
A Diverse Population of Molecular Type VGIII in Southern Californian HIV/AIDS Patients
Cryptococcus gattii infections in southern California have been reported in patients with HIV/AIDS. In this study, we examined the molecular epidemiology, population structure, and virulence attributes of isolates collected from HIV/AIDS patients in Los Angeles County, California. We show that these isolates consist almost exclusively of VGIII molecular type, in contrast to the VGII molecular type isolates causing the North American Pacific Northwest outbreak. The global VGIII population structure can be divided into two molecular groups, VGIIIa and VGIIIb. Isolates from the Californian patients are virulent in murine and macrophage models of infection, with VGIIIa significantly more virulent than VGIIIb. Several VGIII isolates are highly fertile and produce abundant sexual spores that may serve as infectious propagules. The a and α VGIII MAT locus alleles are largely syntenic with limited rearrangements compared to the known VGI (a/α) and VGII (α) MAT loci, but each has unique characteristics including a distinct deletion flanking the 5′ VGIII MATa alleles and the α allele is more heterogeneous than the a allele. Our studies indicate that C. gattii VGIII is endemic in southern California, with other isolates originating from the neighboring regions of Mexico, and in rarer cases from Oregon and Washington state. Given that >1,000,000 cases of cryptococcal infection and >620,000 attributable mortalities occur annually in the context of the global AIDS pandemic, our findings suggest a significant burden of C. gattii may be unrecognized, with potential prognostic and therapeutic implications. These results signify the need to classify pathogenic Cryptococcus cases and highlight possible host differences among the C. gattii molecular types influencing infection of immunocompetent (VGI/VGII) vs. immunocompromised (VGIII/VGIV) hosts.
Published in the journal:
. PLoS Pathog 7(9): e32767. doi:10.1371/journal.ppat.1002205
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002205
Summary
Cryptococcus gattii infections in southern California have been reported in patients with HIV/AIDS. In this study, we examined the molecular epidemiology, population structure, and virulence attributes of isolates collected from HIV/AIDS patients in Los Angeles County, California. We show that these isolates consist almost exclusively of VGIII molecular type, in contrast to the VGII molecular type isolates causing the North American Pacific Northwest outbreak. The global VGIII population structure can be divided into two molecular groups, VGIIIa and VGIIIb. Isolates from the Californian patients are virulent in murine and macrophage models of infection, with VGIIIa significantly more virulent than VGIIIb. Several VGIII isolates are highly fertile and produce abundant sexual spores that may serve as infectious propagules. The a and α VGIII MAT locus alleles are largely syntenic with limited rearrangements compared to the known VGI (a/α) and VGII (α) MAT loci, but each has unique characteristics including a distinct deletion flanking the 5′ VGIII MATa alleles and the α allele is more heterogeneous than the a allele. Our studies indicate that C. gattii VGIII is endemic in southern California, with other isolates originating from the neighboring regions of Mexico, and in rarer cases from Oregon and Washington state. Given that >1,000,000 cases of cryptococcal infection and >620,000 attributable mortalities occur annually in the context of the global AIDS pandemic, our findings suggest a significant burden of C. gattii may be unrecognized, with potential prognostic and therapeutic implications. These results signify the need to classify pathogenic Cryptococcus cases and highlight possible host differences among the C. gattii molecular types influencing infection of immunocompetent (VGI/VGII) vs. immunocompromised (VGIII/VGIV) hosts.
Introduction
The pathogenic Cryptococcus species complex is comprised of two common fungal pathogens of humans and other animals: C. neoformans and C. gattii [1]. The more prevalent C. neoformans is ubiquitously distributed worldwide and a common cause of meningitis in immunocompromised hosts [1], [2], [3]. C. gattii is more geographically restricted to tropical and subtropical regions, associated with eucalypts, Douglas fir, and other trees, and has a greater predilection for infecting immunocompetent hosts [4], [5]. However, the geographic distribution of this species has been expanding, with an outbreak occurring in the Pacific Northwest region of North America [4], [5], [6], [7], [8], [9], [10], [11], [12]. C. gattii can be subdivided into two serotypes (B and C) [13] and four molecular types (VGI, VGII, VGIII, VGIV) that appear to represent genetically isolated cryptic species [14], [15], [16]. VGI and VGII cause the majority of cases in otherwise healthy hosts. VGIII and VGIV appear to more commonly infect immunocompromised patients, including those with HIV/AIDS, similar to C. neoformans [3], [5], [17], [18], [19].
Compared to C. neoformans, less is known about the epidemiology and ecology of C. gattii, especially molecular types VGIII and VGIV. VGIV is rare globally but has been reported to cause infections in sub-Saharan Africa AIDS patients [18], while VGIII has been isolated from a number of regions worldwide [14], [16]. The limited epidemiological data may be due to a lack of laboratory species distinction, particularly in Cryptococcus cases among HIV/AIDS patients, although sporadic C. gattii infections in HIV/AIDS patients have been reported from many global regions [18], [20], [21], [22], [23], [24], [25], [26], [27], [28], [29], [30].
There are limited accounts for HIV/AIDS or VGIII/VGIV associated C. gattii in North America. However, C. gattii has been reported in southern California among an HIV/AIDS patient cohort and one AIDS patient in Mexico, and C. gattii VGIII/VGIV have been reported in clinical cases from Mexico [21], [22], [31]. Additionally, there are three reported VGIII isolates from the Pacific Northwest outbreak [6], [7], [10]. These reports suggest that C. gattii may be endemic in western North America.
Here we compared isolates from HIV/AIDS patients in southern California [22] with a global collection. Based on MLST analyses, >93% (28/30) of the C. gattii isolate cohort are VGIII, and genotypic diversity is much greater than in the Pacific Northwest VGII outbreak. The genotypes cluster into two distinct groups (VGIIIa, VGIIIb). These findings suggest that VGIII may have been endemic in southern California for a longer period of time compared to the more clonal VGII Pacific Northwest population, or that VGIII is more actively recombining and/or mutating. The high levels of diversity, together with results suggesting recombination, support ongoing genetic exchange.
The VGIII lineage is highly fertile compared to other C. gattii molecular types [32]. Isolates from our cohort include both mating types (α and a), unlike the exclusively α mating type Pacific Northwest outbreak [7], [16]. This is noteworthy, as a-α mating can promote recombination and yield infectious spores [33], [34], [35], [36], [37], [38]. Sexual recombination is critical in evolution of eukaryotic microbial pathogens, including both parasites and fungi [39], [40], [41], [42], [43]. For these reasons, we extended the analysis of the VGIII group to include extensive mating assays to determine genotypes associated with high fertility. Additionally, sequenced and characterized the C. gattii VGIII MAT locus alleles. These findings expand on previous studies of the Cryptococcus MAT locus [44], [45], [46], [47], [48] yielding new insights into plasticity of this genomic region involving rearrangements, gene truncation, and loss, which may impact fertility.
While previous studies have shown C. gattii is highly virulent in mice, the focus has been on genotypes causing disease in otherwise healthy hosts [16], [49], [50], with no studies to date comprehensively examining virulence of VGIII. Here we examined murine virulence and macrophage intracellular proliferation for both the VGIIIa and VGIIIb. The isolates were also selected based on mating type criteria to examine what roles, if any, the MAT locus plays in VGIII virulence. This was of interest, as previous studies have shown virulence differences in C. neoformans congenic isolates that differ only at MAT [51], [52]. Our results demonstrate the VGIIIa lineage has increased levels of virulence compared to VGIIIb.
Our studies reveal a complex population structure within the highly fertile VGIII molecular type, suggesting recombination in both the United States and globally via opposite and/or same-sex mating. We posit that genetic exchange and the formation of infectious spores may be contributing to an underlying endemic level of C. gattii infections in southern California. Additionally, we show that these isolates are virulent in both murine and macrophage infection models [10]. Overall, the VGIII isolates from HIV/AIDS patients show decreased levels of virulence in comparison to the Pacific Northwest outbreak VGII isolates. This study highlights the need for isolate typing at a resolution sufficient to distinguish both species and molecular type. Accurate isolate identification will advance understanding of the epidemiology, ecology, and health burden of this emerging pathogen, with potential prognostic and therapeutic benefits for the clinical management of cryptococcal infections.
