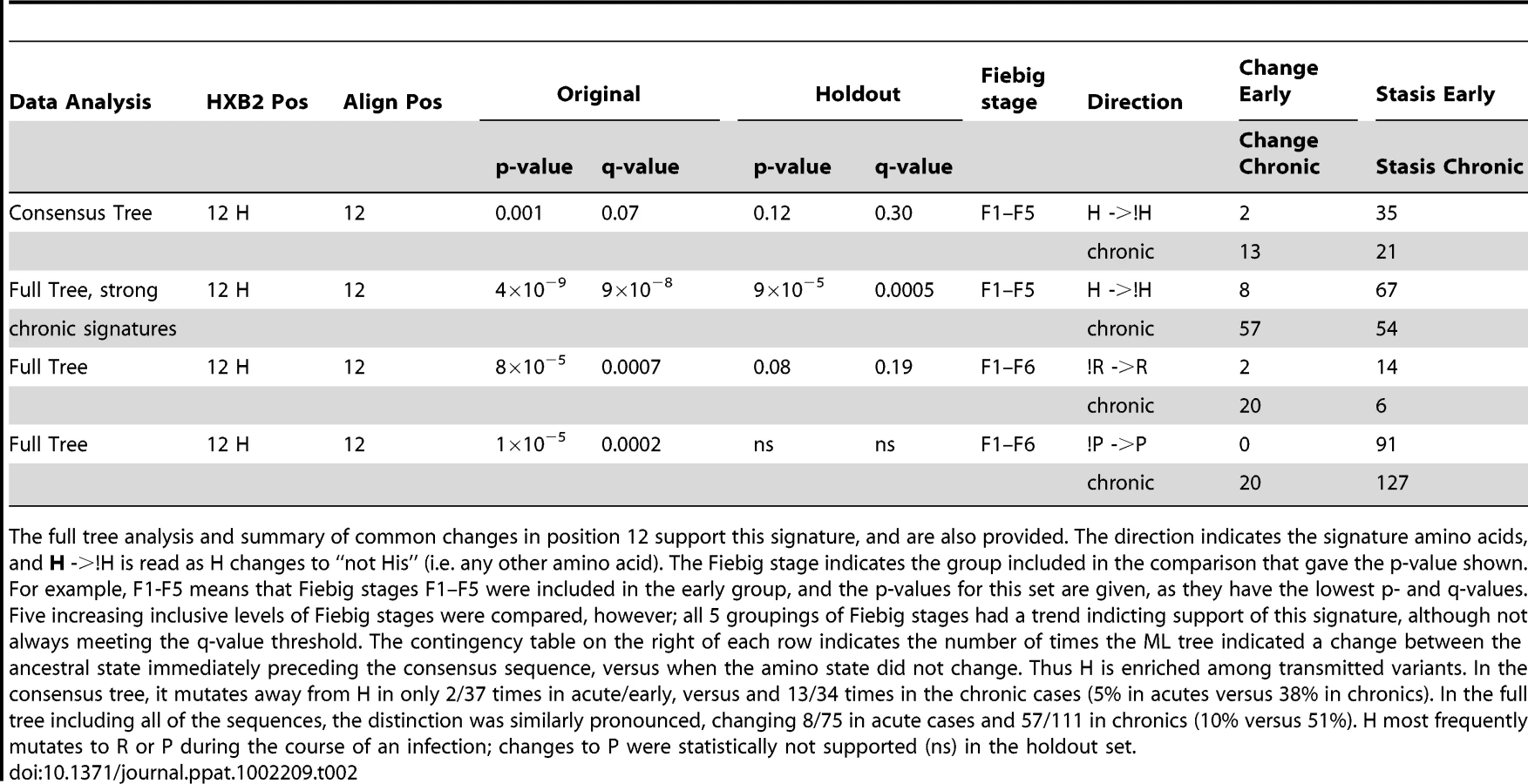
We did not see significant increases in changes towards H12 in early Envs when using a phylogenetic correction, only the reciprocal signature, away from H12 (!H12), in chronics. This could be because these two tests, both based on frequencies of changes from ancestral states, not just simple counts, had different powers in our dataset. The statistic that captures inferred H12 to !H12 changes in the phylogenetic tree in chronic infection was powered by H being the most common amino acid in this position, and so the most commonly inferred ancestral amino acid. In contrast, a statistic looking for changes towards H12 in early sequences required the relatively rare !H12 as an ancestor. In other words, we were statistically better powered to see changes away from His in chronics than towards His in early infection, and this simple explanation may account for the lack of significant association with changes towards H in early infection despite a high level of significance for !H12 in chronic infection.
Identification of a transmission signature at position 415, near the CCR5-binding site
After detecting only a single signature in our first analysis of consensus sequences, we were concerned that we did not have adequate power to detect potentially important but subtle signatures. Thus, to improve our power in the hypothesis-raising context, we extended our original data set with the set of samples from acute and early infection plasma donors, and a set of chronic samples from the Los Alamos database (www.hiv.lanl.gov); our holdout set remained the same (Table 1). A factor complicating our analysis was that although 80% of early patients were productively infected with only one HIV-1 strain, the rest were clearly infected by multiple transmitted viruses. Given that this latter group might have multiple transmissions because of a less restricted transmission bottleneck, we next analyzed only the subset of the early infection cases that were established by a single virus [17]. When one consensus sequence per patient was analyzed after excluding heterogeneous acute infections, a signature pattern of not having a Thr at HXB2 position 415 (!T415), was found to be enriched in acute infection samples (Table 3). This position is part of a PNLG sequon at N413, lies at the end of the flexible part of the gp120 V4 loop, and is in the conformationally conserved part of the outer domain. It is structurally proximal to three regions of potential interest: the binding site of several CD4-binding site (CD4bs) antibodies (Fig. 1a) [42]; two sites that have been implicated in co-receptor binding by mutational studies, positions 419 and 444 [43], [44]; and two key residue for mannose addition for the 2G12 epitope, N295 and N332 [45], [46]. We therefore checked if there was a correlation between the presence or absence of T415 and neutralizing antibody (NAb) IC50 scores that were available for a set of SGA-derived pseudotyped Envs (Table S3). !T415 (Envs lacking the PNLG) was associated with increased b12 neutralization sensitivity (p = 0.0001, Wilcoxon rank test). In contrast, neutralization by sCD4 was not significantly correlated with the !T415 signature (p = 0.2756, Wilcoxon rank test). Detectable neutralization by the CD4-inducible (CD4i) monoclonal antibody 17b, or by 17b with sCD4, was extremely rare in this dataset and observed only 3/113 times. In all three cases, a T415 was present, suggesting that its presence did not inhibit access to the 17b binding site, but this result was not statistically significant. Finally, this site was not significantly correlated with neutralization susceptibility to monoclonal antibody 2G12, which critically depends on other nearby PNLG sites in Env [45], [46].


Analysis combining consensus data from all subjects using cross-validation
In a hypotheses-raising framework, we also did an exploratory signature test on consensus sequences across all positions, combining the subjects listed in Table 1 to further increase our power. For this analysis we compared consensus sequences representing the 135 acute or early infection subjects to the 86 chronic infection subjects sampled and sequenced through this project. To further minimize Type II error and be inclusive in a hypothesis-raising framework, a liberal q-value of 0.5 was used. As stated above, this analysis, with a larger N but without a strict separation of hypothesis generating and validation sets, is not as statistically robust as the original analysis with a distinct validation set. We used a stratified 10-fold cross-validation test as an assessment of the robustness of the predictor. Ten potentially interesting signatures were identified with this strategy, including continuing support for the signatures !H12 and !T415 with a range of cross-validation support, with the signature at position 12 yielding a high degree of support (Table 4). 2 of these 10 associations were early infection signatures (!T415 and F712), the other 8 were chronic. We also performed an additional 10-fold cross validation analysis to reduce the possibility that the observed signatures were the result of an alignment artifact (see methods for alignment details). Our primary alignment for our original analysis was created using the Genecutter alignment tool coupled with a HMMER model [47]; we we repeated the procedures on a second distinct alignment generated with the alignment program MAFFT [48], [49]. 8/10 of the signature sites defined using the HMMER alignment were also found in the MAFFT alignment; the two that were not found in the MAFFT alignment also had only low level support in the cross-validation test.

Identification of signatures using all sequence data from individuals
We also systematically explored the complete Env glycoprotein using all available sequences from individuals, not just the per-individual consensus sequences. To do this, the sequence at the node preceding the ancestral node within each subject in the reconstructed phylogenetic tree was estimated by maximizing the marginal likelihood [39], [50], and the number of times each ancestral amino-acid was estimated to have changed between that node and the sampled sequences within each subject was calculated (See Fig. S1 for an illustration of the strategy). As with our first exploration of the consensus sequences we validated the results from the test data with the holdout data. Position 12 was again found to commonly mutate away from H, most often to R or P, during chronic infection (Fisher's exact p-value of 4×10−9, Table 2). Although we found changes specifically associated with early signatures at a number of positions in the original test set, none of these associations were also supported in the holdout validation set. In contrast, many chronic signatures (specific changes found repeatedly in chronic patients) were supported in both the test and holdout sets. 25 signature patterns were found that were indicative of recurrent change during chronic infection, using the criteria of q<0.2 in the test set and q<0.3 in the holdout set; these signatures are listed in Supplement Table S2. Interestingly, 8 of these 25 chronic signatures, including !H12, were found in either the signal peptide or the cytoplasmic tail, supporting the possibility that modulation of Env expression levels may play a role in selection at transmission, and lowered Env expression levels may be important for immune evasion during chronic infection.
