
Kaposi's Sarcoma Associated Herpesvirus Tegument Protein ORF75 Is Essential for Viral Lytic Replication and Plays a Critical Role in the Antagonization of ND10-Instituted Intrinsic Immunity
Nuclear domain 10 (ND10) components are restriction factors that inhibit herpesviral replication. Effector proteins of different herpesviruses can antagonize this restriction by a variety of strategies, including degradation or relocalization of ND10 proteins. We investigated the interplay of Kaposi's Sarcoma-Associated Herpesvirus (KSHV) infection and cellular defense by nuclear domain 10 (ND10) components. Knock-down experiments in primary human cells show that KSHV-infection is restricted by the ND10 components PML and Sp100, but not by ATRX. After KSHV infection, ATRX is efficiently depleted and Daxx is dispersed from ND10, indicating that these two ND10 components can be antagonized by KSHV. We then identified the ORF75 tegument protein of KSHV as the viral factor that induces the disappearance of ATRX and relocalization of Daxx. ORF75 belongs to a viral protein family (viral FGARATs) that has homologous proteins in all gamma-herpesviruses. Isolated expression of ORF75 in primary cells induces a relocalization of PML and dispersal of Sp100, indicating that this viral effector protein is able to influence multiple ND10 components. Moreover, by constructing a KSHV mutant harboring a stop codon at the beginning of ORF75, we could demonstrate that ORF75 is absolutely essential for viral replication and the initiation of viral immediate-early gene expression. Using recombinant viruses either carrying Flag - or YFP-tagged variants of ORF75, we could further corroborate the role of ORF75 in the antagonization of ND10-mediated intrinsic immunity, and show that it is independent of the PML antagonist vIRF3. Members of the viral FGARAT family target different ND10 components, suggesting that the ND10 targets of viral FGARAT proteins have diversified during evolution. We assume that overcoming ND10 intrinsic defense constitutes a critical event in the replication of all herpesviruses; on the other hand, restriction of herpesviral replication by ND10 components may also promote latency as the default outcome of infection.
Published in the journal:
. PLoS Pathog 10(1): e32767. doi:10.1371/journal.ppat.1003863
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1003863
Summary
Nuclear domain 10 (ND10) components are restriction factors that inhibit herpesviral replication. Effector proteins of different herpesviruses can antagonize this restriction by a variety of strategies, including degradation or relocalization of ND10 proteins. We investigated the interplay of Kaposi's Sarcoma-Associated Herpesvirus (KSHV) infection and cellular defense by nuclear domain 10 (ND10) components. Knock-down experiments in primary human cells show that KSHV-infection is restricted by the ND10 components PML and Sp100, but not by ATRX. After KSHV infection, ATRX is efficiently depleted and Daxx is dispersed from ND10, indicating that these two ND10 components can be antagonized by KSHV. We then identified the ORF75 tegument protein of KSHV as the viral factor that induces the disappearance of ATRX and relocalization of Daxx. ORF75 belongs to a viral protein family (viral FGARATs) that has homologous proteins in all gamma-herpesviruses. Isolated expression of ORF75 in primary cells induces a relocalization of PML and dispersal of Sp100, indicating that this viral effector protein is able to influence multiple ND10 components. Moreover, by constructing a KSHV mutant harboring a stop codon at the beginning of ORF75, we could demonstrate that ORF75 is absolutely essential for viral replication and the initiation of viral immediate-early gene expression. Using recombinant viruses either carrying Flag - or YFP-tagged variants of ORF75, we could further corroborate the role of ORF75 in the antagonization of ND10-mediated intrinsic immunity, and show that it is independent of the PML antagonist vIRF3. Members of the viral FGARAT family target different ND10 components, suggesting that the ND10 targets of viral FGARAT proteins have diversified during evolution. We assume that overcoming ND10 intrinsic defense constitutes a critical event in the replication of all herpesviruses; on the other hand, restriction of herpesviral replication by ND10 components may also promote latency as the default outcome of infection.
Introduction
Human Herpesvirus 8 (HHV8), or Kaposi's Sarcoma-Associated Herpesvirus (KSHV), belongs to the subfamily Gammaherpesvirinae and is grouped together with the closely related prototypic Saimiriine herpesvirus 2, or Herpesvirus saimiri (HVS), into the genus Rhadinovirus. KSHV was first discovered in 1994 from patients with Kaposi's Sarcoma (KS) lesions [1]. KSHV is the etiologic agent of three human diseases; a) KS, a skin tumor of endothelial origin, and two B cell malignancies, b) Primary Effusion Lymphoma and c) Multicentric Castleman's Disease. KS exists in several forms, classical KS, which is endemic in the Mediterranean and Middle East [2], African endemic KS, iatrogenic KS, which is associated with renal transplantation and AIDS-associated KS, which is among the leading causes of death in AIDS patients [3].
The promyelocytic leukemia protein (PML) is the main component of a subnuclear structure called nuclear domain 10 (ND10). ND10 components like PML, Sp100, Daxx and ATRX were identified as cellular restriction factors that are able to inhibit the replication of several DNA viruses, including herpesviruses. The restriction is assumed to be mediated by the recruitment of cellular proteins like heterochromatic protein 1 (HP1), DNA methyltransferases or histone deacetylases that induce a repressive chromatin status on the herpesviral genome [4]. This antiviral function of PML, however, is counteracted by most human herpesviruses through viral effector proteins using a variety of strategies, including degradation or relocalization of PML. So far, herpesviruses were mainly found to target the major ND10 component PML, as exemplified by the herpes simplex virus regulatory protein ICP0, which has been shown to induce the proteasomal degradation of PML either via acting as a SUMO-targeted ubiquitin ligase or via SUMO-independent ubiquitination [5]–[7]. Similarly, the IE1 protein of human cytomegalovirus targets PML via inducing a de-SUMOylation of this major ND10 component [8], [9]. Since SUMOylation of PML is essential for the integrity of ND10, this leads to a dispersal of the subnuclear structure. PML expression is induced by the interferon pathway [10], [11] and the protein is detected in high amounts in KSHV infected PEL cells [12]. KSHV vIRF3 (LANA2, K10.5) has been shown to colocalize with PML and its overexpression is associated with a redistribution of PML [13]. Another KSHV gene, ORF K8 encodes an early lytic protein, which shares homology to the EBV BZLF1 gene. K8 is a SUMO interaction motif (SIM) dependent, SUMO-2/3-specific SUMO E3 ligase [14] that colocalizes with PML [15]. K8 recruits p53 and probably sequesters it to PML bodies but has no function in dispersing PML bodies [12].
We recently demonstrated that infection of fibroblasts with HVS results in restriction of immediate-early gene expression, which can be alleviated by siRNA mediated knock down of PML [16]; PML is thus also a restriction factor for HVS infection. In contrast to all other Herpesviruses, HVS exclusively degrades the cellular ND10 component Sp100 whereas other factors like PML or Daxx remain intact. The ORF3 tegument protein of HVS was identified as being responsible for this effect. ORF3 induced the proteasomal degradation of Sp100, and a mutant HVS lacking the orf3-gene was no longer able to mediate Sp100 degradation [16].
