
Hsp90 Governs Dispersion and Drug Resistance of Fungal Biofilms
Fungal biofilms are a major cause of human mortality and are recalcitrant to most treatments due to intrinsic drug resistance. These complex communities of multiple cell types form on indwelling medical devices and their eradication often requires surgical removal of infected devices. Here we implicate the molecular chaperone Hsp90 as a key regulator of biofilm dispersion and drug resistance. We previously established that in the leading human fungal pathogen, Candida albicans, Hsp90 enables the emergence and maintenance of drug resistance in planktonic conditions by stabilizing the protein phosphatase calcineurin and MAPK Mkc1. Hsp90 also regulates temperature-dependent C. albicans morphogenesis through repression of cAMP-PKA signalling. Here we demonstrate that genetic depletion of Hsp90 reduced C. albicans biofilm growth and maturation in vitro and impaired dispersal of biofilm cells. Further, compromising Hsp90 function in vitro abrogated resistance of C. albicans biofilms to the most widely deployed class of antifungal drugs, the azoles. Depletion of Hsp90 led to reduction of calcineurin and Mkc1 in planktonic but not biofilm conditions, suggesting that Hsp90 regulates drug resistance through different mechanisms in these distinct cellular states. Reduction of Hsp90 levels led to a marked decrease in matrix glucan levels, providing a compelling mechanism through which Hsp90 might regulate biofilm azole resistance. Impairment of Hsp90 function genetically or pharmacologically transformed fluconazole from ineffectual to highly effective in eradicating biofilms in a rat venous catheter infection model. Finally, inhibition of Hsp90 reduced resistance of biofilms of the most lethal mould, Aspergillus fumigatus, to the newest class of antifungals to reach the clinic, the echinocandins. Thus, we establish a novel mechanism regulating biofilm drug resistance and dispersion and that targeting Hsp90 provides a much-needed strategy for improving clinical outcome in the treatment of biofilm infections.
Published in the journal:
. PLoS Pathog 7(9): e32767. doi:10.1371/journal.ppat.1002257
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002257
Summary
Fungal biofilms are a major cause of human mortality and are recalcitrant to most treatments due to intrinsic drug resistance. These complex communities of multiple cell types form on indwelling medical devices and their eradication often requires surgical removal of infected devices. Here we implicate the molecular chaperone Hsp90 as a key regulator of biofilm dispersion and drug resistance. We previously established that in the leading human fungal pathogen, Candida albicans, Hsp90 enables the emergence and maintenance of drug resistance in planktonic conditions by stabilizing the protein phosphatase calcineurin and MAPK Mkc1. Hsp90 also regulates temperature-dependent C. albicans morphogenesis through repression of cAMP-PKA signalling. Here we demonstrate that genetic depletion of Hsp90 reduced C. albicans biofilm growth and maturation in vitro and impaired dispersal of biofilm cells. Further, compromising Hsp90 function in vitro abrogated resistance of C. albicans biofilms to the most widely deployed class of antifungal drugs, the azoles. Depletion of Hsp90 led to reduction of calcineurin and Mkc1 in planktonic but not biofilm conditions, suggesting that Hsp90 regulates drug resistance through different mechanisms in these distinct cellular states. Reduction of Hsp90 levels led to a marked decrease in matrix glucan levels, providing a compelling mechanism through which Hsp90 might regulate biofilm azole resistance. Impairment of Hsp90 function genetically or pharmacologically transformed fluconazole from ineffectual to highly effective in eradicating biofilms in a rat venous catheter infection model. Finally, inhibition of Hsp90 reduced resistance of biofilms of the most lethal mould, Aspergillus fumigatus, to the newest class of antifungals to reach the clinic, the echinocandins. Thus, we establish a novel mechanism regulating biofilm drug resistance and dispersion and that targeting Hsp90 provides a much-needed strategy for improving clinical outcome in the treatment of biofilm infections.
Introduction
In recent decades, fungal pathogens have emerged as a predominant cause of human disease, especially in immunocompromised individuals. The number of acquired fungal bloodstream infections has increased by ∼207% in this timeframe [1], [2], [3]. Although diverse species are capable of causing infection, a few prevail as the most prevalent cause of disease. Candida and Aspergillus species together account for ∼70% of all invasive fungal infections, with Candida albicans and Aspergillus fumigatus prevailing as the leading causal agents of opportunistic mycoses [2]. Candida species are the fourth leading cause of hospital acquired bloodstream infections in the United States with mortality rates estimated at 40% [4], [5]. The profound economic consequences of Candida infections can be demonstrated by the ∼$1.7 billion spent annually on treating candidemia in the United States alone [6]. Further, A. fumigatus is the most common etiological agent of invasive aspergillosis, with a 40–90% mortality rate [7]. In patients with pulmonary disorders such as asthma or cystic fibrosis, A. fumigatus infection can cause allergic bronchopulmonary aspergillosis leading to severe complications. For these fungal species, there are numerous factors that contribute to the pathogenicity and recalcitrance of resulting infections to antifungal treatment, including the ability to evolve and maintain resistance to conventional antifungal therapy [1].
Due to the limited number of drug targets available to exploit in fungal pathogens that are absent or sufficiently divergent in the human host, the vast majority of antifungal drugs in clinical use target ergosterol or its biosynthesis. The azoles are the most widely used class of antifungal in the clinic and function by inhibiting the ergosterol biosynthetic enzyme Erg11, causing a block in the production of ergosterol and the accumulation of the toxic byproduct 14-α-methyl-3,6-diol, culminating in a severe membrane stress [8], [9]. The azoles are generally fungistatic against yeasts, including Candida species, and fungicidal against moulds, such as Aspergillus species. The fungistatic nature of the azoles towards C. albicans culminates in strong directional selection on the surviving population to evolve drug resistance [10], [11]. In fact, high levels of azole resistance in C. albicans clinical isolates often accumulate through multiple mechanisms including: upregulation of drug efflux pumps, overexpression or alteration of Erg11, or modification of stress response pathways that are crucial for resistance [1], [10], [11], [12], [13]. The echinocandins are the only new class of antifungal to reach the clinic in decades. They act as non-competitive inhibitors of β-1,3 glucan synthase, an enzyme involved in fungal cell wall synthesis [9], resulting in the loss of cell wall integrity and a severe cell wall stress. The impact of the echinocandins is generally opposite to that of the azoles, in that they are fungicidal against yeasts and fungistatic against moulds. Resistance of C. albicans clinical isolates to the echinocandins has been reported and is often associated with mutations in the drug target [13], [14], [15].
An additional key factor responsible for the virulence and drug resistance of C. albicans and A. fumigatus is their tendency to form biofilms on medical devices that are highly resistant to antifungal treatment [16], [17], [18], [19], [20]. The use of such medical devices — such as venous catheters, urinary catheters and artificial joints — has dramatically risen to more than 10 million recipients per year [21], [22]. This poses a severe clinical problem as C. albicans is the third leading cause of intravascular catheter-related infections, and has the overall highest crude mortality rate of ∼30% for device-associated infections [17], [22], [23]. Further, A. fumigatus infections have been reported on medical implant devices as well as on bronchial epithelial cells [17], [24]. The inherent drug resistance of biofilms often necessitates surgical removal of the infected medical devices in order to eradicate the fungal infection.
Extensive research has focused on mechanisms of drug resistance in C. albicans biofilms, and it is apparent that cells in a fungal biofilm represent an epigenetic modification of the cellular state compared to their planktonic counterparts, with changes in cellular morphology, cell-to-cell communication, and gene expression, as well as with the production of an extra-cellular matrix [16], [18], [20]. Multiple factors contribute to the elevated drug resistance of C. albicans biofilms. These factors include increased cell density [25], increased expression of drug efflux pumps [26], [27], decreased ergosterol content [27], elevated β-1,3 glucan levels in the cell wall and biofilm matrix [28], [29], as well as signalling mediated by protein kinase C (PKC) [30] and the protein phosphatase calcineurin [31].