Results
Molecular type VGIII forms two distinct lineages that are the predominant cause of C. gattii infection in a southern Californian AIDS patient cohort
To characterize the molecular type of C. gattii isolates collected from the southern California C. gattii cohort [22], we applied multilocus sequence typing (MLST) analysis at eight unlinked genomic loci. In total, the patient cohort consisted of 30 isolates. Of these, one was VGI (3.3%), another isolate was VGII (3.3%), and the remaining 28 isolates were molecular type VGIII (93.3%). Examination of the 28 VGIII isolates revealed a high level of diversity (13 unique genotypes based on seven MLST markers), isolates of the two mating types (α and a) (Figure 1), and no evidence for heterozygosity (consistent with FACS analysis showing they are haploid, data not shown). There are two genetically distinct groups within the VGIII molecular type. One was termed VGIIIa (predominantly orange shading in Figures 1, 2), and the other termed VGIIIb (predominantly green shading in Figures 1, 2). Additionally, two diploid isolates from the cohort (CA1388 and CA2355) were excluded due to high levels of heterozygosity in the majority of sequences, indicating they are likely hybrids. Hybrids between C. neoformans and C. gattii have been previously reported [53] (see discussion).


To further examine the isolates, their MLST profiles were compared to an additional 32 VGIII isolates collected from multiple locations and sources (Figure 2). Based on an examination of 60 VGIII isolates, the VGIIIa and VGIIIb clusters form two distinct groups. While many alleles were VGIIIa or VGIIIb specific, several were shared. Alleles shared between VGIIIa and VGIIIb are colored in fuchsia (Figures 1 and 2). In the VGIIIa cluster, all isolates originate from North America and Australia, while in the VGIIIb cluster, isolates originate from North America, South America, and Asia (Figure 2). Thus while there are possible geographic niches for each individual group, some geographic regions (Mexico and the US) harbor isolates from both groups. This finding suggests that in at least North America, the two groups might occupy similar environmental niches with potential for cross-hybridization.
We next examined the distribution of the sequence types by constructing maximum likelihood (ML) dendrograms (Figure 3A). This analysis also supports two distinct clusters. In total, 28 sequence types are represented in the seven-loci dendrogram 12 in VGIIIa and 16 in VGIIIb. One VGIIIa genotype, ST28, is an intermediate between the two subgroups, and consists of two clinical isolates from Mexico (97/426 and 97/427). This sequence type harbors two alleles shared between the two groups, two alleles common and exclusive to the VGIIIa group, and four alleles unique to this sequence type. The ancestry of this isolate remains unclear; however, both ST28 isolates originate from Mexico, a region that harbors isolates of both VGIIIa and VGIIIb, and thus these isolates may represent VGIIIa/VGIIIb hybrids.

The global population was examined by constructing Neighbor Joining phylograms (Figure 3B, Figure S1). This analysis also illustrates two defined lineages, with the ST28 genotype representing a VGIIIa/b intermediate. Bootstrap support for the separation of the VGIIIb lineage from the other genotypes is robust (100) (Figure 3B). Support for all VGIIIa genotypes other than ST28 is at a level of 86, indicating that ST28 may be a distinct lineage, or conversely be a divergent genotype within the VGIIIa lineage (Figure 3B). To address the ancestry of the VGIII subgroups, we examined the isolates in the context of the three other C. gattii molecular types (VGI, VGII, VGIV) (Figure S1). From this analysis, there is strong support (bootstrap value of 100) that the VGIIIa subgroup is ancestral to the VGIIIb subgroup, with ST28 as the closest genotype to the VGIIIb clade (Figure S1). While the two clusters (VGIIIa/VGIIIb) are distinct from one another, when compared with VGI, VGII, VGIV, all 60 VGIII isolates lie within the VGIII lineage, clearly delineated from the other molecular types with a bootstrap support of 100 (Figure S1).
Speciation dynamics and evidence for recombination in the global VGIII population
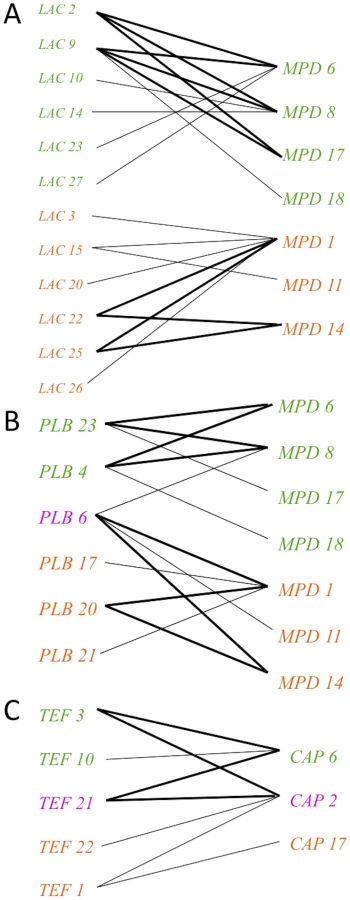
Based on an examination of the 60 total isolates, the VGIIIa and VGIIIb clusters may represent early speciation. To address aspects of the evolutionary history of the groups, haplotype network analysis was applied using TCS phylogenetic estimation for allele ancestry and evolution. The primary alleles focused on in this analysis were those shared between the subgroups. This examination addresses if shared alleles have a probable ancestral origin, or conversely if they were more recently introgressed. Of the shared alleles, SXI2a was not informative as there is only one allele. For the other three loci (CAP10, TEF1, PLB1), TCS analysis revealed that the CAP10 locus and TEF1 locus shared alleles appear ancestral, while the most parsimonious hypothesis is that the PLB1 shared allele was introgressed between VGIIIa and VGIIIb because another allele (PLB1-20) is assigned as the ancestral root (Figure 4A–C).

In addition to markers encompassing shared alleles, haplotype networks were also constructed to examine the evolutionary history of the remaining five markers (Figure S2). For this analysis, four of the five markers (LAC1, MPD1, GPD1, and IGS) showed that the VGIIIa and VGIIIb subgroups are separated (Figure S2). This provides additional evidence for genetic isolation. The remaining marker, SXI1α, shows evidence for separation, but the history is less well resolved, and shared introgressed alleles may remain to be discovered in the population (Figure S2). This marker is highly variable and many of the proposed intermediates have not been found to date. This could be the result of: 1) under-sampling of isolates, 2) missing alleles that are either no longer extant in the population or never existed in cases where mutations occurred simultaneously, or 3) recombination within the allele. Overall, this analysis supports a genetic separation, with several hypothesized but likely rare introgression events having occurred.
To examine the role that recombination may have contributed to population structure, we conducted a paired allele analysis among the global genotypes (Figure 5, Figure S3). The discovery of all four possible allele combinations between two unlinked loci (AB, ab, Ab, aB), in the absence of parallel mutations, serves as evidence for recombination [54], [55]. In total, 18 of 28 different molecular marker pairs showed evidence for recombination, with 25 examples in which all four allele combinations were observed. To further classify the analysis of the VGIIIa and VGIIIb subgroups, we examined how frequently allele combinations indicating recombination were associated with a specific lineage. In total, 1/25 locus pairs involved only the VGIIIa lineage, whereas 20/25 pairs involved only the VGIIIb lineage; the four remaining pairs involved shared alleles. Of the four pairs involving shared alleles, two involved shared and VGIIIa alleles and two involved shared and VGIIIb alleles. No pairs contained both VGIIIa and VGIIIb unique alleles (Figure 5, Figure S3). These studies demonstrate that while recombination may be present within both subgroups, the VGIIIb subgroup may be more actively undergoing recombination in its environmental niche.
To further characterize the role recombination may play in population structure, we employed MultiLocus software to examine the percentages of compatible loci and indices of association (IA). These analyses were completed for the global population, and also individually for the VGIIIa and VGIIIb subgroups. Additionally, the dataset was analyzed using all 60 isolates, and then was analyzed as a clone-corrected set with only unique genotypes represented (n = 28). Clone correction is commonly used to examine fungal populations that are known to often undergo asexual reproduction and clonal blooms, with less frequent sexual reproduction and meiosis [56]. The analysis of both the percentage of compatible loci and IA produce p-values that if significant reject the null hypothesis of recombination. If the p-values are not significant, the null hypothesis cannot be rejected and recombination can be inferred. The analysis illustrates that although the null hypothesis is rejected in the analysis of all isolates, VGIIIa individual, VGIIIb individual, and the overall clone corrected dataset, the null hypothesis cannot be rejected in the analysis of the individual VGIIIa and VGIIIb subgroups when the dataset is clone corrected (Table 1). As we cannot reject the null hypothesis for recombination in the clone corrected subgroup analysis, it suggests recombination may be occurring within each subgroup. These results support the hypothesis of active recombination within the VGIII molecular type and that these events more likely occur between isolates within the same subgroup, rather than between subgroups, similar to studies supporting recombination in VGI and VGII populations in Australia [35], [36].