The ORF3 of HVS shares homology with the cellular formylglycineamid-ribotideamidotransferase enzyme (FGARAT, EC 6.3.5.3). All gammaherpesviruses encode one to three FGARAT-homologous proteins in their genome. Interestingly, recent studies showed that ORF3-homologous proteins from murid gammaherpesvirus 68 (MHV-68) and from Epstein-Barr virus (EBV) modulate the ND10 proteins PML and ATRX, respectively, suggesting that the ND10 targets of the viral FGARAT protein family have diversified during evolution [17], [18]. The KSHV orf75 gene comprises one distinct member of the vFGARAT-family. The putative tegument protein ORF75 has been shown to be involved in NF-kB coactivation with KSHV K13/vFLIP [19].
This study addresses the role of ND10 components in the restriction of lytic replication of KSHV infection. We describe the essential protein ORF75 as a new viral effector, adding a new piece to the intricate puzzle of herpesvirus coevolution with cellular intrinsic immunity.
Results
KSHV infection is restricted by PML
We recently demonstrated that PML acts as a restriction factor for infection with the gammaherpesvirus HVS [16]. To investigate whether ND10 components exert a similar effect on KSHV infection, we analyzed KSHV infection in primary human foreskin fibroblast (HFF) cells carrying an siRNA mediated knock-down of ND10 components. Using recombinant rKSHV.219 [20] we detected significantly more GFP-positive cells in the absence of PML (siPML) or Sp100 (siSp100) compared to control cells (siC) (Figure 1A and 1B). Moreover, we detected a moderate increase in GFP expression from the viral genome after knock-down of Daxx (siDaxx) compared to control cells. However, there was no enhancement of KSHV infection as indicated by GFP expression after depletion of ATRX (siATRX) (Figure 1A and 1B). KSHV-restriction was in a similar range as the restriction of HVS, suggesting that analogous mechanisms are active on these evolutionary closely related viruses (Figure 1C). Accordingly, ND10 components may compromise KSHV infection in primary human cells.

KSHV infection leads to a disappearance of the ND10 component ATRX
We next wanted to elucidate the effect of KSHV de novo infection on major ND10 components. Therefore, epithelial iSLK cells, which promote lytic replication of KSHV [21], [22], were infected with rKSHV.219 and subjected to immunofluorescence analysis. In KSHV infected GFP positive iSLK cells, the dot like staining of ATRX characteristic for ND10 association was lost (Figure 2A). In addition, we observed a loss of dotted Daxx staining pattern in infected cells (Figure 2B). Using Western Blot experiments from iSLK cells infected with high multiplicity of infection (MOI), we could demonstrate that KSHV infection leads to a reduction of ATRX protein levels. However, this coincided only with a minimal decrease of Daxx levels in Western Blot experiments (Figure 2C). Both Western blot and immunofluorescence analysis revealed no effect of KSHV infection on the ND10 components PML and Sp100 in KSHV-infected cells, which were identified by GFP expression. PML and Sp100 were still detectable in numerous foci being characteristic for their assembly in ND10 structures (Figure 2). Thus, KSHV infection leads to the disappearance of ATRX and to the dispersion of Daxx from ND10; total Daxx protein in immunoblots is unchanged, however. In order to further corroborate that the observed effects are due to an incoming tegument protein, cells were infected in the presence of cycloheximide to inhibit de novo protein synthesis, or infected with UV-inactivated virus (Figure 3). ATRX also disappeared under both conditions, underlining that de novo protein synthesis is not necessary and the effect is caused by a viral tegument protein. In an additional set of experiments, treatment with the proteasome inhibitor MG-132 did also not reverse the effect of KSHV infection on ATRX, while the treatment stabilized the phosphorylated form of Cyclin D1 (CCND1-pT286) as expected (Figure 3A, lower panel).


KSHV lytic induction leads to a dispersal of the ND10 components ATRX and Sp100
Next we wanted to know what happens after lytic induction of KSHV. Therefore we used epithelial iSLK.219 cells [21], [22], a cell line that harbors multiple copies of KSHV strain rKSHV.219 per cell. The KSHV strain rKSHV.219 expresses GFP under control of a constitutive EF1a promoter situated in the BACmid backbone, and red fluorescent protein (RFP) under control of the PAN promoter, which is switched on by the KSHV initiator of lytic viral replication RTA encoded by ORF50. Thus, infection per se results in GFP expression, and induction of early lytic gene expression can be easily monitored by RFP expression. Similar to our observation after de novo infection, KSHV lytic induction leads to a decrease of ATRX in cells expressing RFP (Figure 4, panel i to p); it is also able to overcome an apparent initial increase in ATRX expression after sodium-butyrate treatment of cells, which can be seen in cells with GFP-expression alone. Interestingly, we also observed the disappearance of Sp100 from ND10 in lytically infected cells Fig. 4, panels f to k). This indicates that a lytic gene product of KSHV may antagonize Sp100.