The interpretation of chronic signatures identified by analyzing the full-sequence alignment, not just one sequence per person, is complicated by the fact that chronic sequences are inherently more heterogeneous, and hence display more changes than acute sequences, and we can not distinguish between associations arising due to repeated mutations in a small number of very complex chronic infections, and a pattern repeated across multiple patients. Thus we did one further computational experiment to help interpret our observed levels of significance. Since we were interested in identifying recurring patterns across multiple patients, we performed a shuffling test where we randomized the acute/chronic classification categories and redid the signature analysis 10 times (these analyses are extremely computationally intensive, so it was only feasible to do 10 such randomizations for this study). This randomization should maintain significance if it arose as recurrent pattern that was distributed across many distinct infections, but would remove the signal if it was an anomaly resulting from a single or very small set of complex patients. The results of this re-sampling experiment showed that while low p-values did indeed occur even after randomization, p-values of less than 10−8 were not found in the analyses of these randomly classified data (Fig. 2). Four of the chronic mutational signatures were found to both be significant in the test data with p-values of less than 10−8, and also were supported in the holdout data: !H12, !N397, !T399, and !N362 (Tables 2 and 3). Thus, these 4 signatures were singled out as being the most robust. Like the consensus signature analysis, the full tree signature captured the !H12 chronic infection signature (Table 2). Two additional full tree chronic signatures at position 12 shown in Table 2 (12R and 12P); they represent the most common amino acid substitutions in position 12 as it mutates away from His. The other three robust chronic amino acid signature patterns all impact PNLGs: positions 397 and 399 are part of the same PNLG, and 362 is in a PNLG in the C3 region.

Next, associations between the presence or absence of intact PNLG motifs with early versus chronic sequences were examined. Glycans can play an important role in immune escape and immunogenicity, can contribute to transmissibility and impact cell entry [51], [52], [53], and several of the single site signatures already described are part of PNLGs. We identified six PNLG motifs (N-X-[ST], where X is any amino acid other than Pro) that were significantly associated with a repeated pattern of loss during chronic infection (Table 5). These PNLGs spatially mapped on an X-ray structure of gp120 are shown in Figure 1b. The per-subject frequency of one of these patterns, the PNLG motif at position 397–399, is illustrated in Supplement Figure S2B–the PNLG at position 397 was conserved overall (Fig. S2B), although it was more likely to be present early in infection (Table 5, q-value = 3×10−10 in the original data, 0.0001 in the holdout data). One of the PNLG signatures, that enables glycosylation at position 392, is part of the monoclonal antibody 2G12 epitope [45], [46], [54]. Experimental data from Nab IC50 scores 2G12 from 113 clones representing SGA clones from early transmission cases (Table S3), and confirmed that the glycosylation motif at position 392 was highly correlated with 2G12 neutralization (p = 0.006, Wilcoxon rank test).
![Summary statistics using the combined original and PD/DB sets and holdout set to the gain or loss of PNLGs, defined as the motif NX[ST], where N is Asp, X is any amino acid besides Pro, and [ST] is a Ser or Thr.](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/4b53288dbbe63b7f00ae8695f9f8443e.png)
Identification of a complex signature near the CCR5 Coreceptor-binding site (CCR5 CoRbs)
Clearly, analysis of single amino acid positions may miss complex mutational patterns in functionally or conformationally important regions. Given the vast number of combinations of alignment positions and the range of different amino acids at each position, we are limited in our ability to look at arbitrary combinations of sites and amino acids across the full Env sequence, , due to multiple test issues and limited power due to sampling constraints (Table 1) compounded by computational feasibility. Thus, we performed a focused in depth exploration for signatures based on a small number of combinations of sites, including only amino acids within narrowly defined sets of functionally related sites [3] (Table S4). How extensively we searched combinations of sites within these sets was determined dynamically as described in the methods; however, at a minimum, all combinations of up to 3 amino acids at each of 2 positions were searched within each functional region, using a sliding window approach to span different amino acid subsets and combinations within each functional domain. These functional regions included: the CD4bs in gp120; the CCR5 CoRbs region in gp120; positions known to impact R5/X4 tropism; a subset of the V3 loop positions; the b12 binding site in gp120; residues predicted to reside at the gp120/gp41 trimer interface; the gp120 V2 region implicated in binding the gut homing receptor; 2F5/4E10 binding sites in gp41; the lentivirus lytic peptide LLP1 and LLP2 regions of the gp41 cytoplasmic domain; and sites that have been related to membrane fusion, including sites in which changes were shown to result in increased or decreased entry (see Table S4 for positions included). Despite this extensive search, only one statistically significant association with a complex signature was identified and validated in both the test data and holdout data; it was found in a CCR5 CoRbs set and the signature was defined as: L122-[IV]201-N377, with repeated mutation away from this pattern in chronic samples. The statistical summary of this signature pattern is given in Table 3, and the spatial locations of these sites are mapped on gp120 in Fig. 1c. The CCR5 model set contains residues that are proximal to the highly conserved critical residues that take part in the binding to CCR5, but that are clearly amenable to positive selection since they are variable at the population level.
Biochemical patterns in structure-based regional clusters
In our final exploration of this data, we searched for early infection or chronic signatures defined by changes in amino acid chemistry in spatially defined local regions. Our reasoning was that transmission signatures would not necessarily have to involve particular amino acid substitutions at a single site or a collection of sites, but rather might reflect a complicated amino acid substitution pattern that could in turn affect the structure or chemical nature of specific spatial regions within the Env structure. Such regional changes may impact expression or binding to receptors and antibodies. To explore this possibility, we first defined 395 contact sets of spatially defined clusters structurally centered on the amino acids included in the X-ray structure of the gp120 core from the YU2 strain [55], as described in the methods. Each set contained a up to10 amino acids that were less than 10 Å from the center amino acid of the contact set, based on all-atom molecular dynamic simulations. To capture the effects of dynamic interaction between flexible and core regions, no distinction was made for surface residues.
It was not feasible to analyze all neighborhood lists with all combinations of explicit amino acid transitions, so we simplified the data by calculating a regional additive polarity score for the amino acids in each neighborhood cluster (see Methods). Unlike the discrete change-stasis nature of the variables (acute versus chronic) used for the other signature analyses in this study, this score was a continuous variable, so we used the method of phylogenetically independent contrasts [56] to identify changes in polarity that correlated with early or chronic infection sequences. Three statistically significant regions were identified (Table 6), and mapped on the three-dimensional structure of gp120 (Fig. 3). In all three regions, the region became more polar during chronic infection. All three sets have amino acids that share or border the binding sites of CD4, and b12 [57], [58]. The polarity scores did not correlate significantly with sCD4 or b12 neutralization when compared the with experimental binding data (Table S3). Sets 270 and 368 border the highly conserved CD4 binding loop region (HXB2 positions 364–373). Sets 362 and 368 consist of additional residues from β23 strand and V5 loop region that take part in binding to CD4 and b12. All three sets shared a three amino acid segment (465–467) that constitutes part of the binding site for the potent broadly neutralizing monoclonal antibody VRC01 [57], [58].


Hypervariable loop length and number of glycosylation site differences between acute and chronic samples
We tested whether the hypervariable regions V1–V2, V4, or full gp120 revealed a pattern of reduced loop length or number of PNLG sites in the acute/early samples relative to the chronic samples, as would be expected from the literature [59]. When we compared the distributions of all of the within-subject Env consensus sequences in the acute/early versus chronic subjects, fewer PNLG sites overall were found in gp120s from early infection (p = 0.008, Wilcoxon signed rank test). There was also a trend towards fewer PNLG sites in the V1V2 loops (Wilcoxon p = 0.03), as well as a trend toward reduced V4 loop lengths ((Wilcoxon p = 0.03).
Signature analyses methods that did not incorporate a phylogenetic correction
Several other strategies were employed to look for signatures among the sequences by treating the samples as independent, and not accounting for phylogenetic relationships [60]. These methods did not yield any consistent signature patterns between the hypothesis-forming test (with a q-value of <0.2) and hold-out sets (with a q-value of <0.3), although additional support for a signature at position 12 was observed; these methods and results are fully summarized in the Supplement (Text S1, Figs. S3, S4, S5, S6, S7 and Table S8). In these analyses, a lack of concordance between the hypothesis forming and test-sets could arise as a consequence of a lineage effect dominating the signal in the hypothesis-forming set; alternatively, the subjects and sampling may have been too dissimilar to reproduce subtle effects.