In addition to population-based studies, we conducted extensive mating assays among VGIII isolates, showing highly fertile isolates from both subgroups. In total, 182 mating pairs were examined by laboratory mating assays, including all eight mating type a isolates, and representatives from each of the α genotypes. When observed via light microscopy, 138 pairs showed no signs of fertility, while 44 pairs were able to undergo sexual reproduction, with representative high resolution SEM imaging of a VGIII×VGIII cross (NIH312×B4546) shown in Figure 6 (see also Tables 2 and S1). The SEM imaging, similar to previous studies [32], illustrated key hallmarks of mating including hyphae, fused clamp cells, basidia, and elongated basidiospores (Figure 6). When the strains that were fertile with the greatest number of mating partners were separated into the top four from each respective mating type, three of four from both mating types are from the VGIIIb subgroup (Table S1). This shows that while both groups are fertile, levels of fertility (based on the number of fertile partners) may be higher in the VGIIIb subgroup, consistent with the increased number of unique VGIIIb genotypes (n = 16 out of 27 isolates or 59%) compared to VGIIIa (n = 12 out of 33 isolates or 36%). Overall, a higher percentage of a isolates are fertile compared to α isolates (Table 2). When both mating types are combined VGIIIb shows an increased percentage of fertile isolates compared to VGIIIa (Table 2). These findings support the hypothesis that mating may also occur in the environment.


Characterization of the VGIII MAT locus alleles illustrates distinct features
Sequencing of the α and a MAT locus alleles from two representative strains of the VGIII lineage shows that overall, the general structure, size, and characteristics are similar to previously sequenced C. gattii VGI and VGII MAT loci (Figure 7). Both SXI1α or SXI2a and the pheromone receptor and pheromone genes are present in the MAT locus, further supporting that C. gattii VGIII has a bipolar mating system.

Although there are marked similarities for the MAT locus alleles of VGI/VGII with VGIII, there are also distinct characteristics for each VGIII MAT allele. There are two rearrangements in the MATa allele 3′ region compared to the VGI isolate E566. Moreover, a partial deletion of UAP1 and loss of the FCY1 and FAO1 genes from the VGIII MATa allele 5′ flanking region was identified via sequencing of the VGIII a isolate B4546 and confirmed to be present in all eight VGIII a isolates examined, although no phenotypic consequences were observed (Figures 7, S4, and S5). Southern blot analysis of FCY1 and PCR analysis of all three full length genes with primers selected based on regions conserved between C. neoformans and C. gattii shows that they might not be present in the genome, or alternatively that they are rapidly evolving and too diverged to hybridize with probes and primers used (Figure S5). All isolates remain sensitive to the antifungal agent 5-Fluorocytosine (5-FC) indicating that the FCY1 gene is functioning or that another gene acts in a similar manner. Additionally, an ∼1.1 kb truncation of the FAO1 gene, a putative iron oxidoreductase, was found in all VGIII α strains analyzed. Using PCR, we could not detect an intact FAO1 gene elsewhere in the genome of the VGIII α isolates. Four pheromone gene copies were previously found within the VGII and VGI MATα loci, arranged in two pairs of divergently oriented, linked genes. However, although VGIII has been shown to be more fertile than VGI/VGII, based on the sequence assembly for strain NIH312, we found only two copies of the MFα pheromone gene, although one gap remains in this region of the locus. Additionally, we discovered a remnant of the a specific gene SXI2a in the VGIIIα mating type locus, and through sequence comparisons show that it is present in C. gattii molecular types VGI, VGII, and VGIII, but not present in C. neoformans var. neoformans (serotype D) or C. neoformans var. grubii serotype A (Figure S6).
Fingerprinting analysis of the MAT locus alleles revealed no size polymorphisms within MAT for VGIII a isolates, but did reveal diversity within the α allele (Figure 7, Table S2). Distinct alleles for fingerprint products 9 and 18 of the α locus, were identified that are correlated with the VGIIIa and VGIIIb molecular types (Figures 7, Table S2). Additionally, a 120 bp polymorphism within the CID1 and LPD1 intergenic sequence (fingerprint 8 of the α locus) was correlated with VGIIIa and VGIIIb molecular types with the exception of one VGIIIb strain harboring the SXIα allele 38, which contained the VGIIIa genotype at this region and could reflect recombination within MAT as a result of α-α mating (Figure 7, Table S2). Other VGIII isolates with SXI1α allele 38 were also confirmed to have this genotype (data not shown) and fingerprints 2 and 7 of the α locus showed multiple genotypes that were not correlated with the subgroups.
Virulence and histopathology
To address the virulence characteristics of the VGIII isolates, and correlate these with genotype and mating type, we conducted murine intranasal instillation challenges. In total, we chose 11 isolates from the patient cohort and one environmental VGIII control strain isolated in San Diego, California from an E. camaldulensis tree in 1992 [57]. Of the eleven clinical isolates, six were from the VGIIIa subgroup and five from the VGIIIb subgroup, with eight α and three a isolates (Figure 8A). Of the isolates examined, CA1499 was more virulent than all other isolates tested (Figure 8A). Additionally, when we compared overall virulence between the two subgroups, the VGIIIa subgroup showed a significantly higher mortality rate when compared to the VGIIIb subgroup (p<0.025). When isolate CA1089 was excluded from this analysis, the support increased (p<0.005). These results suggest that the molecular differentiation between the two lineages within VGIII is associated with a dichotomy in murine mortality post cryptococcal infection. We also show that the environmental isolate WM161 (VGIIIa) was less virulent than the five clinical VGIIIa isolates, but more virulent than all of the VGIIIb isolates. Isolates with increased levels of mortality were also found to be associated with rapid declines in total body weight during the course of infection (Figure 8B).
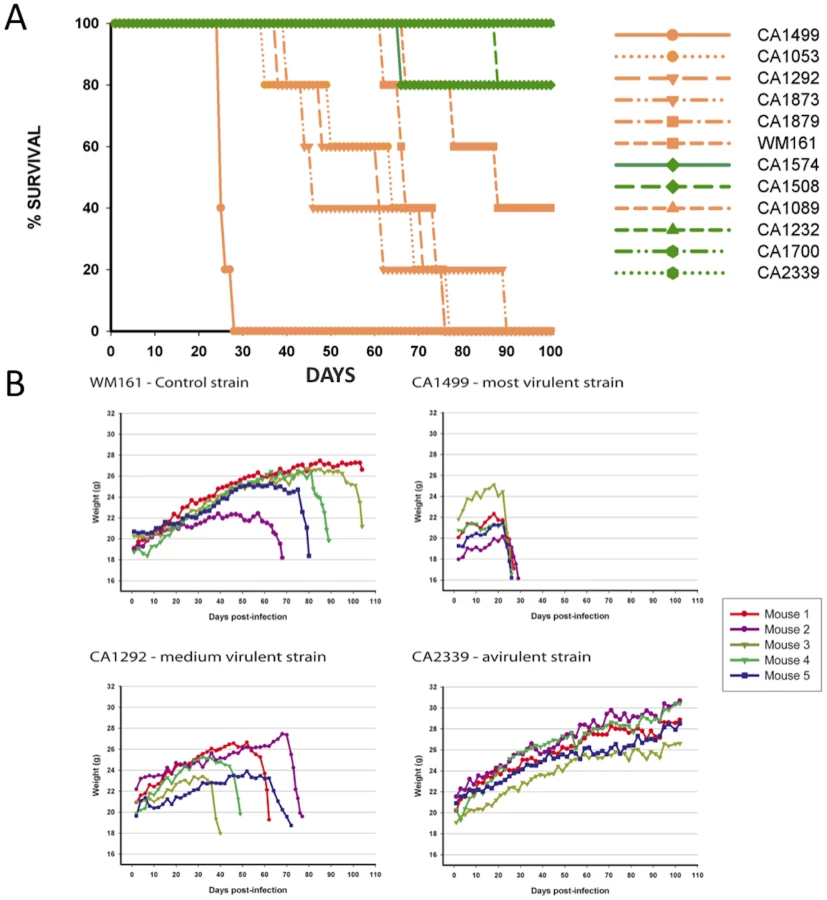
Histological examinations of lung tissues from mice infected with the highly virulent C. gattii strain CA1499 revealed widespread dissemination of cryptococci throughout the alveoli and airways with little to no inflammatory response. The expansion of alveoli with confluent clusters of budding yeasts and breakdown of alveolar walls were seen (Figure 9A–9B). The lungs of mice infected with strain CA1292 (moderate virulence) also revealed minimum inflammation with alveoli engorged with rapidly dividing yeasts. Also evident were a few alveoli with one to two C. gattii yeasts. Despite rapid multiplication, this strain appeared to be less disseminated as some of the alveoli and airways were devoid of any yeast cells (Figure 9C–9D). In contrast, lungs of mice infected with the low virulence strain WM161 revealed influx of inflammatory response surrounding infected alveoli and airways. The majority of the infected alveoli contained only one to two yeasts with a few alveoli containing three or four yeasts (Figure 9E–9F). No yeast cells were seen in any of the lung sections of mice infected with avirulent strain CA2339; however, vigorous tissue response in the form of neutrophil influx was observed throughout the lung section (Figure 9G–9H). The brain sections of all infected mice did not reveal any apparent lesions except for the highly virulent strain (CA1499), which showed occasional single or budding yeasts on the meninges (data not shown).