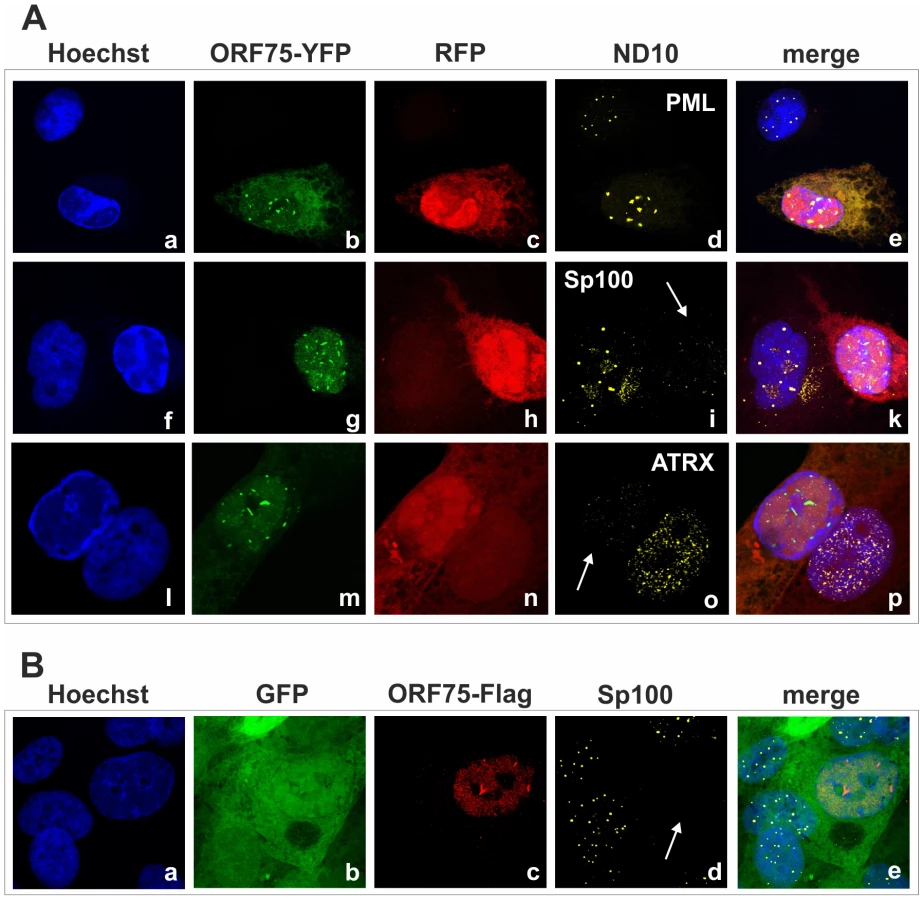
KSHV orf75 induces relocalization and dispersion of multiple ND10 components
We had recently found that the lytic orf3 gene-product of the related primate rhadinovirus HVS induces the proteasomal degradation of Sp100 [16]. Thus, we speculated that the homologous ORF75 protein of KSHV may be involved in the depletion and relocalization of ND10 components. To investigate this, primary human fibroblasts were transfected with expression constructs for ORF75, and the integrity of ND10 components was visualized by multicolor immunofluorescence. The ORF75 protein shows a colocalization with the main ND10 component PML (Figure 5A and B). Moreover, compared to untransfected cells, ORF75 induced the relocalization of PML. However, we never observed a complete loss of spotted PML staining but a decrease in the number of ND10 dots per nucleus, and the remaining dots showed an increase in size (Figure 5). In addition, ORF75 induced the removal of ATRX, which is in accordance with ATRX disappearing after de novo KSHV-infection (Figure 5B, panels f to k and Figure 4). Furthermore, the spotted ND10-associated appearance of Daxx was lost in cells expressing ORF75 compared to untransfected cells (Figure 5A panels f,g). Quantification of ND10-dots per nucleus revealed a significant reduction in the number of PML-, Sp100-, Daxx - and ATRX-dots per cell (Figure 6A). It is worth to note, that the variation in dot number per nucleus in staining of different ND10 components is most likely related to the signal/noise ratio of used antibodies. The loss of ATRX and Daxx dots per nucleus seemed to be dependent on ORF75 levels; cells expressing high amounts of ORF75 showing both nuclear and cytoplasmic staining of ORF75 had a complete loss off ATRX and Daxx dots, whereas cells with ORF75 staining restricted to nuclei showed only reduced numbers of dots (Figure 5). Along this line, by transfection of myc-tagged ORF75 into 293T cells, we could demonstrate that ORF75 leads to a dose dependent loss of ATRX expression as detected by Western Blotting (Figure 6B). In contrast to ATRX, Daxx expression levels remained unaffected (Figures 6B+6C). Moreover, we could demonstrate that transfection of increasing amounts of ORF75-myc had no influence on Sp100 protein levels, indicating that ORF75 does not mediate a degradation of Sp100 (Figure 6C). In cells expressing high amounts of ORF75, we detected a relocalization and frequently also a complete loss of spotted Sp100 staining; PML was relocalized to more intense spots but not dispersed. This suggests that ORF75 is involved in the dispersion of both Sp100 and Daxx during lytic replication but not in Sp100 degradation (Figure 5A, panel e–h; 5B, panels b–d; g–i, Figure 6).


KSHV ORF75 is a tegument protein and part of the virion
The depletion of ATRX in de novo infected human fibroblasts, which are not fully permissive for KSHV, hinted at the involvement of a virion protein. We then wanted to address the question whether ORF75 is truly a tegument protein and therefore part of the virion, as it was assumed in previous studies [23], [24]. To this end, recombinant viruses were generated for further analysis of the role of KSHV ORF75 (Figure 7A). The orf75 gene is located close to the right end of the L-DNA. We generated recombinant KSHV mutants by using recombination-mediated, site-directed mutagenesis of cloned KSHV BACmid Bac16 [25]. Mutant viruses were verified using XhoI restriction enzyme digestion, PCR, PFGE, sequencing of recombination junctions, and Illumina sequencing of the Bac16-75-Stop-revertant and -orf75-Flag (data not shown). Pulsed field gel electrophoresis shows expected restriction patterns of wild type and deletion mutant BACs, including the full maintenance of the terminal repeat sequences (data not shown). All of these experiments proved that the recombinant genomes contained the correct deletion, with no other detectable rearrangements, and were therefore used for further analysis. iSLK cells were transfected with KSHV Bac16 or Bac16-orf75-Flag DNA, selected with hygromycin B, and lytic viral replication induced with doxycyclin and sodium-butyrate. Three days post induction, cells were harvested and Flag-tagged ORF75 was stained by immune electron microscopy (IEM) with a Flag-tag specific antibody and a secondary antibody coupled to 12 nm gold-particles. Electron microscopy imaging revealed a tegument specific gold staining of virions in iSLK-Bac16-Flag cells, which was absent in iSLK-Bac16 wt cells, indicating that the ORF75 protein is truly part of the viral particle (Figure 7B). We could not detect virus particle production by IEM in iSLK-Bac16-orf75-Stop cells after lytic viral induction despite multiple attempts (Figure 7B).

The orf75 gene is essential for lytic replication of KSHV
Recombinant viruses were reconstituted by transfection of BAC DNA into iSLK cells followed by selection with hygromycin B. Cells were treated with sodium-butyrate and doxycyclin to induce lytic virus production. We were able to reconstitute recombinant viruses carrying Flag-tagged or YFP-tagged versions of the ORF75 protein, however, despite multiple attempts we failed to reconstitute virus carrying a STOP codon at the beginning of the orf75 gene. Nine individual iSLK-Bac16-orf75-Stop clones were tested for the production of infectious virus by transfer of supernatant from induced iSLK-Bac16-orf75-Stop clones onto 293T cells; GFP-expression resulting from KSHV infection was measured by flow cytometry two days post infection. While the control showed infection rates above 50%, we could not detect infection of 293T cells using supernatant of any of the nine orf75-Stop clones (Figure 8A). Furthermore, KSHV ORF75 appears to be essential for lytic viral induction, since we could not detect lytic viral gene expression in the absence of ORF75 (Figure 8B). iSLK cells harboring different recombinant Bac16 clones were induced with doxycyclin and sodium-butyrate for 2 days and lytic gene expression determined by Western Blotting. Intriguingly, we could not detect expression of KSHV genes K8, K8.1, and v-IRF3 in iSLK-Bac16-orf75-Stop cells. In contrast, a Bac16-orf75-revertant, constructed by repairing the engineered stop-codon of the orf75-Stop mutant, showed lytic gene expression comparable to that of the wildtype, indicating that the defects observed in the orf75-Stop mutant are in fact due to a lack of orf75 expression. Further support for the essential role of ORF75 comes from knockdown experiments of ORF75 using siRNA and rescue of infectious virus from the iSLK-Bac16-orf75-Stop cells by lentiviral transduction with tet-on ORF75 (Figures S3 and S4). In addition, all cell lines showed regular expression levels of latent genes Lana/orf73 and GFP expressed from the viral genome which increased after lytic induction in all cell lines except iSLK-Bac16-orf75-Stop cells. Finally, when tested for viral DNA replication in iSLK cells by quantitative real time PCR, we could detect only a minimal increase in viral copy numbers per cell in iSLK-Bac16-orf75-Stop cells after lytic viral induction, whereas the controls showed a normal increase in copy numbers corresponding to lytic replication (Figure 8C). This might indicate that, in addition to being a tegument protein important for infectious virion production, ORF75 could have an additional role in generating a permissive environment for viral lytic DNA replication. Accordingly, orf75-mRNA can be detected as early as RTA-mRNA after infection of SLK cells (Figure 8D). In addition, ORF75 protein is present in infected cells during the entire viral replication cycle, starting from 6 h post infection up to 72 h post infection (Figure 8E).