Discussion
In this study we performed a comprehensive analysis of HIV-1 Env sequences to identify signature patterns in proteins that are significantly different in chronic versus early sequences. Here we focus on interpreting the strongly statistically supported signature patterns in the context of what is known about the biological role of these sites.
Signature sites in the signal peptide and cytoplasmic domain
It was intriguing that among the 25 significant signatures identified upon combining all of the data (Table S2), 3 were located in the signal peptide of gp160, and 4 in the cytoplasmic domain. The recurrence of patterns of mutational change in these two regions during chronic infection raises the possibility that they may indirectly influence immune evasion by altering Env protein folding, modification or expression levels. The signal peptide directs Env in its co-translational translocation to the endoplasmic reticulum (ER), where it undergoes further folding, glycosylation, and trimerization [61]; it may also serve as a gatekeeper for the release of correctly folded proteins [62]. It is unusually long (30 amino acids on average), and contains a number of highly charged residues in the N-terminal region [63], [64] spanning position 12, one of our most robust signatures (Table 2). Signal peptides play a role in the efficiency of the protein secretion and in orienting proteins in membrane, influence folding and the exit from the ER [65], [66], and can impact cleavage rates [63], [67]. A slower cleavage rate down-regulates the rates of folding, intracellular transport and secretion [63], [65], [68], [69].
The Env cytoplasmic domain of HIV-1 is also unusually long; at 150 amino acids long, three times longer than that found in typical lentiviruses [70]. It contains three helical fragments called lentivirus lytic peptides (LLPs) [71] that have been implicated in cell surface Env expression [72], [73], incorporation into virus particles [74], [75], fusogenicity [76], [77], and Env's localization in lipid rafts [71]. The chronic infection signatures in the cytoplasmic tail (Table S3) are all concentrated on the LLP-3 segment. This segment has a strong potential to associate with and perturb the membrane [78], and a di-aromatic motif of Y802 W803 in this region has been associated with retrograde transport of Env to the trans-Golgi network [79].
The acute signature site at position 415
!T415 was strongest early sequence signature observed, indicating that the PNLG at 413–415 is selected against at or immediately after transmission. This PNLG is glycosylated when present [80], and is located near the C terminal end of the V4 loop, proximal to both the CCR5 CoRbs and the CD4bs regions that impact both antibody access (Fig. 1a). A highly conserved sequence motif that takes part in CCR5 binding, RIKQ (HXB2 419–422), is just a few residues upstream [43], [44], [81]. The conserved sequence motif PCR (HXB2 417–419) that participates in the binding to monoclonal b12 is also in the neighborhood of this site [58], consistent with our finding that the presence of the PNLG motif at 413–415 is highly correlated with reduced b12 susceptibility. The glycosylation site at 413–415 has repeatedly been singled out as a relevant immune escape site in recent neutralizing antibody studies. Acquisition of a PNLG at 413–15 has been demonstrated to confer escape from autologous antibodies in longitudinal studies of the trajectory of escape in both an HIV-1 infected person (David Montefiori, personal communication), and in a rhesus macaque infected with SIVmac239 [82]. Furthermore, this region in association with the C3 α-2 helical domain is thought to contribute to patterns of neutralization susceptibility [83], [84], [85].
Two studies have found the presence of a glycosylation site 413–415 to be associated with virus isolated from individuals capable of eliciting potent or broadly neutralizing antibodies [40], [86]. This correlation was proposed to either result from a recurrent pattern of escape in people who make potent broad neutralizing antibodies, or as common feature in Envs able to elicit good antibodies [40]. We have tested a strain that has the glycosylation site at 413–415 present (strain CH0219), isolated from an individual who had made very potent broadly neutralizing antibodies in response to infection [40]. This Env was resistant to autologous antibodies in sera from CH0219, supporting its role in antibody escape. Furthermore it was found to be an extremely poor immunogen for eliciting neutralizing antibodies in guinea pigs (BFH, unpublished data). These findings are consistent with the intuitive hypothesis raised by our current signature analysis, that the addition of a glycosylation site at 413–415 provides a common escape mechanism during chronic infection by blocking access to a key epitope, but that it is selected against in early viruses, resulting in the observed !T415 signature pattern.
Implications of the repeated patterns of loss of glycosylation motifs during chronic infection
Changes in glycosylation play a key role in chronic infection, and either the gain or the loss of a particular glycosylation sites can both result in immune escape [32], [87]. As discussed earlier, reduced loops lengths and numbers of PNLGs are characteristic of early viruses, and although the pattern can be subtle and difficult to discern in the B subtype [34], [35], we did find supporting evidence for an overall pattern of reduced numbers of PNLGs after transmission in this data set; this reduction in PNLG sites occurs in the hypervariable loops. In contrast, most of the specific signature PNLGs we have identified are clustered in the outer domain, and these are lost not at transmission but in the course of chronic infection (Fig, 1b). The statistical counterpoint to the chronic loss-of-glycosylation-motif signatures is relative conservation of these PNLG sites at transmission, consistent with a scenario that these specific sites facilitate transmission in early infection, and their loss contributes to immune escape in chronic infection.
Several of the signature PNLGs have known functional roles which support the scenario described above. First, the glycan at N188 facilitates interactions with CD4 and CCR5 [88], and the loss of glycosylation sites in this region have been associated with diminished replicative capacity [55], [57], [58], [89]. Changes in this region have also been associated with immune escape from some of the first neutralizing antibodies in natural infection [33], [85], and a glycan knock-out at position 188 impacts the neutralization potency of the recently isolated broadly neutralizing antibodies PG9 and PG16 [90]. Thus selection for the glycan may occur at transmission, and selection away from in during immune escape from antibodies similar to PG9 and PG16. Similarly, N362 has been shown to contribute specifically to enhanced fusogenicity [91], a property that might be favored during transmission. PNLG 362 and 462 are near the CD4bs, and the b12 and VRC01 monoclonal antibody binding sites [55], [57], [58], and the CD4bs is a common target of neutralizing antibodies in natural infection [92], [93]. Finally, the PNLGs at positions 392, 397 and 356 are all part of the “silent face” of gp120 [94], [95]. The oligomannose glycans that are clustered on the silent face of HIV are ligands for DC-SIGN, a lectin found on the surface of dendritic cells [96]. Dendritic cells encounter HIV soon after mucosal exposure [97], and may have a role in enhancing the efficiency of HIV transmission [88], [91], [98]. A mannose at position 392 is also a critical component of the epitope of the neutralizing antibody 2G12 [45], [54], and our data confirm this previously well-established relationship. Although the 2G12 epitope may not be a common a target of neutralizing antibodies in natural infection [99], antibodies to the 2G12 epitope in neutralizing sera have been found in long-term non-progressors [100], suggesting the glycan shield at the silent face of HIV can be a point of vulnerability in some circumstances. Creating high-density mannose clusters that mimic HIV's glycan shield are being explored as a vaccine strategy [101], [102].
Complex chronic signatures in localized regions of Env
Despite testing for complex multi-site signatures within several functional domains in Env, only one multi-site signature was identified, a chronic signature in the CCR5 CoRbs set (Table 3). The CCR5 CoRbs can be a target for broadly neutralizing antibodies [92], [93], and non-neutralizing antibodies against the CCR5 CoRbs may also be able to impose selection on the virus [103]. Interestingly, the only identified signatures found associated with Env glycoproteins that were isolated from individuals that made broad and potent neutralizing antibody responses were also localized in the CCR5 CoRbs [40]. We also tested for distinctive biochemical patterns in local spatial regions in the gp120 structure, and identified three regions that are proximal to the CD4 binding site [57], [58] that undergo change in polarity (Fig. 3). The regions of gp120 surrounding the CD4bs are the most conserved in Env when considered at a structural level [3], thus providing a vulnerable target for cross-reactive HIV antibodies [57], [58]. Changes in electrostatic potential may enable antibody escape from at least some antibodies in HIV-infected individuals who naturally mount a potent and cross-reactive anti-CD4bs antibody response [93], [99], [104], [105].
A summary view
While the signature patterns we have identified are significantly enriched in terms of association with either early or chronic viruses, still there are exceptions to any given pattern (Table 2–5), and thus the signatures cannot be used to accurately predict whether a given sequence is derived from an acute or chronic infection. This is not surprising, but worth noting. It is a reminder that tests that involve site-directed mutagenesis might fail to result in a phenotypic change even when a site is relevant, because the phenotypic consequences of change in a single amino acid can be context dependent. Furthermore, there may be multiple paths to the same end, and the immune responses that drive repeated patterns of escape in chronic infection are likely to be shared only by a subset of individuals who target a particular Env region. Similarly, reversion in early viruses is likely to be context dependent, depending on the presence of compensatory mutations as well as other selective pressures acting on the virus. It is also of interest that some signature patterns that might have been expected were not observed. We did not see amino acids in the V3 loop that have been noted to be associated with CCR5 co-receptor use predominate in acute infection [17], [23], [26], [37] or those associated with CXCR4 use in chronic infection [106], [107]. We think this is because of inadequate statistical power: CXCR4-using viruses rare among both our early and chronic sequences (Table S4) and there are multiple ways to manifest a CXCR4 phenotype, thus it is likely that no CXCR4-associated substitution was repeated enough to enable identification of a signature.