Although there were some differences in the dissemination pattern of CA1499 (high virulence) and CA1292 (moderate virulence) by histopathology, the quantification of yeasts from infected lungs at weekly intervals did not reveal any significant difference (Figure 10). This could be due to heavy colonization by CA1292 in the infected sites (Figure 9C–9D). When the lung organ loads of these two strains were compared with that of lung organ load of mice infected with low virulent strain WM161, the results were statistically significant (P<0.05). In comparison, the avirulent strain was cleared rapidly by one week post-infection and was not found in the lungs at two weeks post-infection (Figure 10). Overall, these results indicated that C. gattii rapid multiplication, dissemination, and invasion of the host defense of the lungs dictate the outcome of pulmonary cryptococcosis. Only small numbers of yeasts were recovered from brain cultures of mice infected with virulent strains of C. gattii (data not shown). These results indicated that pulmonary cryptococcosis and not CNS dissemination was the primary disease manifestation in this model and the likely cause of mice mortality observed (Figure 8).

Intracellular proliferation rates (IPRs) were determined for each strain examined in the murine model of infection. This assay is correlated positively with murine virulence, based on previous studies Pacific Northwest outbreak isolates [10], [49]. To further examine the virulence characteristics for the VGIII isolates, intracellular proliferation of cells within macrophages was directly tested for the seven VGIIIa and five VGIIIb isolates that were examined in the mouse model (Figure 11 A–B). Similar to the murine experiments, the VGIIIa subgroup showed significantly higher IPR levels than the VGIIIb subgroup (p<0.005, Figure 11 A, C). Additionally, as in the previous studies, IPR values and survival time in vivo showed strongly positive correlations upon regression analysis (Figure 11 B, D). A single VGIIIa isolate, CA1089, was an outlier with high IPR but low virulence. Therefore, the regression analysis was conducted both with (Figure 11 A, B) and without this isolate (Figure 11 C, D). While the significance levels were better supported when CA1089 was excluded, they were statistically significant in both cases (Figure 11 B, D). The IPR analysis combined with the in vivo model shows that the isolates from the Californian patient cohort show virulence differences in which the VGIIIa subgroup was more virulent overall than the VGIIIb subgroup.

Discussion
Globally the burden of cryptococcal disease is significant with approximately one million annual cases [19]. Although >99% of AIDS related infections and >95% of overall cases are attributed to C. neoformans serotype A [1], C. gattii also has been shown to cause disease among AIDS patients in both sub-Saharan Africa and the US [18], [22]. In the study of Litvintseva et al., an African AIDS cohort was found to be infected with C. gattii serotype C VGIV molecular type [18]. Here C. gattii isolates were examined from a previously reported HIV/AIDS patient cohort in southern California [22], and found to be almost exclusively (93%) VGIII molecular type. The finding of C. gattii VGIII and VGIV isolates associated with HIV/AIDS patients is in stark contrast to numerous C. gattii infections among immunocompetent individuals caused by VGI/VGII isolates, and the ongoing VGII outbreak in the North American Pacific Northwest [5], [7], [9], [10], [16], [58], [59], [60], [61], [62].
The Californian HIV/AIDS patient cohort C. gattii strains were analyzed in the context of a global VGIII isolate collection to further characterize this molecular type. Analysis of 60 isolates at eight loci resolved two distinct subgroups designated VGIIIa and VGIIIb. In contrast to the Pacific Northwest VGII outbreak that is largely clonal with one genotype causing the majority of illness [9], [10], [16], [63], based on analysis of seven non sex-linked loci we found a total of 13 unique genotypes in the 28 VGIII isolates from the Californian patients. This increased level of diversity suggests this molecular type was introduced into the region longer ago than the VGII lineage to the Pacific Northwest, or alternatively that the VGIII population in California is more rapidly diverging. Our studies, in addition to a recent study documenting VGIII isolates in Mexico, lead to the hypothesis that the VGIII molecular type may be endemic to a large area of western North America [16], [31]. These findings are significant, as there may be unrecognized cases of C. gattii among HIV/AIDS patients.
Phylogenetic analysis of the molecular types revealed that VGIIIa is basal to VGIIIb (Figure S1). Given that VGIIIa is more virulent in mice and murine macrophages, it may be that VGIIIb has become more specialized to infect hosts with increased susceptibility to infections. It currently remains unclear as to whether these two groups have naturally evolved to display differences in virulence or if there is some form of selection. Regardless, increased surveillance of VGIII cases, aided by clinical studies to determine the types of infection each group causes, should shed further light as to whether these two groups also have distinct clinical features. Clustering and phylogenetic analyses of the VGIII global collection revealed 28 unique genotypes among 60 isolates. Subsequent phylogenetic analysis revealed that the two observed groups were indeed well supported lineages. Haplotype mapping was then conducted, supporting the discrimination into VGIIIa and VGIIIb. Additionally, the mapping of markers harboring shared alleles between the subgroups revealed that two of the three shared alleles are ancestral in origin, while one may result from introgression between VGIIIa and VGIIIb. This type of introgression event is not unprecedented between distinct lineages, cryptic species, or species within the fungal kingdom, with examples from model fungi and both plant and animal pathogens [64], [65], [66], [67].
The ability for microbial pathogens to undergo sexual reproduction involving meiosis is unique to the eukaryotic lineage, including the parasites and fungi. Sex may play a significant role in several aspects of pathogenesis, including the generation of genetic diversity, the formation of invasive hyphae or spores, and direct links between the mating type locus and mating pathways with virulence [34], [35], [37], [39], [52], [68], [69], [70], [71], [72], [73], [74], [75], [76]. For these reasons, and to understand the population dynamics of VGIII, we conducted two types of recombination analyses: informative paired allele graphs and statistical analyses of percentages of compatible loci and IA. Our analyses support the hypothesis of recombination within the distinctive VGIII subgroups. This is consistent with the observed molecular differences between the two groups. Our population results, taken together with an increased number of highly fertile VGIIIb isolates in comparison to VGIIIa, suggest that the VGIIIb group may be more actively recombining. Furthermore, the two excluded hybrid isolates appear to be C. neoformans var. neoformans/C. gattii molecular type VGIIIb hybrids (based on the GPD1 MLST allele specifically amplifying C. gattii sequence and the IGS allele specifically amplifying C. neoformans sequence, while the other MLST loci were highly heterozygous). The enhanced fertility ofn the VGIIIb subgroup may contribute to hybridization with C. neoformans.
The ability to undergo sexual reproduction also has implications for the formation of infectious spores. Recent studies show that spores can initiate disseminated cryptococcal disease in the murine inhalation model of infection [34], [37]. Laboratory studies have also shown that Cryptococcus can complete a full sexual cycle in association with plants, leading to the production of infectious spores [77]. It has been previously shown that the VGIII lineage shows high levels of fertility [32] and our examination of compatible isolate pairs supports that mating and spore formation may play a significant role in the formation of small aerosolized particles that could be readily inhaled. Furthermore, in California, both mating types are present and several of these pairs are fertile under laboratory conditions.