KSHV ORF75 colocalizes with PML in cells undergoing lytic viral replication
In order to further clarify the role of KSHV-ORF75 during lytic replication, we used the recombinant viruses expressing ORF75 either with C-terminal Flag - or YFP-tag. After lytic induction of iSLK cells harboring the respective BACs, ORF75-YFP as well as ORF75-Flag colocalize with PML (Figure 9A and 9B). Moreover, only cells undergoing lytic replication and subsequently showing ORF75 expression exhibited a loss of both ATRX and Sp100 expression, revealing the role of ORF75 in ND10 antagonization. KSHV vIRF3/LANA2 overexpression has previously been associated with redistribution of PML, and a moderate decrease in the number of ND10 dots per cell [13]. Therefore, to separate the effects of ORF75 and vIRF3 on ND10 composition, we generated a recombinant virus expressing ORF75-YFP with additional deletion of the vIRF3 gene. After lytic induction of iSLK-Bac16-Δvirf3-orf75-YFP cells, ORF75-YFP colocalizes with PML (Figure 10). In addition, this experiment recapitulated the loss of both ATRX and Sp100 expression (Figure 10), and clearly shows that vIRF3 is not required for the ORF75-mediated alterations of ND10 composition.

KSHV Bac16-orf75-Stop cells can be complemented by knock-down of ATRX
We demonstrated that KSHV ORF75 mediates the depletion of ATRX and is essential for lytic viral replication. To this end, we addressed the question whether a knock-down of ATRX can re-establish lytic viral replication in the absence of ORF75. iSLK-Bac16-orf75-Stop cells were transduced with a lentiviral shRNA vector targeting ATRX (shATRX) and a respective non-silencing control vector (shC). The ATRX-specific shRNA mediates an almost complete knock-down of ATRX protein in iSLK cells (Figure 11A). After lytic induction of knock-down cells with doxycyclin and sodium-butyrate for three days and transfer of supernatant onto freshly seeded 293T cells, we could not detect infection of cells with supernatant of iSLK-Bac16-ORF75-Stop cells treated with shC (Figure 11B). However, supernatants from iSLK-Bac16-orf75-Stop cells treated with shRNA specific for ATRX contained infectious virus capable of infecting 293T cells, indicating that a viral ORF75 deletion mutant can be rescued by knock down of ATRX (Figure 11B). ATRX knockdown also leads to enhanced lytic viral gene expression. In iSLK-Bac16-orf75-Stop cells, shRNA mediated knockdown of ATRX is able to rescue expression of the lytic gene K8.1, emphasizing the role of ORF75 mediated ATRX counteraction for viral replication (Fig. 11C+D).

Discussion
The counteraction of ND10-instituted intrinsic immunity as a critical step during the replication cycle of DNA viruses and particularly of herpesviruses has become more and more evident over the last decade. Several ND10 components, like PML, Sp100, Daxx and ATRX have been shown to be part of a cellular defense mechanism against viral infection and therefore became targets of viral effector proteins. Recently, members of the viral formylglycineamid-ribotideamidotransferase (vFGARAT) family have been added to the list of herpesviral ND10-antagonists. We demonstrated that one of the two vFGARATs of Herpesvirus saimiri - the ORF3 protein - leads to a selective proteasomal degradation of the ND10 component Sp100 [16]. Moreover, another vFGARAT, the BNRF1 protein of Epstein-Barr virus, has recently been shown to interact with the ND10 component Daxx, resulting in a release of the chromatin remodeling factor ATRX from ND10, and thereby facilitating viral replication [18]. One out of three viral FGARAT proteins of the murid gammaherpesvirus 68 (MHV-68), ORF75c, has been shown to fully disperse cellular ND10 complexes by inducing the proteasomal degradation of PML; it further reduced SP100 isoforms and SUMOylation, and reduced Daxx levels [17], [26]. Thus, although vFGARAT homologs BNRF1, ORF75c and ORF3 all target ND10 components, a comparative analysis of vFGARAT proteins revealed that the ORF3 FGARAT homologs, the rhadinoviral ORF75 homologs, and the lymphocryptoviral BNRF1 homologs cluster within different branches of a phylogenetic tree [16]. Apparently, viral FGARAT homologs have evolved in different ways, probably reflecting different viral survival strategies. Whereas MHV-68 is able to antagonize PML, which drastically enhances viral lytic replication, KSHV and HVS are not able to counteract PML-mediated restriction per se (Figure 1C).
It is notable in this context that the outcome of KSHV infection in vitro is by default latency, whereas the situation in vivo in humans is very difficult to assess. Therefore, it is highly remarkable that there is an increase in viral gene expression after depletion of both PML and Sp100, as shown by de novo GFP expression (Figure 1). According to the current model that ND10 components act as restriction factors by creating a repressive environment for viral gene-expression, this increase in GFP expression in PML - and Sp100-knock-down fibroblasts is most likely due to increased viral lytic gene expression in the absence of ND10 factors. KSHV infection leads to an initial selective depletion of ATRX and relocalization of Daxx, whereas PML and Sp100 remain unaffected (Figures 1D and 2A and B). This is in coincidence with our data of KSHV infection of ATRX knock-down cells: since ATRX is efficiently antagonized by the virus, there is no further enhancement of KSHV infection after ATRX depletion (Figure 1). We could further observe a modest increase in GFP-expression after Daxx depletion, indicating that Daxx can only be partially antagonized by KSHV.