Despite these issues, several interesting and consistent signature patterns emerged through our study. First, multiple signal peptide and cytoplasmic domain signature patterns were found (Table S2), raising the possibility that Env expression levels may be an important generalized aspect of immune escape during chronic infection. Second, two signatures were found near the CCR5 CoRbs region; this domain is emerging as a key region for neutralizing antibody escape and induction of antibodies in a number of studies, and merits close attention as vaccine design and evaluation strategies progress. Third, the recurrent loss of glycosylation sites in key positions during chronic infection suggests that this pattern typifies an essential aspect of immune escape, leaving a profound and recurring trace at the population level. If the loss of these specific glycosylation sites mediates immune escape from common transmitted forms, in may be advantageous to include these sites in vaccines. In contrast, the loss of the PNLG at position 413–415 was enriched among early sequences, so it may be advantageous to also exclude PNLGs at 413–415 from a vaccine immunogen. Thus the signature patterns identified in this study point to post-translational regulation of Env having a role in selection of early sequences, and indicate particular protein modifications that merit consideration for immunogen design and evaluation.
Methods
Ethics statement
Written informed consent was provided by all study participants. The Duke University Health System Institutional Review Board for Clinical Investigations (DUHS IRB), has determined the specific components above under the protocol, “Acute HIV-1 Infection Prospective Cohort Study” (CR3_Pro00006579) to be in compliance with all applicable Health Insurance Portability and Accountability Act ("HIPAA") regulations.
Data sets
The acute samples were collected from individuals sampled at varying time post-infection, and were clinically staged according to Fiebig et al. [17] to estimate the time between infection and sampling [17], [21], [22]. Chronic samples were selected from individuals who were not on anti-retroviral therapy, and infected for a minimum of two years. All represented subtype B infections, and most samples were collected in the United States, although a small number were from Trinidad/Tobago, included to increase our sample size and power (Table S1). This was a retrospective study involving many cohorts, to enable us to get a large enough sample to perform signature analysis. Table S1 includes demographic and clinical information related to these samples, including viral load at the time of sampling, Fiebig stage, year of sample collection, sampling country, primary risk factor for infection, and whether the sequence evidence indicates that the new infections were established by single or multiple strains. All acute and early samples were obtained from people with sexually acquired HIV. Alignments of the full set of 6303 early and chronic SGA Env sequences used are available in the supplement, and GenBank accession numbers are provided in each of the sequence names (Tables S5–S7). As this study involved samples from HIV-1 infected human subjects, informed consent was obtained from all subjects.
The data were originally separated into two sets: the original hypothesis-raising ‘test’ set, the ‘holdout’ hypothesis-validating set. It was critical that the test and holdout sets each had a good representation of early Fiebig stages, so we ensured that the test and holdout sets each had 19 samples with a Fiebig stage of 3 or less. Each set was also matched for samples that were suggested by the data to be consequence of single infection (68% in the test set, and 65% in the holdout). The early and chronic groups within each set were matched in terms of country of origin (the early and the chronic groups each had ∼30% from Trinidad in the test set, and the early and chronic groups each had ∼5% in holdout set); this was important because the Trinidad sequences formed a distinct clade in phylogenetic analysis and such geographically localized clades can have systematically different patterns of mutations in early or chronic infections. Although these were sexual transmission cohorts, the risk factors for infection were not always known; heterosexuals were well represented in each group.
A third set was added to increase our statistical power for hypothesis forming (Table 1). This set was based on adding early infection samples from plasma donors in the United States, and a set of B clade chronic sequences from the Los Alamos HIV database that were from individuals who were documented in the database entry to not be on anti-retroviral therapy and who had been infected for a minimum of two years. This third set was not as well matched in terms of the clinical and geographic origin as other two sets.
Sequencing and sample characterization methods
All sequences were obtained from plasma of infected individuals using single genome amplification (SGA) methods, as previously described [17], [38]. A full alignment of all sequences used in this study is available in Supplement Table S5–S7; all sequences have been submitted to GenBank in conjunction with this paper, or else were previously submitted, and the accession number of each sequence is included in the sequence name, and at the end of this article. The positions numbers in the paper are generally given as HXB2 position numbers (http://www.hiv.lanl.gov/content/sequence/HIV/REVIEWS/HXB2.html), unless it is specified in the text that the numbering refers to the alignment position used in this study. For signature analysis, all sequences were analyzed in maximum likelihood trees, including multiple sequences from each individual; subject-specific phylogenetic clusters were consistently formed, so there were no overt contamination issues in this study.
Sequences were aligned using a HMMER alignment [47] and then codon aligned with GeneCutter (http://www.hiv.lanl.gov/), with hand correction at the borders of the regions with many insertions and deletions to rectify obvious alignment errors. The hand editing was done because the hypervariable region indels in HIV are particularly difficult for multiple alignment programs [48]-ot only do they exhibit extensive length variation, but the insertions are generally comprised of distinctive direct repeats from neighboring regions in the gene [108]. The alignment was done in iterative steps; first each subject was aligned internally, then a majority consensus sequence representing each subject was generated. For within-subject consensus generation, we considered the codons that that bases were imbedded in, and selected the most common codon for the consensus. This step was required because otherwise simple position-wise consensus sequences occasionally created codons that did not exist within the subject, as the most common bases in highly variable codon positions are not always found in combination. The subject consensus sequences were aligned, then the within-subject sequences sets were aligned to their own consensus in the framework of the full population alignment, and then the whole process was iterated. This alignment was 3120 bases long. To test for dependence on the alignment strategy used, we repeated the consensus sequence signature analysis using an unedited MAFFT alignment [48], [49]; this alignment was 3735 bases long, so had many more gaps; a SATe alignment of this same data was even longer, at 3790 bases (http://phylo.bio.ku.edu/software/sate/sate.html).
Phylogenetically-based analysis
To identify signature patterns in HIV that relate to a particular phenotype (in this case, early versus chronic status), sampled viruses cannot be treated as independent samples from a random distribution of genotypes. Any population substructure in the data exacerbates the problem. To correct for this we employed a tree corrected contingency table approach used previously [39], but with the addition of more extensive searching capabilities such as the ability to look for statistically interesting combinations of sites in functional domains and loss and gain of glycosylation site motifs [40]. The phylogeny of all sequences was inferred using a maximum likelihood method, and ancestral states were inferred at the internal nodes in the tree [39]. We used a GTR model and a maximum likelihood assignment of rates per site.
The method we originally developed to study the correlation between HIV genotypic variation and host immunological parameters was used directly to correlate the early/chronic status with the consensus genotype in each patient. This method has been previously shown to enable identification of signature sites that could be experimentally validated as biologically meaningful [39], [40], [109]. In particular, when applied to finding mutational associations with host class I HLA genotyping, the associations identified were in known or predicted cytoxic T cell epitopes with the expected frequency [39], [109], and when applied to neutralizing antibody sensitivity, critical mutational patterns were identified among the natural variants [40]. To fully utilize the availability of multiple sequences per subject in this study, we have adapted the original signature identification method to enable tracking of changes in character states observed within each individual defined relative to the most likely state at the last (closest) ancestral node outside the patient. These changes were correlated with the patient being early or chronic (Fig. S1 illustrates the method). The number of sequences sampled varied widely among the patients (indicated by the heights of the bars in Fig. S2), and the diversity at some positions was much greater than others, so a bootstrap approach was used to determine appropriate significance levels for identification of interesting signals (Fig. 1).
For quantitative signatures of continuous variables (in particular, regional polarity scores) Felsenstein's phylogenetic contrast approach was used [56] to estimate a covariance matrix, and Student's t-statistic was used to obtain significance levels for the differences between the early and chronic patients. Since the variables of interest had a bounded domain, we verified manually that the signatures did not arise from saturation of the bounds where the model was strongly violated.
Statistical testing criteria
Given that we were in a hypothesis raising mode and our expectation was that transmission signatures would be relatively subtle, and we were of necessity in a framework of limited sampling, we decided to require that associations be statistically supported in an initial training data set with a q-value of <0.2, and show at least a trend (q <0.3) towards a consistent pattern of association in a separate analysis of the holdout data. A q-value is a false discovery rate that adjusts for multiple tests [41]. There were many associations with a q<0.2 that were found either only the training or confirmatory data sets that were not supported in both sets, which we do not list here. Retaining a holdout set that is excluded from the initial analysis is not often done in this kind of correlation analysis [109], [110]. Our decision balanced the value of increasing the sample size and the potential for identifying more correlated sites, with the additional level of confidence in our primary findings provided by the holdout set analysis; we opted for the latter to limit our type I false-positive error, although potentially missing interesting signature sites and increasing type II false-negative error. A more comprehensive listing of the non-validated sites provided in the supplement reverses this, and these tables are far more likely to contain false positives, but less likely to miss true positives.