The MAT alleles of C. gattii molecular types VGI and VGII are highly conserved, based on both DNA sequence similarity and gene synteny [48]. The sequenced MATα locus alleles of VGI WM276, VGII R265, and VGIII NIH312 share full synteny (Figure 7). A comparison of the three loci reveals only expansions, contractions, and translocation of intergenic sequences, which account for the majority of the structural variation. Additionally, there is also a truncation of the FAO1 gene in all VGIII α isolates examined. The sequenced MATa locus alleles of VGI E566 and VGIII B4546 show a higher degree of rearrangement in comparison to MATα (Figure 7). Additionally, a partial deletion of the UAP1 gene, along with a complete deletion of the FCY1 and FAO1 genes from the 5′end of the VGIII MATa locus is fixed in the VGIII a population. UAP1 is predicted to encode a candidate uric acid transporter whose closest homolog is the A. nidulans uapA gene [78], [79], [80] and Fcy1 is a cytosine deaminase that has been shown to confer resistance to 5-FC when deleted in C. albicans, S. cerevisiae, and C. neoformans [81], [82]. Although both PCR and southern blot analyses could not detect a copy of the FCY1 gene in the genome of the VGIII MATa isolates, phenotypic tests showed these isolates remain sensitive to 5-FC and they may therefore harbor a more diverged FCY1 gene elsewhere in the genome. Phenotypic assays on media with uric acid as a sole nitrogen source to examine the possible loss of UAP1 also showed no distinct phenotype from control isolates, i.e., all isolates were able to efficiently grow (data not shown). Based on findings in C. neoformans, recombination hotspots may flank the MAT locus alleles and thus have contributed to the flanking region deletions [83]. Overall, analysis of the C. gattii MAT locus alleles showed a more dynamic MATa locus compared to the α locus. Given the significant rearrangement of the MATa locus, and the deletion or exclusion of genes from the locus and flanking region, it appears that the VGIII MATa population has experienced increased genetic variation. Deletion or exclusion of genes from the MAT locus and flanking regions may be an indication of expansion or contraction of the VGIII MATa locus. The finding of a SXI2a remnant conserved in C. gattii may signal a previous tetrapolar mating type state, consistent with the prevailing models and studies in related Cryptococcus species (Figure S6 B) [46], [47], [48], [84], [85], or result from a C. gattii lineage specific gene conversion of SXI2a into the α allele (Figure S6 C). Alternatively, this remnant may be functional and influence mating.
Within VGIII, fingerprinting analysis revealed a monomorphic MATa allele and a mostly biallelic MATα locus correlating with the VGIIIa and VGIIIb lineages. Two fingerprints revealed size polymorphisms correlated with VGIIIa and VGIIIb isolates, serving as additional evidence for ongoing genetic separation between these two groups. VGIIIb strains harboring SXIα allele 38 contained the VGIIIa genotype for the intergenic sequence between CID1 and LPD1, suggesting that these strains harbor either the ancestral VGIII MATα allele or contain a hybrid MATα locus that may be the result of a gene introgression event between the two groups or a recombination event between same-sex isolates of opposite VGIII lineages (i.e., VGIIIa/VGIIIb). However, an additional polymorphic marker within the MATα locus is needed to address the latter hypothesis.
Following the definition of two well-supported lineages including isolates from both mating types, we examined virulence characteristics associated with these properties. We found significant differences between phylogenetic subgroups. In both whole animal murine in vivo intranasal instillation and proliferation assays in murine derived macrophages, the VGIIIa subgroup is more virulent than the VGIIIb subgroup. These findings are significant and serve as a foundation for future studies to determine the molecular basis for these observed phenotypes. Additionally, from a public health and epidemiological standpoint, it may be useful to determine if isolates are VGIIIa or VGIIIb. Clinical studies would have to be conducted in coordination to ascertain if the molecular subgroup is associated with altered clinical manifestations or outcomes. Based on the histological examinations, our findings indicate that host tissue responses in combination with yeast cell multiplication and capsule induction are associated with the outcome of pulmonary cryptococcosis, similar to previous studies documenting that upon serial in vivo passage of C. neoformans, strains increase in virulence with an associated decrease in capsule size [86].
Our study provides a comprehensive molecular and phenotypic overview of the C. gattii VGIII molecular type, which has historically been less studied than both the VGI and VGII molecular types. Our findings support two distinct lineages that might each be recombining, with the VGIIIa lineage showing higher levels of virulence in the models examined. Of significance, many of the isolates examined were from a cohort of HIV/AIDS patients in southern California. The high level of VGIII observed is in stark contrast to the Pacific Northwest VGII outbreak of VGII, in which the vast majority of cases reported are not associated with HIV/AIDS infected patients [7], [9], [10], [16], [63]. This suggests that C. gattii may occur in two general patient settings: VGI/VGII in otherwise healthy hosts (>50%) or those treated with steroids, vs. VGIII/VGIV predominantly in HIV/AIDS patients. Moreover, C. gattii infections may cause a substantial unrecognized health burden. To address these aspects, both retrospective and prospective studies should be conducted to: 1) survey global isolate collections from HIV/AIDS patients and 2) assign species and molecular types to newly collected isolates from HIV/AIDS patients with cryptococcal infections.
Materials and Methods
Phenotypic identification assays
All isolates from southern California and other global isolates were screened to confirm that they were C. gattii. Melanin production was assayed by growth and dark pigmentation on Staib's niger seed medium; urease activity was detected by growth and alkaline pH change on Christensen's agar. These tests established that isolates were Cryptococcus (C. neoformans or C. gattii). Additionally, isolates were assayed for resistance to canavanine and utilization of glycine on L-canavanine, glycine, 2-bromothymol blue (CGB) agar. Growth on CGB agar indicates that isolates are canavanine resistant, and able to use glycine as a sole carbon source, triggering a bromothymol blue color reaction indicative of C. gattii. All CGB positive isolates were then grown under rich culture conditions prior to genomic DNA extraction and storage at −80°C in 25% glycerol. For genomic DNA isolation, the MasterPure Yeast DNA purification kit from Epicentre Biotechnologies was used.
Multilocus sequence typing
Each VGIII isolate examined in the analysis was subjected to multilocus sequence typing (MLST) [87] at a total of eight loci (Table S3). This marker set was selected to include loci that have been validated in other analyses of C. gattii [7], [16], [88], [89], [90]. For each isolate, genomic regions were PCR amplified, purified (ExoSAP-IT, Qiagen), and sequenced. Sequences from both forward and reverse strands were assembled with complete double strand coverage and manually edited using Sequencher version 4.8 (Gene Codes Corporation). Based on BLAST analysis of the GenBank database (NCBI), each allele was assigned a corresponding number, or given a new number if the sequence was not already in the database. GenBank accession numbers with corresponding allele numbers are listed in the supplementary information (Table S4).
Clustering and phylogenetic analyses
For each isolate, a sequence type was defined as a sequence exhibiting a unique sequence profile, based on concatenation of the MLST markers. Each sequence type was confirmed to be unique by BLAST analysis of the NCBI GenBank database [91]. A multiple alignment of the sequences was carried out using Clustal W software [92]. Clustering analysis of the sequences was conducted using PhyML, which applies the maximum likelihood model for analysis [93]. The phylogenetic analysis was conducted using the Neighbor Joining algorithm. Haplotype network modeling was conducted using TCS software (version 1.21) [94]. The statistical recombination analysis was completed using MultiLocus software (version 1.2.2).
Mating assays
Mating assays were conducted on V8 media (pH = 5). Isolates were incubated at room temperature in the dark for 2–4 weeks in dry conditions. Fertility was assessed by light microscopic examination for hyphae, fused clamp cells, basidia, and basidiospore formation at the periphery and surface of the co-incubated mating patch. All mating assays were conducted in duplicate. If there were no signs of fertility after the four-week period, isolate pairs were scored as having no fertility when paired together.
Scanning Electron Microscopy (SEM)
SEM analysis was completed using protocols similar to those previously published [34]. Mating cultures (NIH312×B4546) were excised from V8 medium agar plates and fixed at 4°C in 3% glutaraldehyde that was buffered in 0.1 M Na cacodylate (pH = 6.8). Samples were then washed in triplicate using cold 0.1 M Na cacodylate buffer. This was followed by a graded dehydration series of 1 hr changes in cold 30% and 50% ethanol and held overnight. Dehydration was completed with 1 hr changes of cold 95% and 100% ethanol at 4°C and warming to room temperature in 100% EtOH. Two additional 1 hr changes of room temperature 100% EtOH completed the dehydration series. The samples were then critical point dried in liquid CO2 (Samdri-795; Tousimis Research Corp., Rockville, MD) for 15 min at the critical point. The agar pieces were mounted and sealed with silver paint to ensure good conductivity. The samples were sputter coated with 50 Å of Au/Pd (Hummer 6.2; Anatech U.S.A., Hayward, CA). Samples were held under vacuum conditions until viewed with a Jeol JSM 5900LV scanning electron microscope at 15 kV.