Alpha - and beta-herpesviruses, like herpes simplex virus and human cytomegalovirus, have evolved very efficient counter-mechanisms that dissolve ND10, while primate gammaherpesviruses are lacking the full ability to counteract a central part of the ND10 intrinsic immunity. Therefore, there is good evidence to support the hypothesis that ND10 components like PML may contribute to the efficient establishment of latent infection in target cells, leading to the accumulation of secondary mutations over time and ultimately resulting in oncoprotein mediated cell transformation, as it is the case for most gamma-herpesviruses. On the other side, lytic disease and lytic reactivation is the observed outcome for most alpha - and beta-herpesviruses that can overcome ND10 restriction.
KSHV encodes a single vFGARAT homolog, the ORF75 protein. ORF75 has been described as a tegument protein, due to its homology to other vFGARATs and its presence in preparations of purified virions [23], [24]. We could confirm Flag-tagged ORF75 within KSHV virions by immune electron microscopy (Figure 7B). In addition, in a genome-wide screen for NF-kB activation by KSHV proteins, ORF75 has been shown to activate the NF-kB signaling pathway [19]. The role of ORF75 during lytic viral replication had not been assessed so far. Here we could demonstrate that the ORF75 protein of KSHV is involved in the antagonization of ND10-mediated immunity. Isolated expression of ORF75 in primary human cells led to alterations in ND10 composition reminiscent of the situation during lytic viral replication (Figures 4, 5 and 6): i) relocalization of ND10 components including PML; ii) depletion of ATRX; iii) relocalization and dispersion of Sp100; iv) dispersion of Daxx. Using quantification of immunofluorescent images and immunoblotting we could show that ORF75 acts in a dose dependent manner on ND10 components Daxx and ATRX (Figure 6). In addition we could also observe a strong colocalization of ORF75 and PML. These findings could also be confirmed during lytic viral replication, using either Flag-tagged or YFP-tagged ORF75 protein expressed by recombinant viruses (Figures 9 and 10). Cells undergoing lytic replication showed a relocalization of PML, a colocalization of ORF75-YFP and PML, exclusion of Sp100 and Daxx from ND10, and loss of ATRX protein expression; this further corroborates the role of ORF75 in ND10 antagonization.
For the generation of infectious viruses from recombinant KSHV genomes, we used iSLK cells transfected with BAC DNA. Although it has recently been demonstrated that iSLK cells are not of endothelial origin as it has been assumed previously [22], this cell line can be used for efficient virus production. According to our experience, the internal repeat and multiple TR sequences present in KSHV mandate extra caution. In contrast to other BAC-cloned herpesvirus genomes, which seem to be rather stable during homologous recombination in E. coli, we frequently observed shortening of terminal repeat sequences from Bac16 recombinants. Since this cannot be detected by conventional gel electrophoresis and DNA sequencing, it is notable that we routinely verify KSHV BAC16 recombinants by pulsed field gel electrophoresis (PFGE) as well as Illumina deep sequencing of final constructs. It would have been of great interest to see whether the ablation of ORF75 expression affects the antagonization of ND10 components during infection and lytic replication. Intriguingly, we were unable to reconstitute infectious virus and could detect neither lytic gene expression nor viral DNA replication in the absence of ORF75. The expression of latent genes Lana/orf73 and GFP was not affected in unstimulated iSLK cells. Thus, to our own surprise, ORF75 seems to be essential for lytic replication at multiple levels. We could show for HVS that individual deletion of vFGARATs ORF3 and ORF75, respectively, leads to an impaired viral replication, but did not entirely abrogate viral replication [16]. However, for HVS, we were so far not able to reconstitute a virus with deletion of both ORF3 and ORF75, hinting at the important role of at least one vFGARAT in the genome of a rhadinovirus (Full and Ensser, unpublished data). In this context, it would also be of great interest to elucidate the role of ORF75 of rhesus rhadinovirus (RRV), the closest known relative of KSHV, during lytic replication.
The loss of virus production in the absence of ORF75 hints at multiple functions of the protein within the viral life cycle, as it has also been shown for ORF75c of MHV68 [26]. Since ORF75 is a tegument protein (Figure 7B), it may well be that it is an integral part of the virion and necessary for assembly of viral particles. However, the loss of an important structural component of the virion would not explain the complete absence of lytic gene expression and viral replication. In ORF75c-deleted MHV68 viral capsids are still made and accumulate, while no particles are apparent in orf75-stop KSHV (Figure 7B). It is thus remarkable that we cannot detect lytic gene expression despite overexpression of RTA in induced iSLK cells (Figure 8B). It is also unlikely that the loss of K13/vFLIP-mediated NF-kB (co)activation by ORF75 leads to this effect, as the KSHV genome comprises multiple potent activators of NF-kB signaling [19]. Our data hint at a role of ORF75 in the initiation of lytic gene expression. As a prerequisite, ORF75 protein must be expressed during latency or at least early during initiation of the lytic cycle. This is indeed the case (Figure 8D+E): after infection, ORF75 is released from the viral tegument into the cell. As soon as immediate early gene expression starts, de novo ORF75 protein is made in small amounts and at later timepoints in increasing amounts until new viral particles are released form the cell. Thus ORF75 protein is present for the entire viral replication cycle. The orf75 gene is situated within a region at the right end of the KSHV genome that is transcribed during latency, and genome-wide analysis of epigenetic modifications of latent KSHV genomes revealed histone modification patterns in the putative orf75 promoter region which reflect actively transcribed chromatin [27], [28]. Therefore, it would be very interesting, but beyond the scope of this article, to test for potential epigenetic mechanisms underlying the loss of viral DNA replication in the absence of ORF75, and the role of ORF75 during latency of KSHV.
Concerning the role of ORF75 during lytic replication, we could clearly show that ORF75 is essential for viral replication. Moreover the dispersion of ATRX by ORF75 seems to be crucial for lytic replication of KSHV; a KSHV-orf75 deletion mutant that is not capable of lytic viral replication can at least be partially rescued by knock-down of ATRX (Figure 11), clearly emphasizing the role of ORF75 mediated ATRX relocation. The fate of ATRX after ORF75 mediated removal from ND10 remains elusive. Proteasomal degradation of ATRX can be excluded (Figure 3A+B). Although difficult to prove, we hypothesize that ATRX might be translocated to a highly insoluble nuclear fraction, and therefore can no longer exert its antiviral function.
KSHV infection leads to a dispersion of the ND10 component ATRX (Figures 1D and 2). We could not detect a degradation of Daxx, although KSHV infection also led to a loss of spotted Daxx localization (Figures 1D, 2, 3). Apparently, Daxx is also dispersed from ND10. Whether the Daxx dispersion is a direct or indirect effect of KSHV-induced loss of ATRX could not be demonstrated. Using co-immunprecipitation experiments, we could not detect a direct interaction of ORF75 with Daxx as it has been shown for BNRF1 of EBV [18]. This is also in accordance with findings by Tsai et al [18], indicating that EBV - and KSHV-FGARATs target the same proteins but have evolved different mechanisms in the antagonization of ND10 components. Isolated expression of ORF75 after transfection led to alterations in ND10 composition similar to observations after lytic replication. In order to separate the functions of vIRF3 and ORF75, we constructed a recombinant virus harboring a deletion of the vIRF3 gene in combination with either YFP-tagged ORF75 (Figure 10) or Flag-tagged ORF75 protein (data not shown). It has been shown that vIRF3 is SUMOylated, and that overexpression of vIRF3 leads to a moderate decrease in ND10 foci per cell [13], [29]. However, the phenotype of vIRF3-deletion mutants compared to wildtype virus with respect to ND10 was indistinguishable. Since it is known, that ND10 components can be induced by IFN, the decrease in ND10 dots per cell may also be a result of IFN antagonization by vIRF3 [30]–[32].