The molecular chaperone Hsp90 regulates complex cellular circuitry in eukaryotes by stabilizing regulators of cellular signalling [32], [33]. As a consequence, inhibiting Hsp90 disrupts a plethora of cellular processes and has broad therapeutic potential against diverse eukaryotic pathogens including the protozoan parasites Plasmodium falciparum and Trypanosoma evansi as well as numerous fungal species [34], [35], [36]. In the planktonic state, Hsp90 potentiates the emergence and maintenance of resistance to azoles and echinocandins in C. albicans at least in part via calcineurin [37]; Hsp90 physically interacts with the catalytic subunit of calcineurin, keeping it stable and poised for activation [38]. Recently, Hsp90 was also shown to enable azole and echinocandin resistance in C. albicans via the PKC cell wall integrity pathway [39]. Hsp90 depletion results in the destabilization of the terminal mitogen-activated protein kinase (MAPK) Mkc1, providing the second Hsp90 client protein implicated in drug resistance [39]. Compromising C. albicans Hsp90 function renders drug-resistant isolates susceptible in vitro and improves the therapeutic efficacy of antifungals in a Galleria mellonella model of C. albicans pathogenesis and a murine model of disseminated candidiasis [34]. Compromising A. fumigatus Hsp90 also enhances the efficacy of echinocandins both in vitro and in the G. mellonella model of infection [34]. Notably, Hsp90 regulates not only drug resistance in C. albicans but also the morphogenetic transition between yeast and filamentous growth, a trait important for virulence [40]. Compromising Hsp90 function induces filamentation by relieving Hsp90-mediated repression of cAMP-protein kinase A (PKA) signalling [41]. The ability to transition between morphological states is also critical for biofilm formation and development [42].
Given that Hsp90 governs fungal morphogenesis and drug resistance in planktonic conditions, we sought to investigate if this molecular chaperone also regulates the development and drug resistance of biofilms. We discovered that genetically compromising Hsp90 function reduced but did not block biofilm maturation in vitro and had minimal impact on the ability of C. albicans to form robust biofilms in an in vivo rat catheter model,. Genetic depletion of C. albicans Hsp90 reduced biofilm dispersal, with the few dispersed cells being largely inviable. Moreover, compromising C. albicans Hsp90 function genetically or pharmacologically transformed the azole fluconazole from ineffectual to highly efficacious in eradicating biofilms both in vitro and in a rat catheter model of infection. In stark contrast to planktonic conditions, reduction of C. albicans Hsp90 levels genetically in biofilm conditions did not lead to depletion of the client proteins calcineurin or Mkc1, suggesting that Hsp90 regulates drug resistance through distinct mechanisms in these different cellular states. Genetic depletion of Hsp90 reduced glucan levels in the biofilm matrix, providing a compelling mechanism by which Hsp90 might regulate biofilm drug resistance. Finally, in the most lethal mould, A. fumigatus, compromising Hsp90 function enhanced the efficacy of azoles and echinocandins in an in vitro model. Our results implicate Hsp90 as a novel regulator of biofilm dispersion and drug resistance, and provide strong support for the utility of Hsp90 inhibitors as a therapeutic strategy for biofilm infections caused by diverse fungal species.
Results
Hsp90 is not required for C. albicans biofilm formation in vitro or in vivo
Due to the key roles of Hsp90 in both morphogenesis and drug resistance under planktonic conditions [37], [41], we hypothesized that Hsp90 might also regulate C. albicans biofilm formation and drug resistance. First, we tested whether compromising Hsp90 function affected biofilm growth. To do this, C. albicans biofilms were cultured for 24 hours in static 96 well microtiter plates, washed to remove non-adherent cells, grown for an additional 24 hours with various concentrations of the Hsp90 inhibitor geldanamycin, and growth was quantified by metabolic activity using an XTT reduction assay [43]. The geldanamycin was added at 24 hours rather than at the initial time point as is the standard for biofilm drug studies since the initial cells are planktonic and much more susceptible to drugs than their biofilm counterparts [31], [43]; consistent with this, initial attempts to include geldanamycin during inoculation led to a toxicity profile identical to that of planktonic cells (data not shown). When geldanamycin was added at 24 hours, no significant differences in metabolic activity were observed at a variety of concentrations tested up to 100 µg/mL (P>0.05, ANOVA, Bonferroni's Multiple Comparison Test, Figure 1A). Thus, Hsp90 inhibitors do not compromise biofilm development.

To further explore Hsp90's role in biofilm formation, we exploited a strain of C. albicans in which Hsp90 levels could be depleted by tetracycline-mediated transcriptional repression (tetO-HSP90/hsp90Δ). Biofilms of the wild type and tetO-HSP90/hsp90Δ strain were cultured in static 96 well microtiter plates with or without 20 µg/mL of the tetracycline analog doxycycline from the time of inoculation. Doxycycline was included at this early point given the time required for transcriptional repression to manifest in depletion of Hsp90, and enabled by the absence of toxicity in planktonic cells. Doxycycline-mediated transcriptional repression of Hsp90 decreased biofilm development, but did not block formation of a mature biofilm (Figure 1B, P<0.01). We observed comparable results when biofilms were cultured on silicon elastomer squares, and when biofilm growth was monitored by XTT reduction or by dry weight (Figure S1A and B). To determine if depletion of Hsp90 prior to inoculation had a more profound effect on biofilm formation, we performed a comparable assay but in the presence or absence of doxycycline in the overnight culture. Depletion of Hsp90 prior to inoculation did not further reduce biofilm formation but rather led to a biofilm indistinguishable from the no doxycycline control (Figure S1C). Although Hsp90 is essential, this dose of doxycycline causes reduced growth rate of the tetO-HSP90/hsp90Δ strain in planktonic cultures but has little effect on stationary phase cell density [41]. Western blot analysis validated that Hsp90 levels were dramatically reduced in biofilms formed by the tetO-HSP90/hsp90Δ strain when cultured in the presence of doxycycline (Figure 1C). We note that when biofilms were formed under shaking conditions, the tetO-HSP90/hsp90Δ strain had reduced biofilm growth, which was exacerbated in the presence of doxycycline (Figure S1D). Thus, while Hsp90's impact on biofilm development can vary, under most conditions tested compromising Hsp90 function does not block biofilm formation in vitro.
In order to address the role of Hsp90 in biofilm growth in vivo, biofilm formation was examined using a rat venous catheter model of biofilm-associated candidiasis that mimics central venous catheters in patients [44]. Infection of implanted catheters with C. albicans was performed by intraluminal instillation, catheters were flushed after 6 hours, and biofilm formation was monitored with or without 20 µg/mL doxycycline after 24 hours. The tetO-HSP90/hsp90Δ strain was capable of establishing a biofilm in the rat venous catheter, as visualized by scanning electron microscopy (Figure 2). Further, transcriptional repression of HSP90 with doxycycline did not block the formation of a robust biofilm (Figure 2). These results demonstrate that compromising Hsp90 function does not impair the ability of C. albicans to form mature biofilms in vivo.

Compromising Hsp90 function produces biofilms with altered morphologies
As mentioned above, Hsp90 is a key regulator of the yeast to filament transition in C. albicans [41], a process implicated in virulence and biofilm formation [42]. Therefore, we examined the architecture of geldanamycin treated biofilms cultured on silicon elastomer squares to enable imaging by confocal microscopy. Biofilms treated with geldanamycin had decreased thickness of the bottom yeast layer (30 µm and 45 µm versus 90 µm and 100 µm in the untreated control, P = 0.0237, t-test) without substantial change in the thickness of the upper layer of filaments (Figure 3). That a greater proportion of the biofilm thickness was occupied by filaments compared to yeast suggests that Hsp90 inhibition might lead to enhanced filamentation in biofilms. Moreover, biofilms treated with geldanamycin showed more polarized filaments extending away from the biofilm basal surface compared to the interconnected meshwork of filaments in an untreated control (Figure 3). That biofilms formed upon Hsp90 inhibition had a greater proportion of their total thickness occupied by filaments compared to yeast is consistent with Hsp90's repressive effect on filamentation in planktonic conditions.

Hsp90 function is important for dispersal of C. albicans biofilms
Based on our finding that C. albicans biofilms display altered morphologies upon Hsp90 inhibition, we sought to evaluate the effect of Hsp90 function on biofilm dispersion given that morphogenesis plays a critical role in this process [45], [46]. We monitored dispersion of yeast cells using the only well validated model which involves culturing biofilms on silicon elastomer under conditions of flow [47], [48]. When biofilms were cultured in the absence of doxycycline with the tetO-HSP90/hsp90Δ strain, the number of dispersed cells after 1 hour was 90,000 cells/mL and remained fairly constant over a 24 hour time period (Figure 4A). In contrast, in the presence of 20 µg/mL doxycycline the number of dispersed cells was dramatically reduced to approximately 17,000 cells/mL throughout the 24 hours (P = 0.0022, t-test, Figure 4A). We confirmed that the effects of doxycycline were specifically due to transcriptional repression of HSP90, as doxycycline had no impact on biofilm dispersal of the wild-type strain lacking the tetO promoter (Figure S2A). Intriguingly, the cells that were dispersed upon reduction of Hsp90 levels had major viability defects compared to their untreated counterparts (P = 0.007, t-test) with only 55% viable at 1 hour, 5% viable at 12 hours, and less than 1% viable at 24 hours (Figure 4B). The dramatic reduction in viability was specific to the dispersed cell population with doxycycline-mediated transcriptional repression of HSP90, as the viability of dispersed cells in the untreated control remained close to 50% even at 24 hours (Figure 4B). Viability was unaffected when a wild-type strain lacking the tetO promoter was treated with doxycycline, confirming that the effects observed were due to transcriptional repression of HSP90 (Figure S2B). The reduced viability upon reduction of Hsp90 levels was specific to the dispersed cell population within the biofilm, as there was only a minor defect in overall metabolic activity of the tetO-HSP90/hsp90Δ biofilms in the presence of doxycycline (Figure 1B). Further, under planktonic conditions viability remained>85% when the tetO-HSP90/hsp90Δ strain was grown in the presence of doxycycline for 24 hours. Taken together, Hsp90 plays a critical role in the dispersal step of the biofilm life cycle and is crucial for survival of dispersed cells.