Sequencing and analysis of the MAT locus alleles
The strains used for the construction of the Bacterial Artificial Chromosome (BAC) and fosmid libraries and analysis of the MAT locus alleles were NIH312 (MATα) and B4546 (MATa). Isolates were grown on YPD media at 30°C, and library construction was performed according to previous studies [48], [84]. Sequencing reactions were performed using BigDye 3.1 (Applied Biosystems, Foster City, California, United States) and analyzed on an ABI3100 sequencer. Sequence reads were assembled using the PHRED/PHRAP/CONSED package [95], [96] and Sequencher 4.8 (Gene Codes Corporation). Additional analysis of the data was performed using BLASTn [91]. Based on the initial assembly of sequences, oligonucleotides were selected to close gaps in the sequence coverage by primer walking. Genes on the MAT locus of C. gattii were annotated based on homology to the existing annotation in C. gattii VGII (strain R265) and VGI (strains WM276 and E566), and C. neoformans (H99 and JEC21). Certain gene annotations were modified using the FGENESH program (http://linux1.softberry.com/berry.phtml). Comparison of the MAT locus alleles among VGI, VGII, and VGIII strains was performed using BLASTn. The BLASTn results were parsed using a PERL script and imputed into the ACT program to construct synteny diagrams of the MAT locus alleles among C. gattii strains. In order to evaluate size polymorphisms within the MAT locus, 14 MATα strains representing all SXI1α alleles and 8 MATa strains were amplified using primers complementary to conserved sequences. Primers used for fingerprinting are listed in Table S3. The PCR products were digested using appropriate enzymes selected on the basis of DNA sequences using NEBcutter version 2.0 (http://tools.neb.com/NEBcutter2/index.php). Fingerprints showing variability were sequenced to determine the nature of any size polymorphisms. Sequences from both forward and reverse strands were assembled with complete double strand coverage and manually edited using Sequencher version 4.8 (Gene Codes Corporation).
Murine virulence assays
The pathogenic potential of C. gattii strains belonging to the VGIII genotype was tested in a murine model of pulmonary cryptococcosis [97]. Briefly, C. gattii strains were grown in YPD broth for 16 hours and washed with sterile physiological saline. Cells were counted with a hemocytometer and suspended at a concentration of 3.3×106 cells/ml. Five male BALB/c mice (approximately six-weeks old, 15–20 g; Charles River) in each group were first anesthetized with xylazine-ketamine mixture and then 30 ul of infectious inocula was gently dripped into their nares. The injected animals were observed for any overt signs of illness, and all morbid animals were promptly sacrificed by CO2 inhalation to minimize pain and suffering.
Data from all infected animals were used to determine Kaplan-Meyer survival curves using SAS software (SAS Institute, Inc., Cary, N.C.). For this statistical analysis with genotype information, the non-parametric Mann-Whitney U-test was applied, with further details in the statistical analysis section (http://faculty.vassar.edu/lowry/utest.html). Progression of disease was also determined by weighing infected animals once every alternate day at the start of the infection and then every day until they were moribund. One half portion of the lung and brain tissues from sacrificed animals was cultured on YPD and Staib's niger seed agar for the recovery of cells to determine that infections were cryptococcal in origin.
Organ load and histopathology
To characterize virulence properties, representative C. gattii strains from four virulence groups comprising high virulence strains (CA1499), moderate virulence strains (CA1292), low virulence strains (WM161), and avirulent strains (CA2339), were chosen for further intranasal infection per procedure described previously. A total of six mice were infected for each test strain and groups of three mice were sacrificed at one and two-weeks post infection. The lungs and brains were removed aseptically, one half of each organ was fixed in 10% buffered formalin and Bouin's fixative, processed into paraffin blocks, sectioned, and stained with hematoxylin and eosin (H & E) and Mayer's mucicarmine for histopathological examination. The other halves of the lungs and brains were weighed, homogenized, diluted in PBS, and plated on YPD agar. Colonies were counted after incubation of the plates at 30°C for 4 days, and the results were expressed as colony forming unit (CFU) per gram of infected tissue. Results from organ load experiment were analyzed by student t-test, with significance determined at P≤0.05.
Ethics statement
The animal studies conducted were in full compliance with all of the guidelines set forth by the Wadsworth Center Institutional Animal Care and Use Committee (IACUC) and in full compliance with the United States Animal Welfare Act (Public Law 98–198). The Wadsworth Center IACUC approved all of the vertebrate studies. The studies were conducted in facilities accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC).
Intracellular Proliferation Rate (IPR) determination
A proliferation assay was previously developed to monitor the intracellular proliferation rate (IPR) of individual strains (for a 72-hour period) following phagocytosis [49]. For this assay, J774 macrophage cells were exposed to cryptococcal cells that were opsonized with 18B7 antibody for 2 hr as described previously [98]. Each well was washed with phosphate-buffered saline (PBS) in quadruplicate to remove as many extracellular yeast cells as possible and 1 ml of fresh serum-free DMEM was then added. For time point T = 0, the 1 ml of DMEM was discarded and 200 µl of sterile dH2O was added into wells to lyse macrophage cells. After 30 minutes, the intracellular yeast were released and collected. Another 200 µl dH2O was added to each well to collect the remaining yeast cells. The intracellular yeast were then mixed with Trypan Blue at a 1∶1 ratio and the live yeast cells were counted. For the subsequent five time points (T = 18 hrs, T = 24 hrs, T = 48 hrs, T = 72 hrs), intracellular cryptococcal cells were collected and independently counted with a hemocytometer.
For each strain tested, the time course was repeated at least three independent times, using different batches of macrophages. The IPR value was calculated by dividing the maximum intracellular yeast number by the initial intracellular yeast number at T = 0. We confirmed that Trypan Blue stains 100% of the cryptococcal cells in a heat-killed culture, but only approximately 5% of cells from a standard overnight culture. Compared to a conventional colony counting method, this method was shown to be more sensitive in detecting the clustered yeast population or yeast cells undergoing budding.
Statistical analysis
Time to 50% lethality (LT50) and median IPR values were used to assess the statistical significance between the VGIII subgroups and these respective values. For this statistical analysis, the non-parametric directional Mann-Whitney U-test was applied and values of p<0.025 were considered as statistically significant (http://faculty.vassar.edu/lowry/utest.html). Regression analysis was used to measure the correlation between LT50 and IPR values, and an F-value of p<0.05 was considered to be a significant correlation, with R2, p values, and Pearson correlations also calculated and represented in the analysis.
Strains
All strains used in this study are listed in Table S6, including C. gattii, C. neoformans, and S. cerevisiae isolates.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. CasadevallAPerfectJ 1998 Cryptococcus neoformans Washington DC. ASM Press
2. CarlileMWatkinsonSGoodayG 2001 The Fungi London Academic Press 588
3. PerfectJR 1989 Cryptococcosis. Infect Dis Clin North Am 3 77 102
4. Kwon-ChungKJBennettJE 1984 High prevalence of Cryptococcus neoformans var. gattii in tropical and subtropical regions. Zentralbl Bakteriol Mikrobiol Hyg [A] 257 213 218