In summary, our results clearly revealed that ORF75 is essential for viral replication. Thus, ORF75 could be a new and specific target for antiviral therapy, since inhibition of ORF75 function should abrogate viral lytic replication. This is of particular interest also for therapy of KSHV tumors, since it is known that lytic replication of KSHV is of particular importance for KS tumorigenesis in vivo. KS tumors regularly show lytic viral replication at low levels [33], [34], and a clinical trial demonstrated that the development of new KS lesions in patients can be blocked by administration of ganciclovir, an inhibitor of the viral DNA polymerase which is required for lytic viral replication [35], [36].
Materials and Methods
Cells and viruses
Primary human foreskin fibroblasts (HFF), were prepared from human foreskin-tissue as described previously [37] and HFF with a small interfering RNA-mediated knockdown of PML (siPML cells), Daxx (siDaxx cells), Sp100 (siSp100 cells), ATRX (siATRX cells) and the respective control cells (siC cells) were cultured in Dulbecco's minimal essential medium (DMEM, LifeTechnologies, Germany) supplemented with 10% fetal calf serum and 5 µg/ml puromycin [38]–[40]. HeLa cells and HEK 293T cells were maintained in Eagle's minimal essential medium (LifeTechnologies) supplemented with 10% fetal calf serum. iSLK cells were maintained in DMEM supplemented with 10% fetal calf serum, 2.5 µg/ml puromycin and 250 µg/ml G418. KSHV from BAC-DNA was obtained by reconstitution of infectious viruses after transfection of BAC16 DNA into iSLK cells. iSLK cells were transfected in 6-wells with 5 µg BACmid DNA with 9 µl ExtremeGene (Roche) according to manufacturer's instructions, and selected with 200 µg/ml hygromycin B until all cells were GFP positive.
Lytic replication of KSHV was induced in iSLK harboring KSHV Bac16-DNA cells by plating cells in the absence of antibiotics and adding 900 µM sodium-butyrate and 1 µg/ml doxycyclin. UV inactivation was done by application of 120.000 microjoules/cm2 using the Stratalinker UV Crosslinker 1800 (Stratagene, Amsterdam, the Netherlands). Supernatants containing infectious virus were harvested 3 d post induction and stocks of wildtype and recombinant viruses were stored at −80°C in aliquots. Viral replication in induced iSLK cells was monitored by quantitative PCR of the KSHV orf26 locus and compared to cellular ccr5 DNA.
BAC mutagenesis and plasmids
The KSHV Bac16 clone was used for recombination-mediated genetic engineering of recombinants by a two-step, markerless λ-red-mediated recombination strategy using the kanamycin gene as a first selection marker [41]. Linear fragments for homologous recombination were generated by PCR using Phusion High Fidelity DNA polymerase (Finnzymes); DpnI was added to digest the methylated plasmid template, and the amplification product was purified from an agarose gel with the Nucleobond Gel Extraction Kit (Macherey&Nagel, Düren, Germany). Primers that were used to generate linear fragments for the manipulations were purchased as Ultramers™ from Integrated DNA Technologies.
For homologous recombination, the PCR fragment was then transformed into Escherichia coli strain GS1783 (gift of G. Smith, NW University, USA) [42] harboring BAC16, and Red recombination was performed as described earlier [41], [43]. Cells were then plated on agar plates containing 15 µg/ml kanamycin (first recombination) or 15 µg/ml chloramphenicol and 1% arabinose (second recombination) and incubated at 32°C for 1–2 days. Bacterial colonies growing on these plates were further analyzed. Reconstitution of recombinant KSHV using purified BAC DNA was performed in iSLK cells as described above.
Viral nucleic acid isolation and analysis
BAC DNA was isolated by standard alkaline lysis from 5 ml liquid cultures. Subsequently, the integrity of BACmid DNA was analyzed by digestions with restriction enzyme XhoI and separation in 0.8% PFGE agarose (Bio-Rad) gels and 0.5×TBE buffer by pulse field gel electrophoresis at 6 V/cm, 120 degree field angle, switch time linearly ramped from 1 s to 5 s over 16 h (CHEF DR III, Bio-Rad). For characterization of insertion or deletion of the aphaI selection marker, recombinant BACmids were analyzed by PCR, and the recombined regions within the BAC DNA were controlled by sequence analysis (ABI 3130XL genetic analyzer, Weiterstadt, Germany) in order to confirm the correct deletion sequences and to exclude accidental mutations. Furthermore, the integrity of the complete viral genome of representative Bac16 recombinants was confirmed by next-generation sequencing using Nextera DNA Sample Preparation system and the MiSeq Reagent Kit, 300 Cycles on the Illumina MiSeq system. NGS data were analyzed by Genome Workbench 5 (CLCbio, Aarhus, DK).
Antibodies
Endogenous PML was detected with mouse monoclonal antibody PG-M3 (IgG1, Santa Cruz Biotechnology (SCBT) or 5E10 (gift of R. van Driel, University of Amsterdam, The Netherlands). Human Sp100 was detected with a rabbit polyclonal antiserum (ProteinTech Group, USA) or a mouse polyclonal antibody (MaxPAP, Abnova, USA). A rabbit polyclonal antibody from Sigma-Aldrich (Germany) or the mouse monoclonal antibody MCA2143 (IgG1, Serotec, Germany) were used for detection of hDaxx. ATRX protein expression was detected with mouse monoclonal antibody clone D-5 (IgG2a, SCBT). Rabbit polyclonal antibody for detection of beta-actin, was purchased from Sigma-Aldrich, GFP-specific rabbit polyclonal antibody was purchased from Genescript (Piscataway, NJ). Epitope-tagged proteins were either detected using the anti-FLAG tag antibody M2 (Sigma-Aldrich, Germany) or the anti-myc tag antibodies clone 9E10 (IgG1, Sigma-Aldrich) or clone 9B11 (IgG2a, Cell Signaling Technologies, Germany). Horseradish-peroxidase-conjugated anti-mouse and anti-rabbit secondary antibodies for Western blot analysis were obtained from Dianova (Hamburg, Germany), while Alexa Fluor 488-, Alexa Fluor 555-, and Alexa Fluor 647-conjugated secondary antibodies for indirect immunofluorescence experiments were purchased from LifeTechnologies (Germany). For detection of KSHV proteins, KSHV-antibodies to K8 (clone 8C12G10G1, SCBT, USA), K8.1 (clone 4A4, SCBT, USA), vIRF3 (rat monoclonal, kindly provided by Frank Neipel) and ORF75 KSHV (polyclonal rabbit serum, Genescript) were used.