Hsp90 enables the resistance of C. albicans biofilms to fluconazole in vitro
Genetic or pharmacological compromise of Hsp90 function renders C. albicans susceptible to azoles and echinocandins under planktonic conditions [37], [38], [49]. Since compromising Hsp90 function pharmacologically did not impair biofilm maturation, we investigated whether inhibition of Hsp90 would alter biofilm drug resistance using the standard 96 well microtiter plate static assay that enables testing many drug concentrations. We focused on the azoles, since biofilms are notoriously resistant to this class of drugs, compromising their therapeutic utility [19]. As a positive control, a wild-type C. albicans biofilm was subjected to a gradient of concentrations of the calcineurin inhibitor FK506 in addition to a gradient of fluconazole, a drug combination with established synergistic activity against C. albicans biofilms [31]. We confirmed synergistic activity of FK506 with fluconazole by measuring metabolic activity using the XTT reduction assay (Figure 5A). Biofilms were extremely susceptible to the combination of inhibitors with a calculated FIC index of 0.1093, indicating potent synergy (Table 1). To determine if Hsp90 enables biofilm azole resistance, we used an equivalent experiment but with a gradient of concentrations of the Hsp90 inhibitor geldanamycin and a gradient of fluconazole. Geldanamycin exhibited potent synergy with fluconazole, dramatically reducing azole resistance at only 3.125 µg/mL geldanamycin. Maximal effects were observed with 12.5 µg/mL geldanamycin, which reduced the MIC50 of fluconazole from >1000 µg/mL to 31.25 µg/mL (Figure 5A). Further, FIC indexes as low as 0.125 to 0.156 were calculated for the combination of fluconazole and geldanamycin confirming that inhibition of Hsp90 has a potent synergistic effect with azoles against C. albicans biofilms (Table 1).


Next, we utilized the tetO-HSP90/hsp90Δ strain in order to validate that the synergistic activity of geldanamycin with fluconazole against C. albicans biofilms was indeed due to Hsp90 inhibition. Biofilms of a wild-type strain of C. albicans had a fluconazole MIC50 of over 512 µg/mL (Figure 5B). Deletion of one allele of HSP90 or replacing the promoter of the sole remaining HSP90 allele with the tetracycline-repressible promoter had no impact on fluconazole resistance (Figure 5B). However, upon depletion of Hsp90 by doxycycline-mediated transcriptional repression in the tetO-HSP90/hsp90Δ strain, the fluconazole MIC50 was dramatically reduced to only 8 µg/mL, a >60-fold increase in fluconazole sensitivity (Figure 5B). Hence, both pharmacological and genetic evidence confirms that Hsp90 function is critical for azole resistance of C. albicans biofilms.
The role of downstream effectors of Hsp90 in azole resistance of C. albicans biofilms
To further dissect the mechanism by which Hsp90 regulates azole resistance of C. albicans biofilms, we repeated the drug susceptibility assay with strains lacking specific Hsp90 client proteins. Under planktonic conditions both calcineurin and Mkc1 are important Hsp90 client proteins that regulate the maintenance of azole resistance [37], [38], [39]. Moreover, these client proteins have previously been shown to be important for azole resistance of C. albicans biofilms [30], [31]. We found that biofilms formed by strains lacking the catalytic subunit of calcineurin (cna1Δ/cna1Δ) or the terminal MAPK of the PKC cell wall integrity signalling pathway (mkc1Δ/mkc1Δ) had fluconazole MIC50 values of 32 µg/mL and 128 µg/mL, respectively; their fluconazole resistance levels were intermediate between the robust resistance of the wild-type parental strain and the sensitivity observed upon impairment of Hsp90 function (Figure 5B). The finding that compromise of calcineurin function does not confer as severe a reduction in biofilm fluconazole resistance as compromise of Hsp90 function is intriguing in light of the fact that under all planktonic conditions tested, inhibition of calcineurin phenocopies inhibition of Hsp90 in terms of azole resistance [37], [38], [49]. These results suggest that calcineurin and Mkc1 may be able to partially compensate for the loss of the other client during times of azole-induced stress in a biofilm environment. Alternatively, these findings could be explained by the existence of a novel downstream effector of Hsp90 important for azole resistance of C. albicans biofilms.
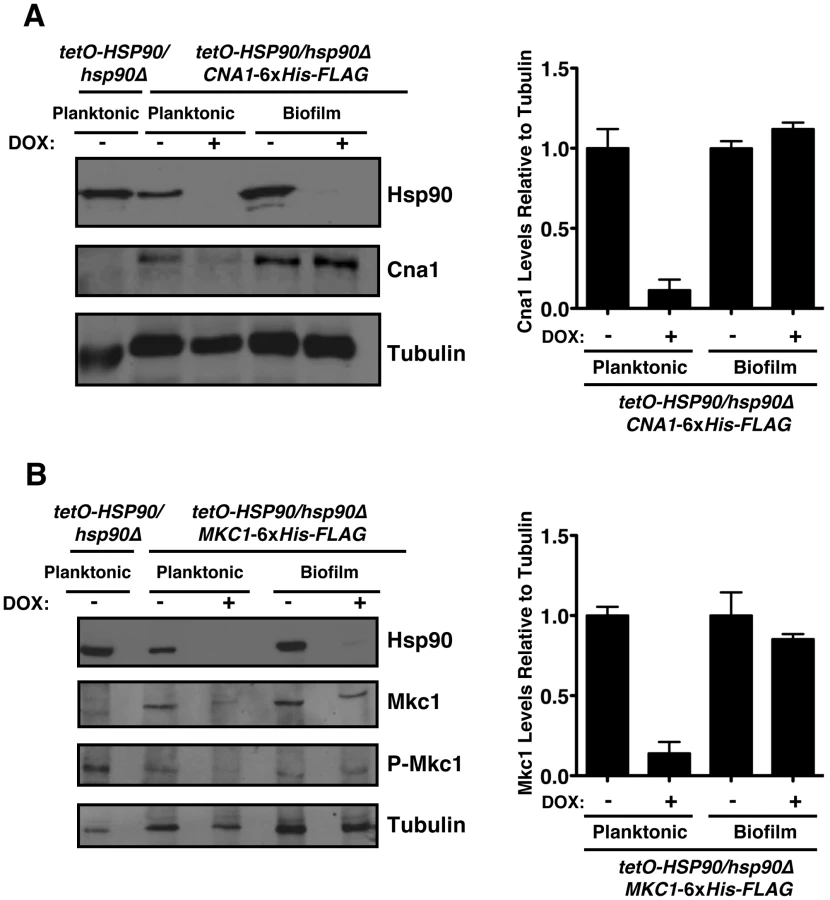
To further investigate the mechanisms by which Hsp90 regulates azole resistance in biofilm conditions, we examined protein levels of the client proteins calcineurin and Mkc1 upon Hsp90 depletion. Strains were cultured in RPMI medium for both planktonic and biofilm growth. Biofilms were cultured on plastic under static conditions, as with our drug studies. We previously established that under planktonic conditions genetic reduction of Hsp90 levels leads to depletion of the catalytic subunit of calcineurin (Cna1) and Mkc1 [38], [39]. Here, the tetO-HSP90/hsp90Δ strain was grown in either planktonic or biofilm conditions in the presence or absence of 20 µg/mL doxycycline for 48 hours. Under both conditions, Hsp90 levels were dramatically reduced in the presence of doxycycline (Figure 6). To monitor calcineurin levels, we used a C-terminal 6xHis-FLAG epitope tag on Cna1 in the tetO-HSP90/hsp90Δ strain. In the tagged strains, Cna1 levels were comparable under planktonic and biofilm conditions in the absence of doxycycline (Figure 6A). Doxycycline-mediated reduction of Hsp90 levels led to an ∼90% reduction in Cna1 in planktonic conditions, however, Cna1 levels remained stable in biofilm conditions (Figure 6A). All strains had comparable amounts of protein loaded, as confirmed with a tubulin loading control. To monitor total Mkc1 levels, we used a C-terminal 6xHis-FLAG epitope tag on Mkc1 in the tetO-HSP90/hsp90Δ strain. Mkc1 levels were comparable in the tagged strains under planktonic and biofilm conditions in the absence of doxycycline (Figure 6B). As with Cna1, doxycycline-mediated reduction of Hsp90 levels led to ∼80% reduction in Mkc1 levels in planktonic conditions, however, Mkc1 levels remained stable in biofilm conditions (Figure 6B). We next addressed whether depletion of Hsp90 affected levels of activated, dually phosphorylated Mkc1. Mkc1 was activated in all strains in the absence of doxycycline. As with total Mkc1 levels, doxycycline-mediated reduction of Hsp90 led to a reduction in levels of activated Mkc1 in planktonic conditions, however, Mkc1 remained activated in biofilm conditions. Taken together, these results suggest that Hsp90 may play different roles in client protein regulation in these distinct cellular states, and also that these client proteins may have other means of maintaining stability in a biofilm environment.