5. SorrellTC 2001 Cryptococcus neoformans variety gattii. Med Mycol 39 155 168
6. IqbalNDebessEEWohrleRSunBNettRJ 2009 Correlation of genotype and in vitro susceptibilities of Cryptococcus gattii from the Pacific Northwest of the United States. J Clin Microbiol 48 539 544
7. ByrnesEJ3rdBildfellRJFrankSAMitchellTGMarrKA 2009 Molecular evidence that the range of the Vancouver Island outbreak of Cryptococcus gattii infection has expanded into the Pacific Northwest in the United States. J Infect Dis 199 1081 1086
8. Kwon-ChungKJBennettJE 1984 Epidemiologic differences between the two varieties of Cryptococcus neoformans. Am J Epidemiol 120 123 130
9. KiddSEHagenFTscharkeRLHuynhMBartlettKH 2004 A rare genotype of Cryptococcus gattii caused the cryptococcosis outbreak on Vancouver Island (British Columbia, Canada). Proc Natl Acad Sci U S A 101 17258 17263
10. ByrnesEJ3rdLiWLewitYMaHVoelzK 2010 Emergence and pathogenicity of highly virulent Cryptococcus gattii genotypes in the northwest United States. PLoS Pathog 6 e1000850
11. SpringerDJChaturvediV 2010 Projecting global occurrence of Cryptococcus gattii. Emerg Infect Dis 16 14 20
12. CDC 2010 Emergence of Cryptococcus gattii – Pacific Northwest, 2004–2010. MMWR Morb Mortal Wkly Rep 59 865 868
13. Kwon-ChungKJVarmaA 2006 Do major species concepts support one, two or more species within Cryptococcus neoformans? FEMS Yeast Res 6 574 587
14. BoversMHagenFKuramaeEEBoekhoutT 2008 Six monophyletic lineages identified within Cryptococcus neoformans and Cryptococcus gattii by multi-locus sequence typing. Fungal Genet Biol 45 400 421
15. BoekhoutTTheelenBDiazMFellJWHopWC 2001 Hybrid genotypes in the pathogenic yeast Cryptococcus neoformans. Microbiology 147 891 907
16. FraserJAGilesSSWeninkECGeunes-BoyerSGWrightJR 2005 Same-sex mating and the origin of the Vancouver Island Cryptococcus gattii outbreak. Nature 437 1360 1364
17. ByrnesEJ3rdHeitmanJ 2009 Cryptococcus gattii outbreak expands into the Northwestern United States with fatal consequences. F1000 Biology Reports 1 62
18. LitvintsevaAPThakurRRellerLBMitchellTG 2005 Prevalence of clinical isolates of Cryptococcus gattii serotype C among patients with AIDS in Sub-Saharan Africa. J Infect Dis 192 888 892
19. ParkBJWannemuehlerKAMarstonBJGovenderNPappasPG 2009 Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS 23 525 530
20. BogaertsJTaelmanHBatungwanayoJVan de PerrePSwinneD 1993 Two cases of HIV-associated cryptococcosis due to the variety gattii in Rwanda. Trans R Soc Trop Med Hyg 87 63 64
21. Castanon-OlivaresLRLopez-MartinezRBarriga-AnguloGRios-RosasC 1997 Crytococcus neoformans var. gattii in an AIDS patient: first observation in Mexico. J Med Vet Mycol 35 57 59
22. ChaturvediSDyavaiahMLarsenRAChaturvediV 2005 Cryptococcus gattii in AIDS patients, southern California. Emerg Infect Dis 11 1686 1692
23. CokerRJ 1992 Cryptococcal infection in AIDS. Int J STD AIDS 3 168 172
24. FernandesOPassosXSSouzaLKMirandaATCerqueiraCH 2003 In vitro susceptibility characteristics of Cryptococcus neoformans varieties from AIDS patients in Goiania, Brazil. Mem Inst Oswaldo Cruz 98 839 841
25. Kapend'aKKomicheloKSwinneDVandepitteJ 1987 Meningitis due to Cryptococcus neoformans biovar gattii in a Zairean AIDS patient. Eur J Clin Microbiol 6 320 321
26. KarstaedtASCrewe-BrownHHDromerF 2002 Cryptococcal meningitis caused by Cryptococcus neoformans var. gattii, serotype C, in AIDS patients in Soweto, South Africa. Med Mycol 40 7 11
27. Lindenberg AdeSChangMRPaniagoAMLazera MdosSMoncadaPM 2008 Clinical and epidemiological features of 123 cases of cryptococcosis in Mato Grosso do Sul, Brazil. Rev Inst Med Trop Sao Paulo 50 75 78
28. RozenbaumRGoncalvesAJWankeBVieiraW 1990 Cryptococcus neoformans var. gattii in a Brazilian AIDS patients. Mycopathologia 112 33 34
29. SeatonRAWembriJPArmstrongPOmbigaJNaraqiS 1996 Symptomatic human immunodeficiency virus (HIV) infection in Papua New Guinea. Aust N Z J Med 26 783 788
30. SteeleKTThakurRNthobatsangRSteenhoffAPBissonGP 2010 In-hospital mortality of HIV-infected cryptococcal meningitis patients with C. gattii and infection in Gaborone, Botswana. Med Mycol 48 1112 1115
31. OlivaresLRMartinezKMCruzRMRiveraMAMeyerW 2009 Genotyping of Mexican Cryptococcus neoformans and C. gattii isolates by PCR-fingerprinting. Med Mycol 1 9
32. FraserJASubaranRLNicholsCBHeitmanJ 2003 Recapitulation of the sexual cycle of the primary fungal pathogen Cryptococcus neoformans var. gattii: implications for an outbreak on Vancouver Island, Canada. Eukaryot Cell 2 1036 1045
33. SukroongreungSKitiniyomKNilakulCTantimavanichS 1998 Pathogenicity of basidiospores of Filobasidiella neoformans var. neoformans. Med Mycol 36 419 424
34. VelagapudiRHsuehYPGeunes-BoyerSWrightJRHeitmanJ 2009 Spores as infectious propagules of Cryptococcus neoformans. Infect Immun 77 4345 4355
35. SaulNKrockenbergerMCarterD 2008 Evidence of recombination in mixed-mating-type and alpha-only populations of Cryptococcus gattii sourced from single eucalyptus tree hollows. Eukaryot Cell 7 727 734
36. CampbellLTCurrieBJKrockenbergerMMalikRMeyerW 2005 Clonality and recombination in genetically differentiated subgroups of Cryptococcus gattii. Eukaryot Cell 4 1403 1409
37. GilesSSDagenaisTRBottsMRKellerNPHullCM 2009 Elucidating the pathogenesis of spores from the human fungal pathogen Cryptococcus neoformans. Infect Immun 77 3491 3500
38. CampbellLTFraserJANicholsCBDietrichFSCarterD 2005 Clinical and environmental isolates of Cryptococcus gattii from Australia that retain sexual fecundity. Eukaryot Cell 4 1410 1419
39. HeitmanJ 2006 Sexual reproduction and the evolution of microbial pathogens. Curr Biol 16 711 725
40. Kwon-ChungKJ 1976 Morphogenesis of Filobasidiella neoformans, the sexual state of Cryptococcus neoformans. Mycologia 68 821 833
41. Kwon-ChungKJ 1976 A new species of Filobasidiella, the sexual state of Cryptococcus neoformans B and C serotypes. Mycologia 68 943 946
42. CampbellLTCarterDA 2006 Looking for sex in the fungal pathogens Cryptococcus neoformans and Cryptococcus gattii. FEMS Yeast Res 6 588 598
43. HeitmanJ 2010 Evolution of eukaryotic microbial pathogens via covert sexual reproduction. Cell Host Microbe 8 86 99
44. LengelerKBFoxDSFraserJAAllenAForresterK 2002 Mating-type locus of Cryptococcus neoformans: a step in the evolution of sex chromosomes. Eukaryot Cell 1 704 718
45. HsuehYPFraserJAHeitmanJ 2008 Transitions in sexuality: recapitulation of an ancestral tri - and tetrapolar mating system in Cryptococcus neoformans. Eukaryot Cell 7 1847 1855
46. FraserJAStajichJETarchaEJColeGTInglisDO 2007 Evolution of the mating type locus: insights gained from the dimorphic primary fungal pathogens Histoplasma capsulatum, Coccidioides immitis, and Coccidioides posadasii. Eukaryot Cell 6 622 629
47. FraserJAHeitmanJ 2003 Fungal mating-type loci. Curr Biol 13 R792 795
48. FraserJADiezmannSSubaranRLAllenALengelerKB 2004 Convergent evolution of chromosomal sex-determining regions in the animal and fungal kingdoms. PLoS Biol 2 e384
49. MaHHagenFStekelDJJohnstonSASionovE 2009 The fatal fungal outbreak on Vancouver Island is characterized by enhanced intracellular parasitism driven by mitochondrial regulation. Proc Natl Acad Sci U S A 106 12980 12985
50. ChengPYShamAKronstadJW 2009 Cryptococcus gattii isolates from the British Columbia cryptococcosis outbreak induce less protective inflammation in a murine model of infection than Cryptococcus neoformans. Infect Immun 77 4284 4294
51. NielsenKCoxGMWangPToffalettiDLPerfectJR 2003 Sexual cycle of Cryptococcus neoformans var. grubii and virulence of congenic a and alpha isolates. Infect Immun 71 4831 4841
52. Kwon-ChungKJEdmanJCWickesBL 1992 Genetic association of mating types and virulence in Cryptococcus neoformans. Infect Immun 60 602 605
53. BoversMHagenFKuramaeEEDiazMRSpanjaardL 2006 Unique hybrids between the fungal pathogens Cryptococcus neoformans and Cryptococcus gattii. FEMS Yeast Res 6 599 607
54. CarterDCampbellLTSaulNKrockenbergerM 2010 Sexual reproduction of Cryptococcus gattii: a population genetics perspective. HeitmanJKozelTKwon-ChungJPerfectJRCasadevallA Cryptococcus: From human pathogen to model yeast ASM Press