Immunofluorescence analysis
For immunofluorescence analysis, iSLK cells or HFF cells were plated onto coverslips. Lytic replication of KSHV in iSLK cells was induced as described above.
Cycloheximide was used at 10 µg/ml, MG132 at 10 µM. At indicated timepoints post induction, infection or transfection, cells were washed three times with phosphate-buffered saline (PBS), followed by fixation with 4%paraformaldehyde for 10 min at room temperature. Then, cells were permeabilized with PBS-0.2% Triton X-100, 5% FCS for 1 hour, followed by incubation with the respective primary or secondary antibodies for 30 min at 37°C. Finally, the cells were mounted by using Mowiol mounting medium (Fluka, Germany) plus 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, USA) or Hoechst (Sigma-Aldrich, Germany). The samples were examined by using a Leica TCS SP5 confocal microscope, with 405 nm, 488 nm, 543 nm or 633 nm laser lines, scanning each channel separately under image capture conditions that eliminated channel overlap. The Fiji distribution of ImageJ was used to count nuclear dots in ORF75 expressing human fibroblasts [44]. Briefly, nuclei were identified by watershedding and the analyze particles function in the DAPI image, then defined as ROI; after setting an appropriate noise tolerance, single point maxima in the channel corresponding to ND10 proteins were then counted within each nuclear ROI and results copied to MS Excel. Statistical analyses were conducted using the unpaired t-test for SP100 and PML, and one way ANOVA test for multiple comparisons for Daxx and ATRX, respectively (GraphPad Prism 6 for Windows, GraphPad Software, USA).
Western blot analysis
Extracts from infected cells were prepared either directly in a sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer or in a RIPA-lysis buffer as described previously, separated either on sodium dodecyl sulfate-containing 8%, 10% 12% polyacrylamide gels or 4–12% bis-tris gradient gels (Life Technologies, Germany), and transferred to polyvinylidene fluoride membranes (Millipore). Enhanced Chemi-luminescence was detected according to the manufacturer's protocol (GE Healthcare).
Flow cytometry
HFF cells or 293T cells were detached using PBS supplemented with 0.1% EDTA, pH 7.4, spun down (300×g, 5 min), supernatant removed and then fixed in PBS supplemented with 2% paraformaldehyde for 30 min. GFP expression of cells was analyzed on a LSRII flow cytometer (BD Biosciences) and data were analyzed with FCS Express 3 software (De Novo Software, Canada). For cell cycle analysis, cells were harvested and fixed in 80% ethanol at least over night. Staining after RNase treatment (50 µg/ml) was done with propidium iodide (20 µg/ml) and analyzed on a BD LSR2 flow cytometer. Cell cycle data were modeled with ModFitLT 3.3 for Windows (Verity Software House, Topsham, ME).
Immunoelectronmicroscopy
Induced iSLK cells were harvested in Dulbeco's modified Eagle's medium (DMEM) 48 h post induction. Cells were fixed with DMEM containing 4% paraformaldehyde, dehydrated at −20°C and embedded in LR-White at −20°C. Polymerization was induced by heat for 2 days at 60°C prior to sectioning using an ultramicrotome (Leica, Wetzlar, Germany). The sections were transferred to nickel grids and immunostaining was performed with anti-FLAG antibody (clone M2, Sigma-Aldrich) and secondary anti-mouse antibody conjugated with 12 nm gold (Dianova). The grids were stained for 10 min with 1% (w/v) uranyl acetate in 40% Ethanol followed by lead citrate staining for additional 10 min prior to analysis by electron microscopy. Specimens were observed using a TecnaiTM G2 Scanning transmission electron microscope (FEI Company, Eindhoven, The Netherlands) operated at 120 kV.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. ChangY, CesarmanE, PessinMS, LeeF, CulpepperJ, et al. (1994) Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266 : 1865–1869.
2. DupinN, GrandadamM, CalvezV, GorinI, AubinJT, et al. (1995) Herpesvirus-like DNA sequences in patients with Mediterranean Kaposi's sarcoma. Lancet 345 : 761–762.
3. GanemD (2010) KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J Clin Invest 120 : 939–949.
4. TavalaiN, StammingerT (2008) New insights into the role of the subnuclear structure ND10 for viral infection. Biochim Biophys Acta 1783 : 2207–2221.
5. MaulGG, GuldnerHH, SpivackJG (1993) Modification of discrete nuclear domains induced by herpes simplex virus type 1 immediate early gene 1 product (ICP0). J Gen Virol 74 (Pt 12) 2679–2690.
6. BoutellC, SadisS, EverettRD (2002) Herpes simplex virus type 1 immediate-early protein ICP0 and is isolated RING finger domain act as ubiquitin E3 ligases in vitro. J Virol 76 : 841–850.
7. BoutellC, Cuchet-LourencoD, VanniE, OrrA, GlassM, et al. (2011) A viral ubiquitin ligase has substrate preferential SUMO targeted ubiquitin ligase activity that counteracts intrinsic antiviral defence. PLoS Pathog 7: e1002245.
8. LeeHR, KimDJ, LeeJM, ChoiCY, AhnBY, et al. (2004) Ability of the human cytomegalovirus IE1 protein to modulate sumoylation of PML correlates with its functional activities in transcriptional regulation and infectivity in cultured fibroblast cells. J Virol 78 : 6527–6542.
9. MullerS, DejeanA (1999) Viral immediate-early proteins abrogate the modification by SUMO-1 of PML and Sp100 proteins, correlating with nuclear body disruption. J Virol 73 : 5137–5143.
10. Chelbi-AlixMK, PelicanoL, QuignonF, KokenMH, VenturiniL, et al. (1995) Induction of the PML protein by interferons in normal and APL cells. Leukemia 9 : 2027–2033.
11. GrotzingerT, SternsdorfT, JensenK, WillH (1996) Interferon-modulated expression of genes encoding the nuclear-dot-associated proteins Sp100 and promyelocytic leukemia protein (PML). Eur J Biochem 238 : 554–560.
12. KatanoH, Ogawa-GotoK, HasegawaH, KurataT, SataT (2001) Human-herpesvirus-8-encoded K8 protein colocalizes with the promyelocytic leukemia protein (PML) bodies and recruits p53 to the PML bodies. Virology 286 : 446–455.
13. Marcos-VillarL, Lopitz-OtsoaF, GallegoP, Munoz-FontelaC, Gonzalez-SantamariaJ, et al. (2009) Kaposi's sarcoma-associated herpesvirus protein LANA2 disrupts PML oncogenic domains and inhibits PML-mediated transcriptional repression of the survivin gene. J Virol 83 : 8849–8858.