Hsp90 regulates matrix glucan levels in C. albicans biofilms
Given our findings that Hsp90 client proteins remain stable in a biofilm, irrespective of Hsp90 levels, and that deletion of these client proteins does not phenocopy Hsp90 depletion in terms of biofilm azole resistance, we hypothesized that Hsp90 also regulates biofilm drug resistance through a mechanism independent of calcineurin and Mkc1 signalling. Recent studies established that glucan present in the biofilm matrix is critical for azole resistance due its capacity to sequester fluconazole, preventing it from reaching its intracellular target [29]. Consequently, we investigated whether Hsp90 affects glucan levels in the biofilm matrix. Biofilms were cultured on plastic in static conditions in the presence or absence of 20 µg/mL doxycycline for 48 hours, matrix material was harvested from biofilms with equivalent metabolic activity, and β-1,3 glucan levels were quantified. In the tetO-HSP90/hsp90Δ strain, the level of glucan in the biofilm matrix was ∼6,000 pg/mL in the absence of doxycycline (Figure 7). Transcriptional repression of HSP90 with 20 µg/mL doxycycline led to reduced glucan levels of only ∼3,700 pg/mL (P<0.01, ANOVA, Bonferroni's Multiple Comparison Test, Figure 7). Doxycycline had no impact on matrix glucan levels of a wild-type strain lacking the tetO promoter, confirming that Hsp90 depletion leads to reduced glucan levels (Figure 7). The ∼40% reduction in matrix glucan upon Hsp90 depletion is likely to have made a major contribution to azole susceptibility, given that reduction of biofilm matrix glucan levels of ∼60% in an FKS1/fks1Δ mutant abrogates biofilm drug resistance [29]. These results provide the first link of Hsp90 to glucan production in C. albicans and mechanistic insight as to how Hsp90 regulates biofilm drug resistance.

Hsp90 is required for C. albicans biofilm azole resistance in vivo
Due to the robust synergy observed between Hsp90 inhibition and fluconazole in vitro, we sought to address whether synergy was also observed in vivo in the rat venous catheter model of C. albicans biofilm infection using the tetO-HSP90/hsp90Δ strain. Addition of fluconazole alone (250 µg/mL) after 24 hours of biofilm growth did not affect the biofilm formed by the tetO-HSP90/hsp90Δ strain (Figure 8A). Doxycycline was delivered during both the biofilm formation and drug treatment phases, and also had no major effect on the biofilm formed by the tetO-HSP90/hsp90Δ strain (Figure 2). However, the combination of fluconazole and doxycycline destroyed the biofilm as observed by scanning electron microscopy (Figure 8A). Thus, Hsp90 is required for the resistance of C. albicans biofilms to fluconazole in a mammalian host.

In order to further explore the therapeutic potential of targeting Hsp90 for C. albicans biofilm infections in vivo, we explored the efficacy of combining fluconazole with an Hsp90 inhibitor structurally related to geldanamycin and in clinical development as an anti-cancer agent, 17-(allylamino)-17-demethoxygeldanamycin (17-AAG). Central venous rat catheters were infected with C. albicans and biofilm formation proceeded over a 24-hour period. At this point, fluconazole alone (250 µg/mL), 17-AAG alone (100 µg/mL), or the drug combination was instilled and allowed to dwell in the catheter for an additional 24 hours. Serial dilutions of the catheter fluid were then plated in order to assess viable colony forming units. We found that the combined drug treatment significantly reduced fungal burden compared to the individual drug treatments alone (P<0.001, ANOVA, Bonferroni's Multiple Comparison Test, Figure 8B). In fact, catheters from the animals undergoing the combination therapy were completely sterile (Figure 8B). These experiments in a mammalian model provide compelling evidence that clinically relevant Hsp90 inhibitors may prove to be extremely valuable in combating C. albicans biofilm infections.
Hsp90 is required for drug resistance of A. fumigatus biofilms
We previously established that Hsp90 inhibitors increase the efficacy of the echinocandins against A. fumigatus under standard culture conditions [34], motivating these studies to determine if Hsp90 inhibitors also affect drug resistance of A. fumigatus biofilms. After 24 hours of growth, A. fumigatus biofilms were subjected to a gradient of concentrations of the echinocandins caspofungin or micafungin, or the azoles voriconazole or fluconazole, in addition to a gradient of concentrations of the Hsp90 inhibitor geldanamycin in 96 well microtiter plates under static conditions. Metabolic activity was assessed using the XTT reduction assay after an additional 24 hours. The biofilms were completely resistant to all the antifungal drugs tested and geldanamycin individually, though the combination of geldanamycin with many of the antifungals was effective in reducing biofilm development. Geldanamycin displayed robust synergy with both caspofungin (Figure 9A) and micafungin (Figure S3A), with an FIC value of 0.375 for both drugs (Table 2). Geldanamycin also enhanced voriconazole activity (Figure 9A), with more potent effects observed when drugs were added to biofilms after only 8 hours of growth (Figure S3B). Geldanamycin did not enhance the efficacy of fluconazole under any conditions tested (data not shown). These patterns of drug synergy observed with A. fumigatus biofilms are consistent with those patterns observed with Aspergillus in planktonic conditions [37].


Next, given Hsp90's role in regulating fungal morphogenesis we explored the impact of drug treatment on morphology of A. fumigatus biofilms. Scanning electron microscopy revealed striking architectural changes of A. fumigatus biofilms upon drug treatment. The control biofilms appeared robust and healthy, however, upon Hsp90 inhibition increased hyphal and matrix production was observed (Figure 9B). Treating biofilms with caspofungin alone resulted in minimal damage, however, the addition of both caspofungin and geldanamycin caused numerous burst and broken hyphae throughout the biofilm (Figure 9B). Finally, voriconazole treatment resulted in a flat ribbon-like morphology, and the addition of geldanamycin induced further cell damage (Figure 9B). Taken together, these results indicate that inhibition of Hsp90 induces changes in morphology of A. fumigatus biofilms, in addition to enhancing the efficacy of azoles and echinocandins against these otherwise recalcitrant cellular structures.
Discussion
Our results establish a novel role for Hsp90 in dispersion and drug resistance of fungal biofilms, with profound therapeutic potential. Resistance of C. albicans biofilms to many antifungal drugs including the azoles, often necessitates surgical removal of the infected catheter or substrate demanding new therapeutic strategies. Here, we demonstrate that compromising the function of C. albicans Hsp90 blocks biofilm dispersal, potentially reducing their ability to serve as reservoirs for persistent infection (Figure 4). Further, we show that compromising Hsp90 function genetically or pharmacologically in C. albicans renders biofilms exquisitely susceptible to azoles, such that fluconazole is transformed from inefficacious to highly effective in destroying biofilms both in vitro (Figure 5 and Table 1) and in a mammalian model of infection (Figure 8). Finally, in A. fumigatus we found that compromising Hsp90 function dramatically improves the efficacy of antifungals (Figure 9). Thus, inhibition of Hsp90 enhances the efficacy of antifungals against biofilms formed by the two leading fungal pathogens of humans separated by ∼1 billion years of evolution, suggesting that this combinatorial therapeutic strategy could have a broad spectrum of activity against diverse fungal pathogens.