Signatures were sought comparing sequences classified as early by combining data sets incrementally from Fiebig stage 2 up to 6, such that all sequences up to a given stage were combined and then analyzed, and then contrasted with chronic data. The reason we explored the data in this increasingly inclusive fashion was to balance the increasing power that is a consequence of including additional sequences from later Fiebig stages, against the possibility that as samples are taken at progressively later Fiebig stages, transmission signatures may no longer be evident in the sample due to early immune or fitness selection pressures [23], [30]. The Fiebig stage of the data combination that produces the most significant signature associations for a given amino acid pattern is provided in the Tables manuscript to simplify presentation; the use of q-values for statistical significance guards against increasing Type I errors by this procedure.
Cross-validation strategies can provide reasonably unbiased accuracy estimates for classifiers [111], but their use in hypothesis testing suffers from the absence of reliable estimates of their variance [112], [113], [114]. In particular, they are known to have inflated type 1 error rates [115], [116] and can sometimes lead to incorrect choice [117] when used for model/feature selection; we have encountered such issues in a previous study [40], hence we did not use this approach initially for this study, rather we used the strategy described above involving a strictly maintained holdout set. When very few signatures were evident by this conservative approach, however, we turned to cross-validation; even though it has limitations, it often works well in practice [118] and is commonly used for data mining. We used a stratified 10-fold cross validation approach [119] to check robustness of our findings when analyzing the combined test and holdout datasets, to raise hypotheses for further work. We stratified by the early/chronic status, as well as by the sample's geographic origin (i.e. whether the sample originated in the United State or in Trinidad and Tobago, given that Trinidad and Tobago B subtype viruses formed a distinct lineage relative to the B subtype US viruses). 90% of the sample was selected randomly for a training set, and 10% was retained as a test set. As with the full data set analysis of all patient consensus sequences, a q-value of 0.5 was used for the training set criteria of positive, and the test was considered a match if the direction of the odds ratio was preserved (<1 or > = 1).
Grouping of positions and amino acids for signature analysis based on alignment positions
Our primary analysis was concerned with single site signatures. In addition to the single site signatures we considered the loss or gain of aligned PNLG motifs, where the motif is: NX[T/S], and N is Asn and T/S is either Thr or Ser [120]. Regions of the alignment that could not be reliably aligned due to insertion/deletion events were essentially excluded, by systematically excluding positions where more than 10% gaps had been included to maintain the alignment. One important consequence of this is the exclusion of hypervariable domains where we did not feel confident of the alignment, so associations would be missed in these regions as they could not be reliably identified.
Signature analysis of combinations of sites in functional domains of HIV-1 Env
We also defined sets of amino acid based on the computed structure and presumed function of the envelope protein. Three sources were used to define these sets, as described in Korber and Gnanakaran [3]. A search of the literature provided critical residues obtained through site directed mutational experiments that probed sites within functional domains as well as antibody binding motifs in gp120 and gp41. We compiled those sites classified according to corresponding functional activities and antibody epitopes. Second, x-ray crystal structures of gp120 are available with different binding partners, including neutralizing monoclonal antibodies. In these cases, we identified the set of relevant key sites based on spatial contacts.
The amino acid positions included in these sets, and the references used to select them, are provided in the supplement (Table S4). The functional domains in gp120 that we considered included CD4 and co-receptor (CCR5 and CXCR4) binding sites, sites that correlate with CCR5 and CXCR4 co-receptor usage, exclusive sites within V3 loop that take part in binding to co-receptor, and V2 gut mucosal homing receptor binding sites. In gp41, we included sites in LLP1 associated with virion incorporation, LLP2 sites associated with Tyrosine-dependent sorting signal and exposure of CD4 binding site, and an additional set of sites associated with modulating entry during fusion process. We also included a set of amino acid positions in both gp120 and gp41 thought important to maintain the Env trimer, and those sites that lie on the interface between gp120 and gp41. The gp120 epitope sites included the binding sites of monoclonal antibodies b12 and 17b. In gp41, the epitope sets included two sets in MPER region covering 4E10 and 2F5 binding sites.
We looked as exhaustively as was feasible given our data and computational constraints for early or chronic signatures in functional domains. How extensively the combinations of sites and amino acids in a given functional domain were explored was determined dynamically. All sets were initially explored based on combinations of 3 positions in the functional domain and up to three amino acids per position; if this resulted in more than 5 million patterns, we then considered only 2 positions and 3 amino acid combinations in the first series of tests. We then tested incrementally more combinations of amino acids at the each of the positions until we reached 5 million patterns per domain, a limit based on computational feasibility; however, if at this point the p - and q-values were still improving, we increased this to up to 10 million tests. We then repeated the incremental iterations including more sites rather than more amino acids per site. When this was done, the combinations with the best p - and q-values were compared between the test and holdout sets; then end result was that essentially only one complex combination signature, in the CCR5 model set, was supported in both sets.
Contact matrix based signature analysis
A third kind of amino acid set analyzed was based on spatial proximity; we called these ‘contact’ sets. These sets were created from the contact matrix obtained from long timescale molecular dynamics simulations of liganded gp120. The gp120 structure of YU2 strain with modeled loops was carried out with molecular dynamics simulations in explicit aqueous solvent [79], [80], thus incorporating into our model the dynamics, the influence of solvent, relative flexibility of both flexible and conserved regions and the interaction between core and variable regions. Contact profiles were obtained from the simulation trajectories. For each residue in the simulated structure a contact set was generated such that it contained at most the 10 closest contact amino acids, and all amino acids included were within 10 Å of the center. We made a total of 395 contact sets corresponding to the total number of residues in the simulated gp120 molecule informed by the crystal structure. The definition of these contact sets was based both on the distance between amino acids obtained during the entire dynamics and the duration in the dynamical conformation. Sets were excluded from consideration if they contained regions of alignment uncertainty caused by insertion/deletion events.
It was not computationally feasible to analyze all contact lists with all explicit amino acid substitutions. Therefore a few contact sets were chosen for an in-depth analysis based on the full tree single amino acid scan identifying an amino acid within the contact set as potentially interesting. Combinations of positions in the sets containing these positions were analyzed in the same manner as the functional domains; this yielded no complex signatures that were supported in both the test and the holdout sets. We then simplified the information in the contact sets by grouping amino acids into standard side-chain chemically motivated equivalence classes, J = [A I L M F W V], X = [S Y T Q N H], Z = [K R], O = [D E] and U = [G P C], and their unions, and used this to test of complex signatures within all contact sets; this effort identified no new signatures in both test and holdout analysis. We then computed a polarity score for each of the contact sets, a single number representing the chemistry of each local spatial region in gp120. To do this, we used the Hopp and Woods scale, which has been used previously to identify antigenic sites [121], to assign scores to individual amino acids, and then summed these scores over the contact sets. In this case three contact sets yielded statistically interesting correlations in both the test and hold out sets. Because this score could vary almost continuously through small changes in amino acid composition, we used the method of phylogenetically independent contrasts [56] to identify changes in polarity that either correlated with transmission or were recurrent during chronic infection based on the full dataset. Though the range of polarity is finite, violating the assumptions of the method, however we found the observed signatures did not arise from saturating the bounds.
Correlation of signature sites with neutralization by antibodies and sCD4
For each of a panel of MAbs, or sCD4, concentrations required for 50% neutralization (IC50) were determined for 113 SGA-derived Envs expressed as pseudovirions [122] from 73 individuals sampled either in early or chronic infection (Table S3). This represents an extension of the set previously reported in Keele et al. [17], using the same experimental methods. To determine if there were significant correlations between the presence or absence of signatures patterns and neutralization phenotypes we used non-parametric Wilcoxon rank statistics as implemented in the R project for statistical computing http://www.r-project.org/).
Testing for correlations of between lengths and number of glycosylation sites in hypervariable loops and early versus chronic sampling
Because of pre-existing literature on this subject leading to an expectation that early Env hypervariable loops would be shorter with few glycosylation sequons, we grouped all early and all chronic samples for this study, and did not separate our data into a hypothesis forming and holdout sets. Furthermore, since we have no good models to reconstruct ancestral states for the variable loops that are subject to rapid within-subject insertions and deletions, in this study we did not correct for phylogenetic relationship between the sequences. Instead, we compared the tallies of number of glycosylation sites or loop lengths based on a single consensus sequence from each subject in early versus chronic infections using a Wilcoxon rank statistic; this simple test revealed there were less glycosylation sites overall in gp120 among early infections, supporting previous findings. We next compared the spectrum of variants found in each subject. Because the within-subject sequences are not independent and the number of such samples varied widely from patient-to-patient, we re-sampled the sequences from each subject 1,000 times to create sets with a constant sample size across subjects, which we chose to be the smallest number of sequences obtained from a single subject in the real data. We then compared the distributions found in the early versus chronic data with a Wilcoxon test, and then did a Monte Carlo test shuffling the early/chronic designations 1000 times based on each of the re-samplings, to see how often the level of distinction based on the real data was found in the randomized data.