14. ChangPC, IzumiyaY, WuCY, FitzgeraldLD, CampbellM, et al. (2010) Kaposi's sarcoma-associated herpesvirus (KSHV) encodes a SUMO E3 ligase that is SIM-dependent and SUMO-2/3-specific. J Biol Chem 285 : 5266–5273.
15. WuFY, AhnJH, AlcendorDJ, JangWJ, XiaoJ, et al. (2001) Origin-independent assembly of Kaposi's sarcoma-associated herpesvirus DNA replication compartments in transient cotransfection assays and association with the ORF-K8 protein and cellular PML. J Virol 75 : 1487–1506.
16. FullF, ReuterN, ZielkeK, StammingerT, EnsserA (2012) Herpesvirus Saimiri Antagonizes Nuclear Domain 10-Instituted Intrinsic Immunity via an ORF3-Mediated Selective Degradation of Cellular Protein Sp100. J Virol 86 : 3541–3553.
17. LingPD, TanJ, SewatanonJ, PengR (2008) Murine gammaherpesvirus 68 open reading frame 75c tegument protein induces the degradation of PML and is essential for production of infectious virus. J Virol 82 : 8000–8012.
18. TsaiK, ThikmyanovaN, WojcechowskyjJA, DelecluseHJ, LiebermanPM (2011) EBV tegument protein BNRF1 disrupts DAXX-ATRX to activate viral early gene transcription. PLoS Pathog 7: e1002376.
19. KonradA, WiesE, ThurauM, MarquardtG, NaschbergerE, et al. (2009) A systems biology approach to identify the combination effects of human herpesvirus 8 genes on NF-kappaB activation. J Virol 83 : 2563–2574.
20. VieiraJ, O'HearnPM (2004) Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology 325 : 225–240.
21. MyoungJ, GanemD (2011) Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: maintenance of tight latency with efficient reactivation upon induction. J Virol Methods 174 : 12–21.
22. StürzlM, GausD, DirksWG, GanemD, JochmannR (2013) Kaposi's sarcoma-derived cell line SLK is not of endothelial origin, but is a contaminant from a known renal carcinoma cell line. Int J Cancer 132 : 1954–1958.
23. BechtelJT, WinantRC, GanemD (2005) Host and viral proteins in the virion of Kaposi's sarcoma-associated herpesvirus. J Virol 79 : 4952–4964.
24. RozenR, SathishN, LiY, YuanY (2008) Virion-wide protein interactions of Kaposi's sarcoma-associated herpesvirus. J Virol 82 : 4742–4750.
25. BruloisKF, ChangH, LeeAS, EnsserA, WongLY, et al. (2012) Construction and manipulation of a new Kaposi's sarcoma-associated herpesvirus bacterial artificial chromosome clone. J Virol 86 : 9708–9720.
26. GasparM, GillMB, LosingJB, MayJS, StevensonPG (2008) Multiple functions for ORF75c in murid herpesvirus-4 infection. PLoS ONE 3: e2781.
27. GüntherT, GrundhoffA (2010) The epigenetic landscape of latent Kaposi sarcoma-associated herpesvirus genomes. PLoS Pathog 6: e1000935.
28. TothZ, MaglinteDT, LeeSH, LeeHR, WongLY, et al. (2010) Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog 6: e1001013.
29. Marcos-VillarL, CampagnaM, Lopitz-OtsoaF, GallegoP, Gonzalez-SantamariaJ, et al. (2011) Covalent modification by SUMO is required for efficient disruption of PML oncogenic domains by Kaposi's sarcoma-associated herpesvirus latent protein LANA2. J Gen Virol 92 : 188–194.
30. JooCH, ShinYC, GackM, WuL, LevyD, et al. (2007) Inhibition of interferon regulatory factor 7 (IRF7)-mediated interferon signal transduction by the Kaposi's sarcoma-associated herpesvirus viral IRF homolog vIRF3. J Virol 81 : 8282–8292.
31. JacobsSR, DamaniaB (2011) The viral interferon regulatory factors of KSHV: immunosuppressors or oncogenes? Front Immunol 2 : 19.
32. SchmidtK, WiesE, NeipelF (2011) Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor 3 inhibits gamma interferon and major histocompatibility complex class II expression. J Virol 85 : 4530–4537.
33. OrensteinJM, AlkanS, BlauveltA, JeangKT, WeinsteinMD, et al. (1997) Visualization of human herpesvirus type 8 in Kaposi's sarcoma by light and transmission electron microscopy. AIDS 11: F35–F45.
34. StaskusKA, ZhongW, GebhardK, HerndierB, WangH, et al. (1997) Kaposi's sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J Virol 71 : 715–719.
35. GrundhoffA, GanemD (2004) Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J Clin Invest 113 : 124–136.
36. MartinDF, KuppermannBD, WolitzRA, PalestineAG, LiH, et al. (1999) Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. Roche Ganciclovir Study Group. N Engl J Med 340 : 1063–1070.
37. JahnG, KnustE, SchmollaH, SarreT, NelsonJA, et al. (1984) Predominant immediate-early transcripts of human cytomegalovirus AD 169. J Virol 49 : 363–370.
38. AdlerM, TavalaiN, MüllerR, StammingerT (2011) Human cytomegalovirus immediate-early gene expression is restricted by the nuclear domain 10 component Sp100. J Gen Virol 92 : 1532–1538.
39. TavalaiN, PapiorP, RechterS, LeisM, StammingerT (2006) Evidence for a role of the cellular ND10 protein PML in mediating intrinsic immunity against human cytomegalovirus infections. J Virol 80 : 8006–8018.
40. TavalaiN, AdlerM, SchererM, RiedlY, StammingerT (2011) Evidence for a dual antiviral role of the major nuclear domain 10 component Sp100 during the immediate-early and late phases of the human cytomegalovirus replication cycle. J Virol 85 : 9447–9458.
41. TischerBK, von EinemJ, KauferB, OsterriederN (2006) Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40 : 191–197.
42. TischerBK, SmithGA, OsterriederN (2010) En passant mutagenesis: a two step markerless red recombination system. Methods Mol Biol 634 : 421–430.
43. ZhangY, BuchholzF, MuyrersJP, StewartAF (1998) A new logic for DNA engineering using recombination in Escherichia coli. Nat Genet 20 : 123–128.
44. SchindelinJ, Arganda-CarrerasI, FriseE, KaynigV, LongairM, et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9 : 676–682.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 1
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Lyme Disease: Call for a “Manhattan Project” to Combat the Epidemic
- Origin, Migration Routes and Worldwide Population Genetic Structure of the Wheat Yellow Rust Pathogen f.sp.
- IFNγ/IL-10 Co-producing Cells Dominate the CD4 Response to Malaria in Highly Exposed Children
- Human and Plant Fungal Pathogens: The Role of Secondary Metabolites