Hsp90 exerts pleiotropic effects on cellular circuitry in eukaryotes by stabilizing diverse regulators of cellular signalling [32], [33], [50]. Hsp90 regulates the temperature-dependent morphogenetic transition from yeast to filamentous growth in C. albicans, such that compromise of Hsp90 function by elevated temperature relieves Hsp90-mediated repression of Ras1-PKA signalling and induces filamentous growth [41]. While compromise of Hsp90 function could have impaired biofilm development by enhancing filamentous growth, we found negligible impact on biofilm development in vivo (Figure 2); in vitro, compromise of Hsp90 function did reduce biofilm maturation under static conditions with more severe effects under shaking conditions (Figures 1 and S1). Biofilms formed in the presence of Hsp90 inhibitor had a greater proportion of their total thickness occupied by filaments compared to yeast (Figure 3), suggesting that Hsp90's role in repressing the yeast to filament transition in planktonic cells [41] is conserved in the biofilm state. Consequently, we investigated the impact of compromising Hsp90 function on dispersion, a stage of the biofilm life cycle intimately coupled to morphogenetic transitions, with the majority of dispersed cells being in the yeast form [45], [46]. We found that compromising Hsp90 function dramatically reduces the dispersed cell population (Figure 4), consistent with previous findings with hyperfilamentous C. albicans mutants [45], [46]. Strikingly, the majority of cells that disperse from biofilms with reduced levels of Hsp90 are inviable (Figure 4), which likely reflects an enhanced dependence of this cell population on Hsp90. Given that the dispersed cell population is thought to be responsible for device-associated candidemia and the establishment of disseminated infection, inhibition of C. albicans Hsp90 function in individuals suffering from biofilm infections may assist in the prevention of the invasive forms of disease. In the broader sense, it is striking that depletion of Hsp90 blocks the production of yeast in C. albicans in planktonic conditions [41] as well as throughout the biofilm lifecycle, creating a constitutively filamentous program characteristic of the strictly filamentous lifestyle of the vast majority of fungi.
Hsp90 potentiates the emergence and maintenance of C. albicans drug resistance through multiple client proteins. A key mediator of Hsp90-dependent drug resistance is the protein phosphatase calcineurin [37], [38], [49]. In planktonic cells, Hsp90 stabilizes the catalytic subunit of calcineurin, Cna1, thereby enabling calcineurin-dependent cellular signalling required for survival of drug-induced cellular stress [38]. Hsp90 also regulates drug resistance by stabilizing the MAPK Mkc1, thereby enabling additional stress responses important for resistance [39]. In planktonic conditions, inhibition of calcineurin phenocopies inhibition of Hsp90 reducing drug resistance of diverse mutants, though deletion of MKC1 has a less severe effect on resistance under specific conditions [37], [38], [39]. In biofilms, homozygous deletion of either CNA1 or MKC1 causes an intermediate increase in sensitivity to azoles compared to reduction of HSP90 levels (Figure 5). Genetic depletion of Hsp90 reduces the fluconazole MIC50 from >512 µg/mL to 8 µg/mL, whereas deletion of CNA1 reduces resistance to 32 µg/mL and deletion of MKC1 reduces resistance only to 128 µg/mL (Figure 5). Thus, both calcineurin and Mkc1 have reduced impact on azole resistance of biofilms compared to Hsp90, suggesting differences in the Hsp90-dependent cellular circuitry between the biofilm and planktonic cellular states.
Hsp90 regulates circuitry required for fungal drug resistance largely by stabilizing key regulators of cellular signalling. In planktonic conditions, reduction of Hsp90 levels leads to depletion of both Cna1 and Mkc1 [38], [39]. In stark contrast, Cna1 and Mkc1 remain stable in biofilms, despite reduction of Hsp90 levels (Figure 6). In both planktonic and biofilm conditions, Hsp90 levels were reduced by doxycycline-mediated transcriptional repression in the tetO-HSP90-hsp90Δ strain and levels of Hsp90 were reduced sufficiently to abrogate drug resistance in both conditions. The reduced dependence of Cna1 and Mkc1 on Hsp90 in biofilms suggests that these proteins have altered stability in this cellular state. These Hsp90 client proteins may assume an alternate conformation in biofilms that is inherently more stable, or they may interact with other proteins or chaperones that confer increased stability and reduced dependence upon Hsp90. Consistent with the possibility of altered chaperone balance in biofilm cells, the Hsp70 family member SSB1 is overexpressed six-fold in biofilms compared to their planktonic counterparts [51]. While it is possible that Hsp90 may still regulate Cna1 and Mkc1 function through a mechanism distinct from protein stability, we note that Mkc1 is still activated upon Hsp90 depletion in biofilms (Figure 6). Given Hsp90's high degree of connectivity in diverse signalling cascades, it could also affect biofilm drug resistance in a multitude of other ways, such as by regulating remodeling of the cell wall and cell membrane [27], [28], signalling cascades important for matrix production [29], [52], or the function of contact-dependent signalling molecules that initiate responses to surfaces [30]. Future studies will determine on a more global scale the impact of cellular state on Hsp90 client protein stability, and the complex circuitry by which Hsp90 regulates biofilm drug resistance.
Our results suggest that Hsp90 is a novel regulator of matrix glucan levels. For C. albicans the reduction in matrix glucan levels upon Hsp90 depletion provides a mechanism by which Hsp90 might govern biofilm azole resistance. C. albicans biofilms possess elevated cell wall β-1,3 glucan content compared to their planktonic counterparts [28], and matrix glucan sequesters fluconazole, preventing it from reaching its intracellular target [28], [29]. The ∼40% reduction in matrix glucan we observed upon Hsp90 depletion (Figure 7) likely contributes to reduced azole resistance, given that a reduction of matrix glucan levels of ∼60% in an FKS1/fks1Δ mutant abrogates biofilm drug resistance [29]. Hsp90 could regulate glucan levels by directly or indirectly affecting β-1,3 glucan synthase, Fks1, a protein important for the production of matrix glucan and for antifungal resistance [28], [29]. Alternatively, Hsp90 could regulate matrix production by directly or indirectly affecting Zap1, or its downstream targets Gca1 and Gca2, which play an important role in matrix production, likely through the hydrolytic release of β-glucan fragments from the environment [52]. We note that in A. fumigatus, inhibition of Hsp90 appears to increase matrix production (Figure 9), though glucan levels remain unknown. Future studies will dissect the molecular mechanisms by which Hsp90 regulates biofilm matrix production and if there is divergent circuitry between these fungal pathogens.
This work establishes that targeting Hsp90 may provide a powerful therapeutic strategy for biofilm infections caused by the leading fungal pathogens of humans. Compromising Hsp90 function genetically or pharmacologically reduces azole resistance of C. albicans biofilms both in vitro and in the rat venous catheter model of infection (Figures 5 and 8). Importantly, inhibition of Hsp90 with 17-AAG, an Hsp90 inhibitor that has advanced in clinical trials for the treatment of cancer [53], [54] and is synergistic with antifungals in planktonic conditions [34], transforms fluconazole from ineffective to highly efficacious in a mammalian model of biofilm infection (Figure 8). There may in fact be a multitude of benefits of inhibiting Hsp90 in the context of C. albicans biofilm infections given a recent report that treatment of in vitro C. albicans biofilms with voriconazole induces resistance to micafungin in an Hsp90-dependent manner [55]. The therapeutic potential of Hsp90 inhibitors against fungal biofilms extends beyond C. albicans to the most lethal mould, A. fumigatus. Pharmacological inhibition of Hsp90 enhances the efficacy of both azoles and echinocandins against A. fumigatus biofilms (Figure 9). The synergy between Hsp90 inhibitors and echinocandins is more pronounced than that with azoles, consistent with findings in the planktonic cellular state [34]. Thus, targeting Hsp90 may provide a much-needed strategy to enhance the efficacy of antifungal drugs against biofilms formed by diverse fungal pathogens.
Our results provide a new facet to the broader therapeutic paradigm of Hsp90 inhibitors in the treatment of infectious disease caused by fungi and other pathogenic eukaryotes. In addition to the profound effects on biofilm drug resistance and dispersal, compromising Hsp90 function enhances the efficacy of azoles and echinocandins against disseminated disease caused by the leading fungal pathogens of humans in invertebrate and mammalian models of infection [34], [38]. Beyond enhancing antifungal activity, Hsp90 also provides an attractive antifungal target on its own given that depletion of fungal Hsp90 results in complete clearance of a kidney fungal burden in a mouse model of disseminated candidiasis [41]. Hsp90 inhibitors also exhibit potent activity against malaria and Trypanosoma infections, thus extending their spectrum of activity to the protozoan parasites Plasmodium falciparum and Trypanosoma evansi [35], [36]. The development of Hsp90 as a therapeutic target for infectious disease may benefit from the plethora of structurally diverse Hsp90 inhibitors that have been developed, many of which are in advanced phase clinical trials for cancer treatment, with substantial promise due to the depletion of a myriad of oncoproteins upon inhibition of Hsp90 [56]. Given the importance of Hsp90 in chaperoning key regulators of cellular signalling in all eukaryotes, the challenge of advancing Hsp90 as a target for infectious disease lies in avoiding host toxicity issues. Indeed, although well tolerated in the mammalian host individually or in combination therapies [56], Hsp90 inhibitors have toxicity in the context of an acute disseminated fungal infection [34]. This toxicity may be due to Hsp90's role in regulating host immune and stress responses during infection. Toxicity was not observed in our studies of biofilm infections in the mammalian model, perhaps owing to both the localized infection and drug delivery, suggesting that this therapeutic strategy could rapidly translate from the laboratory bench to the patients' bedside. In the broader context, the challenge for further development of Hsp90 as a therapeutic target for infectious disease lies in developing pathogen-selective inhibitors or drugs that target pathogen-specific components of the Hsp90 circuitry governing drug resistance and virulence.