GenBank accession mumbers
GenBank accession numbers for sequences used in this study. New sequence accession numbers for newly introduced sequences in this study: HQ216367-HQ218052, HQ238279-HQ238288. Previously published CHAVI sequence accession numbers: EU574937-EU575065, EU575067-EU575212, EU575214-EU575231, EU575233,EU575235-EU575251, EU575253-EU575265, EU575267-EU575272, EU575274-EU575441, EU575443-EU575468, EU575470-EU575552, EU575554-EU575636, EU575638-EU575704, EU575706-EU575775, EU575777-EU575852, EU575854-EU575943, EU575945-EU575980, EU575982-EU575990, EU575992-EU576064, EU576066-EU576089,EU576091-EU576237, EU576239-EU576292, EU576294-EU576296, EU576298-EU576619, EU576621-EU576642, EU576644, EU576646-EU576774, EU576776-EU576799, EU576801-EU576814, EU576816-EU576817, EU576819-EU576840, EU576842-EU576936, EU576938-EU577005, EU577007-EU577100, EU577102-EU577114, EU577116-EU577310, EU577312-EU577350, EU577352-EU577433, EU577435-EU577440, EU577442-EU577478, EU577480-EU577662, EU577664-EU578089, EU578091-EU578109, EU578111-EU578174, EU578176-EU578239, EU578241-EU578292, EU578294-EU578307, EU578309-EU578321, EU578323-EU578328, EU578330-EU578331, EU578333-EU578375, EU578377-EU578512, EU578514-EU578559, EU578561-EU578576, EU578578, EU578580-EU578636, EU578638-EU578677, EU578679-EU578686, FJ495818, FJ495937, FJ496000-FJ496001, GU330247-GU330646, GU330648-GU330861, GU330938-GU331030, GU331032-GU331095, GU331098-GU331102, GU331114, GU331116, GU331121-GU331122, GU331127, GU331129-GU331133, GU331183-GU331217, GU331634 GU331721. Previously published database chronics selected from the Los Alamos based on their annotation clearly indicating chronic infection, including one representative sequence chosen per subject sample: AY223734, AY223785, AY535433, AY535511, AY535480, AY535461, AY842807, AY842816, AY842830, DQ410613, DQ410520, DQ410533, DQ410542, DQ410569, DQ410586, DQ410599, AY357342, DQ410219, DQ410057, DQ410068, DQ410075, DQ410099, DQ410105, DQ410120, DQ410177, DQ410196, DQ222216, DQ410454, DQ410483, DQ410623, DQ410280, DQ410322, DQ410338, DQ410384, DQ410412, AY423385, DQ410232, DQ976380, DQ976394, AJ535593, AJ535602, DQ853433, DQ976430, AY314044.
Supporting Information
Zdroje
1. HaynesBFShattockRJ 2008 Critical issues in mucosal immunity for HIV-1 vaccine development. J Allergy Clin Immunol 122 3 9
2. Rerks-NgarmSPitisuttithumPNitayaphanSKaewkungwalJChiuJ 2009 Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med 361 2209 2220
3. KorberBGnanakaranS 2009 The implications of patterns in HIV diversity for neutralizing antibody induction and susceptibility. Curr Opin HIV AIDS 4 408 417
4. BarouchDHKorberB 2010 HIV-1 vaccine development after STEP. Annu Rev Med 61 153 167
5. GaschenBTaylorJYusimKFoleyBGaoF 2002 Diversity considerations in HIV-1 vaccine selection. Science 296 2354 2360
6. DerdeynCADeckerJMBibollet-RucheFMokiliJLMuldoonM 2004 Envelope-constrained neutralization-sensitive HIV-1 after heterosexual transmission. Science 303 2019 2022
7. LearnGHMuthuiDBrodieSJZhuTDiemK 2002 Virus population homogenization following acute human immunodeficiency virus type 1 infection. J Virol 76 11953 11959
8. DelwartELSheppardHWWalkerBDGoudsmitJMullinsJI 1994 Human immunodeficiency virus type 1 evolution in vivo tracked by DNA heteroduplex mobility assays. J Virol 68 6672 6683
9. FurutaYBergstromTNorkransGHoralP 1994 HIV type 1 V3 sequence diversity in contact-traced Swedish couples at the time of sexual transmission. AIDS Res Hum Retroviruses 10 1187 1189
10. ShankarappaRMargolickJBGangeSJRodrigoAGUpchurchD 1999 Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J Virol 73 10489 10502
11. WolinskySMWikeCMKorberBTHuttoCParksWP 1992 Selective transmission of human immunodeficiency virus type-1 variants from mothers to infants. Science 255 1134 1137
12. ZhangLQMacKenziePClelandAHolmesECBrownAJ 1993 Selection for specific sequences in the external envelope protein of human immunodeficiency virus type 1 upon primary infection. J Virol 67 3345 3356
13. ZhuTMoHWangNNamDSCaoY 1993 Genotypic and phenotypic characterization of HIV-1 patients with primary infection. Science 261 1179 1181
14. RitolaKPilcherCDFiscusSAHoffmanNGNelsonJA 2004 Multiple V1/V2 env variants are frequently present during primary infection with human immunodeficiency virus type 1. J Virol 78 11208 11218
15. WolfsTFZwartGBakkerMGoudsmitJ 1992 HIV-1 genomic RNA diversification following sexual and parenteral virus transmission. Virology 189 103 110
16. AbrahamsMRAndersonJAGiorgiEESeoigheCMlisanaK 2009 Quantitating the multiplicity of infection with human immunodeficiency virus type 1 subtype C reveals a non-poisson distribution of transmitted variants. J Virol 83 3556 3567
17. KeeleBFGiorgiEESalazar-GonzalezJFDeckerJMPhamKT 2008 Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci U S A 105 7552 7557
18. LeeHYGiorgiEEKeeleBFGaschenBAthreyaGS 2009 Modeling sequence evolution in acute HIV-1 infection. J Theor Biol 261 341 360
19. KearneyMMaldarelliFShaoWMargolickJBDaarES 2009 Human immunodeficiency virus type 1 population genetics and adaptation in newly infected individuals. J Virol 83 2715 2727
20. HaalandREHawkinsPASalazar-GonzalezJJohnsonATichacekA 2009 Inflammatory genital infections mitigate a severe genetic bottleneck in heterosexual transmission of subtype A and C HIV-1. PLoS Pathog 5 e1000274
21. FiebigEWWrightDJRawalBDGarrettPESchumacherRT 2003 Dynamics of HIV viremia and antibody seroconversion in plasma donors: implications for diagnosis and staging of primary HIV infection. AIDS 17 1871 1879
22. McMichaelAJBorrowPTomarasGDGoonetillekeNHaynesBF 2009 The immune response during acute HIV-1 infection: clues for vaccine development. Nat Rev Immunol 10 11 23
23. Salazar-GonzalezJFSalazarMGKeeleBFLearnGHGiorgiEE 2009 Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J Exp Med 206 1273 1289
24. KeeleBFLiHLearnGHHraberPGiorgiEE 2009 Low-dose rectal inoculation of rhesus macaques by SIVsmE660 or SIVmac251 recapitulates human mucosal infection by HIV-1. J Exp Med 206 1117 1134
25. LiuJKeeleBFLiHKeatingSNorrisPJ 2010 Low-dose mucosal simian immunodeficiency virus infection restricts early replication kinetics and transmitted virus variants in rhesus monkeys. J Virol 84 10406 10412
26. LiHBarKJWangSDeckerJMChenY 2010 High Multiplicity Infection by HIV-1 in Men Who Have Sex with Men. PLoS Pathog 6 e1000890
27. BorrowPLewickiHHahnBHShawGMOldstoneMB 1994 Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J Virol 68 6103 6110
28. BorrowPLewickiHWeiXHorwitzMSPefferN 1997 Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat Med 3 205 211
29. FischerWGanusovVVGiorgiEEHraberPTKeeleBF 2010 Transmission of single HIV-1 genomes and dynamics of early immune escape revealed by ultra-deep sequencing. PLoS ONE 5 e12303
30. GoonetillekeNLiuMKSalazar-GonzalezJFFerrariGGiorgiE 2009 The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J Exp Med 206 1253 1272
31. TurnbullELWongMWangSWeiXJonesNA 2009 Kinetics of expansion of epitope-specific T cell responses during primary HIV-1 infection. J Immunol 182 7131 7145
32. RongRLiBLynchRMHaalandREMurphyMK 2009 Escape from autologous neutralizing antibodies in acute/early subtype C HIV-1 infection requires multiple pathways. PLoS Pathog 5 e1000594
33. MoorePLRanchobeNLambsonBEGrayESCaveE 2009 Limited neutralizing antibody specificities drive neutralization escape in early HIV-1 subtype C infection. PLoS Pathog 5 e1000598
34. ChohanBLangDSagarMKorberBLavreysL 2005 Selection for human immunodeficiency virus type 1 envelope glycosylation variants with shorter V1-V2 loop sequences occurs during transmission of certain genetic subtypes and may impact viral RNA levels. J Virol 79 6528 6531
35. FrostSDLiuYPondSLChappeyCWrinT 2005 Characterization of human immunodeficiency virus type 1 (HIV-1) envelope variation and neutralizing antibody responses during transmission of HIV-1 subtype B. J Virol 79 6523 6527