55. CarterDSaulNCampbellLTTienBKrockenbergerM 2007 Sex in natural populations of C. gattii. HeitmanJKronstadJWTaylorJCasseltonL 477 488 Sex in Fungi: Molecular Determination and Evolutionary Implications: ASM Press
56. GiraudTGladieuxPByrnesEJIIIFisherMAguiletaG 2011 Epidemiology and evolution of fungal pathogens in plants and animals. TibayrencM Genetics and Evolution of Infectious Diseases New York Elsevier 59 106
57. KiddSEGuoHBartlettKHXuJKronstadJW 2005 Comparative gene genealogies indicate that two clonal lineages of Cryptococcus gattii in British Columbia resemble strains from other geographical areas. Eukaryot Cell 4 1629 1638
58. LindbergJHagenFLaursenAStenderupJBoekhoutT 2007 Cryptococcus gattii risk for tourists visiting Vancouver Island, Canada. Emerg Infect Dis 13 178 179
59. DattaKBartlettKHBaerRByrnesEGalanisE 2009 Spread of Cryptococcus gattii into Pacific Northwest region of the United States. Emerg Infect Dis 15 1185 1191
60. OkamotoKHatakeyamaSItoyamaSNukuiYYoshinoY 2010 Cryptococcus gattii genotype VGIIa infection in man, Japan, 2007. Emerg Infect Dis 16 1155 1157
61. HagenFAssenSVLuijckxGJBoekhoutTKampingaGA 2009 Activated dormant Cryptococcus gattii infection in a Dutch tourist who visited Vancouver Island (Canada): a molecular epidemiological approach. Med Mycol 48 528 531
62. GeorgiASchneemannMTintelnotKCalligaris-MaibachRCMeyerS 2009 Cryptococcus gattii meningoencephalitis in an immunocompetent person 13 months after exposure. Infection 37 370 373
63. MacDougallLKiddSEGalanisEMakSLeslieMJ 2007 Spread of Cryptococcus gattii in British Columbia, Canada, and detection in the Pacific Northwest, USA. Emerg Infect Dis 13 42 50
64. LitiGBartonDBLouisEJ 2006 Sequence diversity, reproductive isolation and species concepts in Saccharomyces. Genetics 174 839 850
65. Le GacMHoodMEFournierEGiraudT 2007 Phylogenetic evidence of host-specific cryptic species in the anther smut fungus. Evolution 61 15 26
66. KavanaughLAFraserJADietrichFS 2006 Recent evolution of the human pathogen Cryptococcus neoformans by intervarietal transfer of a 14-gene fragment. Mol Biol Evol 23 1879 1890
67. NeafseyDEBarkerBMSharptonTJStajichJEParkDJ 2010 Population genomic sequencing of Coccidioides fungi reveals recent hybridization and transposon control. Genome Res 20 938 946
68. OkagakiLHStrainAKNielsenJNCharlierCBaltesNJ 2010 Cryptococcal cell morphology affects host cell interactions and pathogenicity. PLoS Pathog 6 e1000953
69. NgamskulrungrojPSorrellTCChindampornAChaiprasertAPoonwanN 2008 Association between fertility and molecular sub-type of global isolates of Cryptococcus gattii molecular type VGII. Med Mycol 1 9
70. BakkerenGKamperJSchirawskiJ 2008 Sex in smut fungi: Structure, function and evolution of mating-type complexes. Fungal Genet Biol 45 Suppl 1 S15 21
71. NielsenKCoxGMLitvintsevaAPMylonakisEMalliarisSD 2005 Cryptococcus neoformans alpha strains preferentially disseminate to the central nervous system during coinfection. Infect Immun 73 4922 4933
72. NielsenKHeitmanJ 2007 Sex and virulence of human pathogenic fungi. Adv Genet 57 143 173
73. DanielsKJSrikanthaTLockhartSRPujolCSollDR 2006 Opaque cells signal white cells to form biofilms in Candida albicans. EMBO J 25 2240 2252
74. SahniNYiSDanielsKJHuangGSrikanthaT 2010 Tec1 mediates the pheromone response of the white phenotype of Candida albicans: insights into the evolution of new signal transduction pathways. PLoS Biol 8 e1000363
75. YiSSahniNPujolCDanielsKJSrikanthaT 2009 A Candida albicans-specific region of the alpha-pheromone receptor plays a selective role in the white cell pheromone response. Mol Microbiol 71 925 947
76. ZhaoRDanielsKJLockhartSRYeaterKMHoyerLL 2005 Unique aspects of gene expression during Candida albicans mating and possible G(1) dependency. Eukaryot Cell 4 1175 1190
77. XueCTadaYDongXHeitmanJ 2007 The human fungal pathogen Cryptococcus can complete its sexual cycle during a pathogenic association with plants. Cell Host Microbe 1 263 273
78. PapageorgiouIGournasCVlantiAAmillisSPantazopoulouA 2008 Specific interdomain synergy in the UapA transporter determines its unique specificity for uric acid among NAT carriers. J Mol Biol 382 1121 1135
79. DiallinasGGorfinkielLArstHNJrCecchettoGScazzocchioC 1994 Genetic and molecular characterisation of purine permease genes of Aspergillus nidulans reveals a novel family of transporters conserved in prokaryotes and eukaryotes. Folia Microbiol (Praha) 39 513 514
80. DiallinasGScazzocchioC 1989 A gene coding for the uric acid-xanthine permease of Aspergillus nidulans: inactivational cloning, characterization, and sequence of a cis-acting mutation. Genetics 122 341 350
81. ErbsPExingerFJundR 1997 Characterization of the Saccharomyces cerevisiae FCY1 gene encoding cytosine deaminase and its homologue FCA1 of Candida albicans. Curr Genet 31 1 6
82. WhelanWL 1987 The genetic basis of resistance to 5-fluorocytosine in Candida species and Cryptococcus neoformans. Crit Rev Microbiol 15 45 56
83. HsuehYPIdnurmAHeitmanJ 2006 Recombination hotspots flank the Cryptococcus mating-type locus: implications for the evolution of a fungal sex chromosome. PLoS Genet 2 e184
84. FindleyKRodriguez-CarresMMetinBKroissJFonsecaA 2009 Phylogeny and phenotypic characterization of pathogenic Cryptococcus species and closely related saprobic taxa in the Tremellales. Eukaryot Cell 8 353 361
85. MetinBFindleyKHeitmanJ 2010 The mating type locus (MAT) and sexual reproduction of Cryptococcus heveanensis: insights into the evolution of sex and sex-determining chromosomal regions in fungi. PLoS Genet 6 e1000961
86. McClellandEEPerrineWTPottsWKCasadevallA 2005 Relationship of virulence factor expression to evolved virulence in mouse-passaged Cryptococcus neoformans lines. Infect Immun 73 7047 7050
87. MaidenMCBygravesJAFeilEMorelliGRussellJE 1998 Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A 95 3140 3145
88. ByrnesEJ3rdBildfellRJDearingPLValentineBAHeitmanJ 2009 Cryptococcus gattii with bimorphic colony types in a dog in western Oregon: additional evidence for expansion of the Vancouver Island outbreak. J Vet Diagn Invest 21 133 136
89. ByrnesEJ3rdLiWLewitYPerfectJRCarterDA 2009 First reported case of Cryptococcus gattii in the Southeastern USA: implications for travel-associated acquisition of an emerging pathogen. PLoS One 4 e5851
90. MeyerWAanensenDMBoekhoutTCogliatiMDiazMR 2009 Consensus multi-locus sequence typing scheme for Cryptococcus neoformans and Cryptococcus gattii. Med Mycol 47 561 570
91. AltschulSFMaddenTLSchafferAAZhangJZhangZ 1997 Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25 3389 3402
92. LarkinMABlackshieldsGBrownNPChennaRMcGettiganPA 2007 Clustal W and Clustal X version 2.0. Bioinformatics 23 2947 2948
93. GuindonSDelsucFDufayardJFGascuelO 2009 Estimating maximum likelihood phylogenies with PhyML. Methods Mol Biol 537 113 137
94. ClementMPosadaDCrandallKA 2000 TCS: a computer program to estimate gene genealogies. Mol Ecol 9 1657 1659
95. EwingBGreenP 1998 Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 8 186 194
96. EwingBHillierLWendlMCGreenP 1998 Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 8 175 185
97. NarasipuraSDChaturvediVChaturvediS 2005 Characterization of Cryptococcus neoformans variety gattii SOD2 reveals distinct roles of the two superoxide dismutases in fungal biology and virulence. Mol Microbiol 55 1782 1800
98. MaHCroudaceJELammasDAMayRC 2006 Expulsion of live pathogenic yeast by macrophages. Curr Biol 16 2156 2160
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 9
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- HTLV-1 Propels Thymic Human T Cell Development in “Human Immune System” Rag2 gamma c Mice
- Hostile Takeover by : Reorganization of Parasite and Host Cell Membranes during Liver Stage Egress
- Exploiting and Subverting Tor Signaling in the Pathogenesis of Fungi, Parasites, and Viruses
- A Viral Ubiquitin Ligase Has Substrate Preferential SUMO Targeted Ubiquitin Ligase Activity that Counteracts Intrinsic Antiviral Defence