Materials and Methods
Ethics statement
All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Wisconsin according to the guidelines of the Animal Welfare Act, The Institute of Laboratory Animal Resources Guide for the Care and Use of Laboratory Animals, and Public Health Service Policy.
Strains and culture conditions
Archives of C. albicans strains were maintained at −80°C in 25% glycerol. Strains were routinely maintained and grown in YPD liquid medium (1% yeast extract, 2% bactopeptone, 2% glucose) at 30°C. Strains used in this study are listed in Table S1. Strain construction is described in the Supplemental Material.
Biofilm growth conditions
Multiple in vitro assays were used to assess C. albicans biofilm growth and antifungal drug susceptibility. In the first model, biofilms were developed in 96-well polystyrene plates, as previously described [28], [43]. Briefly, strains were grown overnight in YPD at 37°C. Subsequently, cultures were resuspended in RPMI medium buffered with HEPES or MOPS, in the presence or absence of doxycycline (631311, BD Biosciences) to a final concentration of 106 cells/mL. An aliquot of 100 µl was added to each well of a 96-well flat-bottom plate, followed by incubation at 37°C. After 24 hours, the wells were gently washed twice with phosphate-buffered saline (PBS) to remove non-adherent cells, and fresh medium was added with or without a gradient of geldanamycin (ant-gl-5, Cedarlane). After 24 hours, non-adherent cells were washed away with PBS and biofilm cell metabolic activity was measured using the XTT reduction assay as previously described [28], [43]. Briefly, 90 µl of XTT (X4251, Sigma) at 1 mg/mL and 10 µl phenazine methosulfate (P9625, Sigma) at 320 µg/mL were added to each well, followed by incubation at 37°C for 2 hours. Absorbance of the supernatant transferred to a fresh plate was measured at 490 nm using an automated plate reader, and experiments were carried out in a minimum of 5 replicates for each strain.
In the second model, biofilms were developed on silicon elastomer (SE) surfaces as has been described previously [57]. C. albicans wild-type cells were grown overnight in YPD medium at 30°C and diluted to an optical density at 600 nm of 0.5 in RPMI medium. The suspension was added to a sterile 12-well plate containing bovine serum (B-9433, Sigma)-treated SE (Cardiovascular Instrument silicon sheets; PR72034-06N) and incubated at 37°C for 90 min at 150 rpm agitation for initial adhesion. The SE were washed with PBS, transferred to fresh plates containing either fresh RPMI medium in the absence of drug, or RPMI with 10 µg/mL geldanamycin or 20 µg/mL doxycycline. Plates were incubated at 37°C for 48 hours at 150 rpm agitation to allow biofilm formation, followed by visualization by microscopy or by monitoring biofilm growth by XTT reduction or dry weight, as previously described [31], [43].
C. albicans biofilm dispersion
For obtaining cells dispersed from biofilms, C. albicans biofilms were cultured in a simple flow biofilm model, as described previously [47], [48]. Briefly, this model involves a controlled flow of fresh medium via Tygon tubing (Cole-Parmer, Vernon Hills, IL) into a 15 mL polypropylene conical tube (BD, Franklin, NJ) holding a SE strip. Medium flow is controlled at 1 mL/minute, by connecting the tubing to a peristaltic pump (Masterflex L/S Easy-Load II, Cole-Parmer). The whole apparatus is placed inside an incubator to facilitate biofilm development at 37°C. SE strips (1×9 cm, Cardiovascular instrument Corp, Wakefield, MA), were sterilized by autoclaving and pre-treated for 24 hours with bovine serum. C. albicans was grown overnight at 30°C, washed, and diluted to an optical density at 600 nm of 0.5 in Yeast Nitrogen base (YNB) medium (BD Biosciences, San Jose, CA) with 50 mM glucose. The SE strips were incubated with the diluted C. albicans suspension at 37°C for 90 min at 100 rpm agitation for the initial adhesion of cells. Next, the strip was inserted into the conical tube and the peristaltic pump was turned on. At various time points during biofilm development, cells released from the biofilm in the flow-through were collected from the bottom of the conical tube. The dispersed cells were enumerated by a hemocytometer to obtain cell counts and there were no differences observed in the degree of clumping or morphological state of the dispersed cells, which were in the yeast form. Viability of the dispersed cells was assessed by plating and by colony counts on YPD agar.
C. albicans in vitro biofilm drug susceptibility
Drug susceptibility assays were performed on biofilms formed in wells of 96-well plates. Fresh medium (RPMI/HEPES) and drugs were added to wells containing biofilms grown for 24 hours. Dilutions of fluconazole (Sequoia Research Products) were from 1000 µg/ml down to 0 with the following concentration steps in µg/ml: 1000, 500, 250, 125, 62.5, 31.25, 15.625, 7.8125, 3.90625, 1.953125, 0.9765625. FK506 (AG Scientific) gradients were from 75 µg/mL down to 0 with the following concentration steps in µg/ml: 75, 37.5, 18.75, 9.375, 4.6875, 2.3475, 1.171875. Geldanamycin gradients were from 100 µg/mL to 0 with the following concentration steps in µg/ml: 100, 50, 25, 12.5, 6.25, 3.125, 1.5625. Drug combinations were examined alone or in combination in a checkerboard format. After incubation at 37°C for 24 hours, biofilms were washed twice with PBS and metabolic activity was measured using the XTT assay, as described above. The drug concentration associated with 50% reduction in optical density compared to the drug-free control wells (MIC50) was determined. The fractional inhibitory concentration (FIC) was calculated as follows: [(MIC 50 of drug A in combination)/(MIC 50 of drug A alone)] + [(MIC50 of drug B in combination)/(MIC50 of drug B alone)]. Values of <0.5 indicates synergy, those of >0.5 but <2 indicate no interaction, and those of >2 show antagonism [28].
C. albicans in vivo biofilm model
In order to evaluate biofilm formation in vivo, a rat central venous catheter infection model was employed [44]. Specific-pathogen-free Sprague-Dawley rats weighing ∼400 g were used (Harlan Sprague-Dawley, Indianapolis, IN). A heparinized (100 U/mL) polyethylene catheter was surgically inserted into the jugular vein and advanced 2 cm to a site above the right atrium. After the catheter was secured to the vein, the proximal end was tunneled subcutaneously to the midscapular space and externalized through the skin. The catheters were implanted 24 hours prior to inoculation with C. albicans to allow a conditioning period for deposition of host protein on the catheter surface. Infection was performed by intraluminal instillation of 500 µl of C. albicans (106 cells/mL). After 6 hours, the catheters were flushed and maintained with heparinized 0.85% NaCl for 24 hours to allow for biofilm formation. While one end of the catheter is open to the venous blood, most of the fluid contents remain within the catheter unless pushed into the bloodstream with additional fluid from the external end. For drug treatment studies, fluconazole (250 µg/mL), 17-AAG (A-6880, LC Laboratories, 100 µg/mL), or saline was instilled and allowed to dwell in the catheter for an additional 24 hours [28]. For doxycycline studies, doxycycline (20 µg/mL) was delivered during both the biofilm formation and the drug treatment phases. At the end of the observation period, the animals were sacrificed and the catheters were removed. In order to quantify fungal biofilm formation in the catheter, the contents were drained to remove blood and non-adherent organisms. The distal 2 cm of catheter was cut from the entire catheter length and the segment was placed in 1 mL of 0.85% NaCl. Following sonication for 10 minutes (FS 14 water bath sonicator and 40-kHz transducer [Fisher Scientific]) and vigorous vortexing for 30 seconds, serial dilutions of the catheter fluid were plated on Sabouraud Dextrose Agar (SDA) for viable fungal colony counts. Results are expressed as the mean colony forming unit (CFU) per milliliter.
A. fumigatus in vitro biofilm drug susceptibility
Aspergillus fumigatus Af293 was maintained on SDA slopes at 4°C. For conidial preparation Af293 was propagated on SAB agar for 72 hours and conidia harvested in PBS containing 0.025% (v/v) Tween 20 and quantified as previously described [58]. Commercially available voriconazole (Pfizer Pharmaceuticals, NY, USA), micafungin (Astellas Pharma Inc, Ibaraki, Japan) and caspofungin (Merck Sharp Dohme Ltd, NJ, USA) were used throughout this study. Each antifungal drug was prepared at stock concentrations of 10 mg/mL in sterile water and used within 24 hours of reconstitution.