36. SagarMLaeyendeckerOLeeSGamielJWawerMJ 2009 Selection of HIV variants with signature genotypic characteristics during heterosexual transmission. J Infect Dis 199 580 589
37. MargolisLShattockR 2006 Selective transmission of CCR5-utilizing HIV-1: the ‘gatekeeper’ problem resolved? Nat Rev Microbiol 4 312 317
38. Salazar-GonzalezJFBailesEPhamKTSalazarMGGuffeyMB 2008 Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J Virol 82 3952 3970
39. BhattacharyaTDanielsMHeckermanDFoleyBFrahmN 2007 Founder effects in the assessment of HIV polymorphisms and HLA allele associations. Science 315 1583 1586
40. GnanakaranSDanielsMGBhattacharyaTLapedesASSethiA 2010 Genetic signatures in the envelope glycoproteins of HIV-1 that associate with broadly neutralizing antibodies. PLoS Comput Biol 6 e1000955
41. StoreyJDTibshiraniR 2003 Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 100 9440 9445
42. ChenLKwonYDZhouTWuXO'DellS 2009 Structural basis of immune evasion at the site of CD4 attachment on HIV-1 gp120. Science 326 1123 1127
43. RizzutoCSodroskiJ 2000 Fine definition of a conserved CCR5-binding region on the human immunodeficiency virus type 1 glycoprotein 120. AIDS Res Hum Retroviruses 16 741 749
44. RizzutoCDWyattRHernandez-RamosNSunYKwongPD 1998 A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science 280 1949 1953
45. SandersRWVenturiMSchiffnerLKalyanaramanRKatingerH 2002 The mannose-dependent epitope for neutralizing antibody 2G12 on human immunodeficiency virus type 1 glycoprotein gp120. J Virol 76 7293 7305
46. ScanlanCNPantophletRWormaldMROllmann SaphireEStanfieldR 2002 The broadly neutralizing anti-human immunodeficiency virus type 1 antibody 2G12 recognizes a cluster of alpha1-->2 mannose residues on the outer face of gp120. J Virol 76 7306 7321
47. EddySR 1995 Multiple alignment using hidden Markov models. Proc Int Conf Intell Syst Mol Biol 3 114 120
48. GolubchikTWiseMJEastealSJermiinLS 2007 Mind the gaps: evidence of bias in estimates of multiple sequence alignments. Mol Biol Evol 24 2433 2442
49. KatohKAsimenosGTohH 2009 Multiple alignment of DNA sequences with MAFFT. Methods Mol Biol 537 39 64
50. KorberBMuldoonMTheilerJGaoFGuptaR 2000 Timing the ancestor of the HIV-1 pandemic strains. Science 288 1789 1796
51. KongLSheppardNCStewart-JonesGBRobsonCLChenH 2010 Expression-system-dependent modulation of HIV-1 envelope glycoprotein antigenicity and immunogenicity. J Mol Biol 403 131 147
52. SandersRWvan AnkenENabatovAALiscaljetIMBontjerI 2008 The carbohydrate at asparagine 386 on HIV-1 gp120 is not essential for protein folding and function but is involved in immune evasion. Retrovirology 5 10
53. ReitterJNMeansREDesrosiersRC 1998 A role for carbohydrates in immune evasion in AIDS. Nat Med 4 679 684
54. TrkolaAPurtscherMMusterTBallaunCBuchacherA 1996 Human monoclonal antibody 2G12 defines a distinctive neutralization epitope on the gp120 glycoprotein of human immunodeficiency virus type 1. J Virol 70 1100 1108
55. KwongPDWyattRMajeedSRobinsonJSweetRW 2000 Structures of HIV-1 gp120 envelope glycoproteins from laboratory-adapted and primary isolates. Structure 8 1329 1339
56. FelsensteinJ 1985 Phylogenies and the comparative method. Am Nat 125 1 12
57. ZhouTGeorgievIWuXYangZYDaiK 2010 Structural Basis for Broad and Potent Neutralization of HIV-1 by Antibody VRC01. Science. 329 811 817
58. ZhouTXuLDeyBHessellAJVan RykD 2007 Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature 445 732 737
59. LiuYCurlinMEDiemKZhaoHGhoshAK 2008 Env length and N-linked glycosylation following transmission of human immunodeficiency virus Type 1 subtype B viruses. Virology 374 229 233
60. GilbertPBWuCJobesDV 2008 Genome scanning tests for comparing amino acid sequences between groups. Biometrics 64 198 207
61. LandABraakmanI 2001 Folding of the human immunodeficiency virus type 1 envelope glycoprotein in the endoplasmic reticulum. Biochimie 83 783 790
62. LandAZonneveldDBraakmanI 2003 Folding of HIV-1 envelope glycoprotein involves extensive isomerization of disulfide bonds and conformation-dependent leader peptide cleavage. FASEB J 17 1058 1067
63. LiYLuoLThomasDYKangCY 1994 Control of expression, glycosylation, and secretion of HIV-1 gp120 by homologous and heterologous signal sequences. Virology 204 266 278
64. PancinoGEllerbrokHSitbonMSonigoP 1994 Conserved framework of envelope glycoproteins among lentiviruses. Curr Top Microbiol Immunol 188 77 105
65. von HeijneG 1984 Analysis of the distribution of charged residues in the N-terminal region of signal sequences: implications for protein export in prokaryotic and eukaryotic cells. EMBO J 3 2315 2318
66. BoydDBeckwithJ 1990 The role of charged amino acids in the localization of secreted and membrane proteins. Cell 62 1031 1033
67. EllerbrokHD'AuriolLVaqueroCSitbonM 1992 Functional tolerance of the human immunodeficiency virus type 1 envelope signal peptide to mutations in the amino-terminal and hydrophobic regions. J Virol 66 5114 5118
68. RehmASternPPloeghHLTortorellaD 2001 Signal peptide cleavage of a type I membrane protein, HCMV US11, is dependent on its membrane anchor. EMBO J 20 1573 1582
69. LiYBergeronJJLuoLOuWJThomasDY 1996 Effects of inefficient cleavage of the signal sequence of HIV-1 gp 120 on its association with calnexin, folding, and intracellular transport. Proc Natl Acad Sci U S A 93 9606 9611
70. HunterESwanstromR 1990 Retrovirus envelope glycoproteins. Curr Top Microbiol Immunol 157 187 253
71. YangPAiLSHuangSCLiHFChanWE 2009 The cytoplasmic domain of human immunodeficiency virus type 1 transmembrane protein gp41 harbors lipid raft association determinants. J Virol 84 59 75
72. EdwardsTGWyssSReevesJDZolla-PaznerSHoxieJA 2002 Truncation of the cytoplasmic domain induces exposure of conserved regions in the ectodomain of human immunodeficiency virus type 1 envelope protein. J Virol 76 2683 2691
73. BultmannAMuranyiWSeedBHaasJ 2001 Identification of two sequences in the cytoplasmic tail of the human immunodeficiency virus type 1 envelope glycoprotein that inhibit cell surface expression. J Virol 75 5263 5276
74. MurakamiTFreedEO 2000 The long cytoplasmic tail of gp41 is required in a cell type-dependent manner for HIV-1 envelope glycoprotein incorporation into virions. Proc Natl Acad Sci U S A 97 343 348
75. PillerSCDubayJWDerdeynCAHunterE 2000 Mutational analysis of conserved domains within the cytoplasmic tail of gp41 from human immunodeficiency virus type 1: effects on glycoprotein incorporation and infectivity. J Virol 74 11717 11723
76. JiangJAikenC 2007 Maturation-dependent human immunodeficiency virus type 1 particle fusion requires a carboxyl-terminal region of the gp41 cytoplasmic tail. J Virol 81 9999 10008
77. KaliaVSarkarSGuptaPMontelaroRC 2003 Rational site-directed mutations of the LLP-1 and LLP-2 lentivirus lytic peptide domains in the intracytoplasmic tail of human immunodeficiency virus type 1 gp41 indicate common functions in cell-cell fusion but distinct roles in virion envelope incorporation. J Virol 77 3634 3646
78. KligerYShaiY 1997 A leucine zipper-like sequence from the cytoplasmic tail of the HIV-1 envelope glycoprotein binds and perturbs lipid bilayers. Biochemistry 36 5157 5169
79. BlotGJanvierKLe PanseSBenarousRBerlioz-TorrentC 2003 Targeting of the human immunodeficiency virus type 1 envelope to the trans-Golgi network through binding to TIP47 is required for env incorporation into virions and infectivity. J Virol 77 6931 6945
80. IrunguJGoEPZhangYDalpathadoDSLiaoHX 2008 Comparison of HPLC/ESI-FTICR MS versus MALDI-TOF/TOF MS for glycopeptide analysis of a highly glycosylated HIV envelope glycoprotein. J Am Soc Mass Spectrom 19 1209 1220
81. CormierEGTranDNYukhayevaLOlsonWCDragicT 2001 Mapping the determinants of the CCR5 amino-terminal sulfopeptide interaction with soluble human immunodeficiency virus type 1 gp120-CD4 complexes. J Virol 75 5541 5549