Af293 conidial inoculum (1×105 conidia/mL) was dispensed into flat bottomed 96-well microtitre plates and incubated for 8 or 24 hours at 37°C as previously described [58]. Biofilms were gently washed twice with PBS and each antifungal agent and geldanamycin were diluted to working concentrations in RPMI, which were tested either alone or in combination in a checkerboard format. Antifungal agent dilutions were from 512 µg/ml down to 0 with the following concentration steps in µg/ml: 512, 256, 128, 64, 32, 16, 8, 4, 2, 1, 0.5. Geldanamycin dilutions were from 100 µg/ml down to 0 with the following concentration steps in µg/ml: 100, 25, 12.5, 6.25, 3.125, 1.5625. The biofilms were then treated and processed as described for C. albicans.
Confocal microscopy
Biofilms were stained with 25 µg/mL concanavalin A–Alexa Fluor 594 conjugate (C-11253; Molecular Probes, Eugene, OR) for 1 hour in the dark at 37°C. Confocal scanning laser microscopy (CSLM) was performed with a ZeissLSM 510 upright confocal microscope using a Zeiss Achroplan 40X, 0.8-W objective. Stained biofilms were observed using a HeNe1 laser with an excitation wavelength of 543 nm. The Zeiss LSM Image Browser v4.2 software was used to assemble images into side and depth views. Artificially coloured depth view images represent cell depth using a colour gradient, where cells closest to the SE are represented in blue and the cells farthest away are represented in red.
Scanning electron microscopy
Biofilms formed in vitro were placed overnight in a fixative (4% formaldehyde v/v, 1% glutaraldehyde v/v in PBS), rinsed in 0.1 M phosphate buffer and air dried in desiccators. Notably, harsh dehydration steps were not performed to minimize the damage to the original biofilm structure. The samples were coated with gold/palladium (40%/60%) and observed under a scanning electron microscope (Leo 435 VP) in high vacuum mode at 15 kV. The images were assembled using Photoshop software (Adobe, Mountain View, CA.).
Catheter segments were processed for scanning electron microscopy as previously described [44]. Following overnight fixation (4% formaldehyde, 1% glutaraldehyde in PBS), catheter segments were washed with PBS and treated with osmium tetroxide (1% in PBS) for 30 minutes. Drying was accomplished using a series of alcohol washes followed by critical point drying. Catheter segments were mounted and gold coated. Images were obtained with a scanning electron microscope (JEOL JSM-6100) in the high-vacuum mode at 10 kV. The images were assembled using Adobe Photoshop 7.0.1.
Immune blot analysis
For the protein stability assay, planktonic cultures were grown in RPMI buffered with MOPS and treated as described previously [39]. For biofilm cultures, C. albicans was grown overnight in YPD medium at 30°C and diluted to an optical density at 600 nm of 0.5 in RPMI medium. The suspension was added to a bovine serum (16190; Gibco)-treated sterile 6-well plate and incubated at 37°C for 90 minutes for initial adhesion. The plates were washed with PBS, and fresh RPMI medium was added with or without 20 µg/mL doxycycline. Plates were incubated at 37°C for 48 hours.
Cells were harvested by centrifugation and were washed with sterile water. Cell pellets were resuspended in lysis buffer containing 50 mM HEPES pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 1 mM PMSF, and protease inhibitor cocktail (complete, EDTA-free tablet, Roche Diagnostics). Cells suspended in lysis buffer were mechanically disrupted by adding acid-washed glass beads and bead beating for 3 minutes. Protein concentrations were determined by Bradford analysis. Protein samples were mixed with one-sixth volume of 6X sample buffer containing 0.35 M Tris-HCl, 10% (w/v) SDS, 36% glycerol, 5% β-mercaptoethanol, and 0.012% bromophenol blue for SDS-PAGE. Samples were boiled for 5 minutes and then separated by SDS-PAGE using an 8% acrylamide gel. Proteins were electrotransferred to PVDF membranes (Bio-Rad Laboratories, Inc.) and blocked with 5% skimmed milk in phosphate buffered saline (PBS) with 0.1% tween. Blots were hybridized with antibodies against CaHsp90 (1∶10000), generously provided by Brian Larsen [59], FLAG (1∶10000, Sigma Aldrich Co.), His6 (1∶10, P5A11, generously provided by Elizabeth Wayner), phospho-p44/42 MAPK (Thr202/Tyr204) (1∶2000, Cell Signaling), or against alpha-tubulin (1∶1000; AbD Serotec, MCA78G).
Biofilm matrix collection and matrix β-1,3 glucan measurements
Matrix β-1,3 glucan content was measured using a limulus lysate based assay, as previously described [28], [60]. Matrix was collected from C. albicans biofilms growing in the wells of 6-well polystyrene plates with or without 20 µg/mL doxycycline for 48 hours. The method for culturing biofilms was as described above for the immune blot analysis with the exception that all reagents were glucan-free. Biofilms were dislodged using a sterile spatula, washed with PBS, sonicated for 10 minutes, and centrifuged 3 times at 4500 x g for 20 minutes to separate cells from soluble matrix material [28], [61]. Samples were stored at -20°C and glucan concentrations were determined using the Glucatell (1,3)-Beta-D-Glucan Detection Reagent Kit (Associates of Cape Cod, MA) as per the manufacturer's directions.
Accession numbers for genes and proteins mentioned in text (NCBI Entrez Gene ID number)
C. albicans: PKC1 (3635298); HSP90 (3637507); CNA1 (3639406); CNB1 (3636463); MKC1 (3639710); ERG11 (3641571); FKS1 (3637073); SSB1 (3642206); GCA1 (3635124); ZAP1 (3641162).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. CowenLESteinbachWJ 2008 Stress, drugs, and evolution: the role of cellular signaling in fungal drug resistance. Eukaryot Cell 7 747 764
2. PfallerMADiekemaDJ 2010 Epidemiology of invasive mycoses in North America. Crit Rev Microbiol 36 1 53
3. McNeilMMNashSLHajjehRAPhelanMAConnLA 2001 Trends in mortality due to invasive mycotic diseases in the United States, 1980–1997. Clin Infect Dis 33 641 647
4. ZaoutisTEArgonJChuJBerlinJAWalshTJ 2005 The epidemiology and attributable outcomes of candidemia in adults and children hospitalized in the United States: a propensity analysis. Clin Infect Dis 41 1232 1239
5. PfallerMADiekemaDJ 2007 Epidemiology of invasive candidiasis: a persistent public health problem. Clin Microbiol Rev 20 133 163
6. WilsonLSReyesCMStolpmanMSpeckmanJAllenK 2002 The direct cost and incidence of systemic fungal infections. Value Health 5 26 34
7. LinSJSchranzJTeutschSM 2001 Aspergillosis case-fatality rate: systematic review of the literature. Clin Infect Dis 32 358 366
8. LupettiADanesiRCampaMDel TaccaMKellyS 2002 Molecular basis of resistance to azole antifungals. Trends Mol Med 8 76 81
9. Ostrosky-ZeichnerLCasadevallAGalgianiJNOddsFCRexJH 2010 An insight into the antifungal pipeline: selected new molecules and beyond. Nat Rev Drug Discov 9 719 727
10. AndersonJB 2005 Evolution of antifungal-drug resistance: mechanisms and pathogen fitness. Nat Rev Microbiol 3 547 556
11. CowenLE 2008 The evolution of fungal drug resistance: modulating the trajectory from genotype to phenotype. Nat Rev Microbiol 6 187 198
12. PereaSLopez-RibotJLKirkpatrickWRMcAteeRKSantillanRA 2001 Prevalence of molecular mechanisms of resistance to azole antifungal agents in Candida albicans strains displaying high-level fluconazole resistance isolated from human immunodeficiency virus-infected patients. Antimicrob Agents Chemother 45 2676 2684
13. ShapiroRSRobbinsNCowenLE 2011 Regulatory circuitry governing fungal development, drug resistance, and disease. Microbiol Mol Biol Rev 75 213 267
14. BalashovSVParkSPerlinDS 2006 Assessing resistance to the echinocandin antifungal drug caspofungin in Candida albicans by profiling mutations in FKS1. Antimicrob Agents Chemother 50 2058 2063
15. ParkSKellyRKahnJNRoblesJHsuMJ 2005 Specific substitutions in the echinocandin target Fks1p account for reduced susceptibility of rare laboratory and clinical Candida sp. isolates. Antimicrob Agents Chemother 49 3264 3273
16. FinkelJSMitchellAP 2011 Genetic control of Candida albicans biofilm development. Nat Rev Microbiol 9 109 118
17. RamageGMowatEJonesBWilliamsCLopez-RibotJ 2009 Our current understanding of fungal biofilms. Crit Rev Microbiol 35 340 355
18. BlankenshipJRMitchellAP 2006 How to build a biofilm: a fungal perspective. Curr Opin Microbiol 9 588 594
19. d'EnfertC 2006 Biofilms and their role in the resistance of pathogenic Candida to antifungal agents. Curr Drug Targets 7 465 470
20. NobileCJMitchellAP 2006 Genetics and genomics of Candida albicans biofilm formation. Cell Microbiol 8 1382 1391
21. RamageGMartinezJPLopez-RibotJL 2006 Candida biofilms on implanted biomaterials: a clinically significant problem. FEMS Yeast Res 6 979 986
22. KojicEMDarouicheRO 2004 Candida infections of medical devices. Clin Microbiol Rev 17 255 267
23. ViudesAPemanJCantonEUbedaPLopez-RibotJL 2002 Candidemia at a tertiary-care hospital: epidemiology, treatment, clinical outcome and risk factors for death. Eur J Clin Microbiol Infect Dis 21 767 774
24. SeidlerMJSalvenmoserSMüllerF-MC 2008 Aspergillus fumigatus forms biofilms with reduced antifungal drug susceptibility on bronchial epithelial cells. Antimicrob Agents Chemother 52 4130 4136
25. PerumalPMekalaSChaffinWL 2007 Role for cell density in antifungal drug resistance in Candida albicans biofilms. Antimicrob Agents Chemother 51 2454 2463
26. RamageGBachmannSPattersonTFWickesBLLopez-RibotJL 2002 Investigation of multidrug efflux pumps in relation to fluconazole resistance in Candida albicans biofilms. J Antimicrob Chemother 49 973 980
27. MukherjeePKChandraJKuhnDMGhannoumMA 2003 Mechanism of fluconazole resistance in Candida albicans biofilms: phase-specific role of efflux pumps and membrane sterols. Infect Immun 71 4333 4340
28. NettJLincolnLMarchilloKMasseyRHoloydaK 2007 Putative role of beta-1,3 glucans in Candida albicans biofilm resistance. Antimicrob Agents Chemother 51 510 520
29. NettJESanchezHCainMTAndesDR 2010 Genetic basis of Candida biofilm resistance due to drug-sequestering matrix glucan. J Infect Dis 202 171 175
30. KumamotoCA 2005 A contact-activated kinase signals Candida albicans invasive growth and biofilm development. Proc Natl Acad Sci U S A 102 5576 5581
31. UppuluriPNettJHeitmanJAndesD 2008 Synergistic effect of calcineurin inhibitors and fluconazole against Candida albicans biofilms. Antimicrob Agents Chemother 52 1127 1132
32. TaipaleMJaroszDFLindquistS 2010 HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol 11 515 528
33. PearlLHProdromouC 2006 Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu Rev Biochem 75 271 294
34. CowenLESinghSDKöhlerJRCollinsCZaasAK 2009 Harnessing Hsp90 function as a powerful, broadly effective therapeutic strategy for fungal infectious disease. Proc Natl Acad Sci USA 106 2818 2823
35. PallaviRRoyNNageshanRKTalukdarPPavithraSR 2010 Heat shock protein 90 as a drug target against protozoan infections: biochemical characterization of HSP90 from Plasmodium falciparum and Trypanosoma evansi and evaluation of its inhibitor as a candidate drug. J Biol Chem 285 37964 37975
36. ShahinasDLiangMDattiAPillaiDR 2010 A repurposing strategy identifies novel synergistic inhibitors of Plasmodium falciparum heat shock protein 90. J Med Chem 53 3552 3557
37. CowenLELindquistS 2005 Hsp90 potentiates the rapid evolution of new traits: drug resistance in diverse fungi. Science 309 2185 2189
38. SinghSDRobbinsNZaasAKSchellWAPerfectJR 2009 Hsp90 governs echinocandin resistance in the pathogenic yeast Candida albicans via calcineurin. PLoS Pathog 5 e1000532
39. LaFayetteSLCollinsCZaasAKSchellWSBetancourt-QuirozM 2010 PKC signaling regulates drug resistance of the fungal pathogen Candida albicans via circuitry comprised of Mkc1, calcineurin, and Hsp90. PLoS Pathog 6 e1001069
40. NobleSMFrenchSKohnLAChenVJohnsonAD 2010 Systematic screens of a Candida albicans homozygous deletion library decouple morphogenetic switching and pathogenicity. Nat Genet 42 590 598
41. ShapiroRSUppuluriPZaasAKCollinsCSennH 2009 Hsp90 orchestrates temperature-dependent Candida albicans morphogenesis via Ras1-PKA signaling. Curr Biol 19 621 629
42. RamageGVandeWalleKLopez-RibotJLWickesBL 2002 The filamentation pathway controlled by the Efg1 regulator protein is required for normal biofilm formation and development in Candida albicans. FEMS Microbiol Lett 214 95 100
43. RamageGVande WalleKWickesBLLopez-RibotJL 2001 Standardized method for in vitro antifungal susceptibility testing of Candida albicans biofilms. Antimicrob Agents Chemother 45 2475 2479
44. AndesDNettJOschelPAlbrechtRMarchilloK 2004 Development and characterization of an in vivo central venous catheter Candida albicans biofilm model. Infect Immun 72 6023 6031
45. UppuluriPPierceCGThomasDPBubeckSSSavilleSP 2010 The transcriptional regulator Nrg1p controls Candida albicans biofilm formation and dispersion. Eukaryot Cell 9 1531 1537
46. UppuluriPChaturvediAKSrinivasanABanerjeeMRamasubramaniamAK 2010 Dispersion as an important step in the Candida albicans biofilm developmental cycle. PLoS Pathog 6 e1000828
47. UppuluriPChaturvediAKLopez-RibotJL 2009 Design of a simple model of Candida albicans biofilms formed under conditions of flow: development, architecture, and drug resistance. Mycopathologia 168 101 109
48. UppuluriPLopez-RibotJL 2011 An easy and economical in vitro method for the formation of Candida albicans biofilms under continuous conditions of flow. Virulence 1 483 487
49. CowenLECarpenterAEMatangkasombutOFinkGRLindquistS 2006 Genetic architecture of Hsp90-dependent drug resistance. Eukaryot Cell 5 2184 2188
50. YoungJCMoarefiIHartlFU 2001 Hsp90: a specialized but essential protein-folding tool. J Cell Biol 154 267 273
51. Garcia-SanchezSAubertSIraquiIJanbonGGhigoJM 2004 Candida albicans biofilms: a developmental state associated with specific and stable gene expression patterns. Eukaryot Cell 3 536 545
52. NobileCJNettJEHerndayADHomannORDeneaultJS 2009 Biofilm matrix regulation by Candida albicans Zap1. PLoS Biol 7 e1000133
53. UsmaniSZBonaRLiZ 2009 17 AAG for HSP90 inhibition in cancer–from bench to bedside. Curr Mol Med 9 654 664
54. KimYSAlarconSVLeeSLeeMJGiacconeG 2009 Update on Hsp90 inhibitors in clinical trial. Curr Top Med Chem 9 1479 1492
55. KanekoYOhnoHFukazawaHMurakamiYImamuraY 2010 Anti-Candida-biofilm activity of micafungin is attenuated by voriconazole but restored by pharmacological inhibition of Hsp90-related stress responses. Med Mycol 48 606 612
56. TrepelJMollapourMGiacconeGNeckersL 2010 Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer 10 537 549
57. RichardMLNobileCJBrunoVMMitchellAP 2005 Candida albicans biofilm-defective mutants. Eukaryot Cell 4 1493 1502
58. MowatEButcherJLangSWilliamsCRamageG 2007 Development of a simple model for studying the effects of antifungal agents on multicellular communities of Aspergillus fumigatus. J Med Microbiol 56 1205 1212
59. BurtETDalyRHogansonDTsirulnikovYEssmannM 2003 Isolation and partial characterization of Hsp90 from Candida albicans. Ann Clin Lab Sci 33 86 93
60. OdabasiZMattiuzziGEsteyEKantarjianHSaekiF 2004 Beta-D-glucan as a diagnostic adjunct for invasive fungal infections: validation, cutoff development, and performance in patients with acute myelogenous leukemia and myelodysplastic syndrome. Clin Infect Dis 39 199 205
61. McCourtieJDouglasLJ 1985 Extracellular polymer of Candida albicans: isolation, analysis and role in adhesion. J Gen Microbiol 131 495 503
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 9
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- HTLV-1 Propels Thymic Human T Cell Development in “Human Immune System” Rag2 gamma c Mice
- Hostile Takeover by : Reorganization of Parasite and Host Cell Membranes during Liver Stage Egress
- Exploiting and Subverting Tor Signaling in the Pathogenesis of Fungi, Parasites, and Viruses
- A Viral Ubiquitin Ligase Has Substrate Preferential SUMO Targeted Ubiquitin Ligase Activity that Counteracts Intrinsic Antiviral Defence