82. SatoSYusteELauerWAChangEHMorganJS 2008 Potent antibody-mediated neutralization and evolution of antigenic escape variants of simian immunodeficiency virus strain SIVmac239 in vivo. J Virol 82 9739 9752
83. RongRGnanakaranSDeckerJMBibollet-RucheFTaylorJ 2007 Unique mutational patterns in the envelope alpha 2 amphipathic helix and acquisition of length in gp120 hypervariable domains are associated with resistance to autologous neutralization of subtype C human immunodeficiency virus type 1. J Virol 81 5658 5668
84. GnanakaranSLangDDanielsMBhattacharyaTDerdeynCA 2007 Clade-specific differences between human immunodeficiency virus type 1 clades B and C: diversity and correlations in C3-V4 regions of gp120. J Virol 81 4886 4891
85. MoorePLGrayESChogeIARanchobeNMlisanaK 2008 The c3-v4 region is a major target of autologous neutralizing antibodies in human immunodeficiency virus type 1 subtype C infection. J Virol 82 1860 1869
86. KirchherrJLHamiltonJLuXGnanakaranSMuldoonM 2011 Identification of amino acid substitutions associated with neutralization phenotype in the human immunodeficiency virus type-1 subtype C gp120. Virology 409 163 174
87. WeiXDeckerJMWangSHuiHKappesJC 2003 Antibody neutralization and escape by HIV-1. Nature 422 307 312
88. LyAStamatatosL 2000 V2 loop glycosylation of the human immunodeficiency virus type 1 SF162 envelope facilitates interaction of this protein with CD4 and CCR5 receptors and protects the virus from neutralization by anti-V3 loop and anti-CD4 binding site antibodies. J Virol 74 6769 6776
89. AuwerxJFrancoisKOCovensKVan LaethemKBalzariniJ 2008 Glycan deletions in the HIV-1 gp120 V1/V2 domain compromise viral infectivity, sensitize the mutant virus strains to carbohydrate-binding agents and represent a specific target for therapeutic intervention. Virology 382 10 19
90. DooresKJBonomelliCHarveyDJVasiljevicSDwekRA 2010 Envelope glycans of immunodeficiency virions are almost entirely oligomannose antigens. Proc Natl Acad Sci U S A 107 13800 13805
91. SterjovskiJChurchillMJEllettAGrayLRRocheMJ 2007 Asn 362 in gp120 contributes to enhanced fusogenicity by CCR5-restricted HIV-1 envelope glycoprotein variants from patients with AIDS. Retrovirology 4 89
92. LiYMiguelesSAWelcherBSvehlaKPhogatA 2007 Broad HIV-1 neutralization mediated by CD4-binding site antibodies. Nat Med 13 1032 1034
93. LiYSvehlaKLouderMKWycuffDPhogatS 2009 Analysis of neutralization specificities in polyclonal sera derived from human immunodeficiency virus type 1-infected individuals. J Virol 83 1045 1059
94. McCaffreyRASaundersCHenselMStamatatosL 2004 N-linked glycosylation of the V3 loop and the immunologically silent face of gp120 protects human immunodeficiency virus type 1 SF162 from neutralization by anti-gp120 and anti-gp41 antibodies. J Virol 78 3279 3295
95. WyattRKwongPDDesjardinsESweetRWRobinsonJ 1998 The antigenic structure of the HIV gp120 envelope glycoprotein. Nature 393 705 711
96. GeijtenbeekTBKwonDSTorensmaRvan VlietSJvan DuijnhovenGC 2000 DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell 100 587 597
97. HirbodTKaldensjoTLopalcoLKlareskogEAnderssonS 2009 Abundant and superficial expression of C-type lectin receptors in ectocervix of women at risk of HIV infection. J Acquir Immune Defic Syndr 51 239 247
98. WuLKewalRamaniVN 2006 Dendritic-cell interactions with HIV: infection and viral dissemination. Nat Rev Immunol 6 859 868
99. BinleyJMLybargerEACrooksETSeamanMSGrayE 2008 Profiling the specificity of neutralizing antibodies in a large panel of plasmas from patients chronically infected with human immunodeficiency virus type 1 subtypes B and C. J Virol 82 11651 11668
100. BraibantMBrunetSCostagliolaDRouziouxCAgutH 2006 Antibodies to conserved epitopes of the HIV-1 envelope in sera from long-term non-progressors: prevalence and association with neutralizing activity. AIDS 20 1923 1930
101. AstronomoRDKaltgradEUditAKWangSKDooresKJ 2010 Defining criteria for oligomannose immunogens for HIV using icosahedral virus capsid scaffolds. Chem Biol 17 357 370
102. WangSKLiangPHAstronomoRDHsuTLHsiehSL 2008 Targeting the carbohydrates on HIV-1: Interaction of oligomannose dendrons with human monoclonal antibody 2G12 and DC-SIGN. Proc Natl Acad Sci U S A 105 3690 3695
103. GrayESMoorePLChogeIADeckerJMBibollet-RucheF 2007 Neutralizing antibody responses in acute human immunodeficiency virus type 1 subtype C infection. J Virol 81 6187 6196
104. DhillonAKDonnersHPantophletRJohnsonWEDeckerJM 2007 Dissecting the neutralizing antibody specificities of broadly neutralizing sera from human immunodeficiency virus type 1-infected donors. J Virol 81 6548 6562
105. WuXYangZYLiYHogerkorpCM Schief WR, et al. 2010 Rational Design of Envelope Identifies Broadly Neutralizing Human Monoclonal Antibodies to HIV-1. Science 329 856 861
106. BrummeZLGoodrichJMayerHBBrummeCJHenrickBM 2005 Molecular and clinical epidemiology of CXCR4-using HIV-1 in a large population of antiretroviral-naive individuals. J Infect Dis 192 466 474
107. CoetzerMNedellecRSalkowitzJMcLaughlinSLiuY 2008 Evolution of CCR5 use before and during coreceptor switching. J Virol 82 11758 11766
108. WoodNBhattacharyaTKeeleBFGiorgiELiuM 2009 HIV evolution in early infection: selection pressures, patterns of insertion and deletion, and the impact of APOBEC. PLoS Pathog 5 e1000414
109. BrummeZLBrummeCJHeckermanDKorberBTDanielsM 2007 Evidence of differential HLA class I-mediated viral evolution in functional and accessory/regulatory genes of HIV-1. PLoS Pathog 3 e94
110. TreurnichtFKSeoigheCMartinDPWoodNAbrahamsMR 2009 Adaptive changes in HIV-1 subtype C proteins during early infection are driven by changes in HLA-associated immune pressure. Virology 396 213 225
111. EfronB 1983 Estimating the error rate of a prediction rule: improvement on cross-validation. J Am Stat Assoc 78 316 331
112. BengioYGrandvaletY 2004 No unbiased estimator of the variance of K-fold cross-validation. J Mach Learn Res 5 1089 1105
113. EfronBTibshiraniR 1997 Improvements on cross-validation: The .632+Bootstrap Method. J Am Stat Assoc 92 548 560
114. StoneM 1977 Asymptotics for and against cross-validation. Biometrika 64 29 35
115. BouckaertRR 2003 Choosing between two learning algorithms based on calibrated tests. 51-58 In: Proceedings of the 20th International Conference on Machine Learning; August 21-24, 2003; Washington DC, United States. Available: http://www.hpl.hp.com/conferences/icml2003/
116. SalzbergS 1997 On comparing classifiers: pitfalls to avoid and a recommended approach. Data Min Knowl Disc 1 317 328
117. XhuHRohwerR 1196 No Free Lunch for Cross-Validation. Neural Computation 8 1421 1426
118. RefaeilzadehPTangLLiuH 2009 Cross Validation. In: Liu L, Tamer Ozsu M, editors. Encyclopaedia of Database Systems. Springer, 532-538
119. KohaviR 1995 A study of cross-validation and bootstrap for accuracy estimation and model selection. The Proceedings of International Joint Conference on AL 1137 1145
120. MellquistJLKasturiLSpitalnikSLShakin-EshlemanSH 1998 The amino acid following an asn-X-Ser/Thr sequon is an important determinant of N-linked core glycosylation efficiency. Biochemistry 37 6833 6837
121. HoppTPWoodsKR 1983 A computer program for predicting protein antigenic determinants. Mol Immunol 20 483 489
122. LiMGaoFMascolaJRStamatatosLPolonisVR 2005 Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J Virol 79 10108 10125
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 9
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- HTLV-1 Propels Thymic Human T Cell Development in “Human Immune System” Rag2 gamma c Mice
- Hostile Takeover by : Reorganization of Parasite and Host Cell Membranes during Liver Stage Egress
- Exploiting and Subverting Tor Signaling in the Pathogenesis of Fungi, Parasites, and Viruses
- A Viral Ubiquitin Ligase Has Substrate Preferential SUMO Targeted Ubiquitin Ligase Activity that Counteracts Intrinsic Antiviral Defence