
Phosphopyruvate Carboxylase Identified as a Key Enzyme in Erythrocytic Carbon Metabolism
Phospoenolpyruvate carboxylase (PEPC) is absent from humans but encoded in the Plasmodium falciparum genome, suggesting that PEPC has a parasite-specific function. To investigate its importance in P. falciparum, we generated a pepc null mutant (D10Δpepc), which was only achievable when malate, a reduction product of oxaloacetate, was added to the growth medium. D10Δpepc had a severe growth defect in vitro, which was partially reversed by addition of malate or fumarate, suggesting that pepc may be essential in vivo. Targeted metabolomics using 13C-U-D-glucose and 13C-bicarbonate showed that the conversion of glycolytically-derived PEP into malate, fumarate, aspartate and citrate was abolished in D10Δpepc and that pentose phosphate pathway metabolites and glycerol 3-phosphate were present at increased levels. In contrast, metabolism of the carbon skeleton of 13C,15N-U-glutamine was similar in both parasite lines, although the flux was lower in D10Δpepc; it also confirmed the operation of a complete forward TCA cycle in the wild type parasite. Overall, these data confirm the CO2 fixing activity of PEPC and suggest that it provides metabolites essential for TCA cycle anaplerosis and the maintenance of cytosolic and mitochondrial redox balance. Moreover, these findings imply that PEPC may be an exploitable target for future drug discovery.
Published in the journal:
. PLoS Pathog 10(1): e32767. doi:10.1371/journal.ppat.1003876
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1003876
Summary
Phospoenolpyruvate carboxylase (PEPC) is absent from humans but encoded in the Plasmodium falciparum genome, suggesting that PEPC has a parasite-specific function. To investigate its importance in P. falciparum, we generated a pepc null mutant (D10Δpepc), which was only achievable when malate, a reduction product of oxaloacetate, was added to the growth medium. D10Δpepc had a severe growth defect in vitro, which was partially reversed by addition of malate or fumarate, suggesting that pepc may be essential in vivo. Targeted metabolomics using 13C-U-D-glucose and 13C-bicarbonate showed that the conversion of glycolytically-derived PEP into malate, fumarate, aspartate and citrate was abolished in D10Δpepc and that pentose phosphate pathway metabolites and glycerol 3-phosphate were present at increased levels. In contrast, metabolism of the carbon skeleton of 13C,15N-U-glutamine was similar in both parasite lines, although the flux was lower in D10Δpepc; it also confirmed the operation of a complete forward TCA cycle in the wild type parasite. Overall, these data confirm the CO2 fixing activity of PEPC and suggest that it provides metabolites essential for TCA cycle anaplerosis and the maintenance of cytosolic and mitochondrial redox balance. Moreover, these findings imply that PEPC may be an exploitable target for future drug discovery.
Introduction
Plasmodium falciparum is the causative agent of malaria, a disease claiming an estimated 660,000 lives annually, primarily among children in Africa [1]. The spread of drug resistance and the lack of an efficacious vaccine hinder the control of the disease, and new targets for chemotherapies or vaccine development need to be identified and exploited [2]. The development of the parasite and its modification of the host's red blood cells (RBC) cause the pathogenesis of malaria, but the biochemical adaptations enabling this remain to be fully elucidated.
Energy generation in P. falciparum asexual erythrocytic stages depends upon glucose being primarily converted to lactate by anaerobic glycolysis, which is then excreted as the major metabolic end product [3]. This is consistent with the finding that in P. falciparum the pyruvate dehydrogenase complex (PDH) is solely present in a plastid-like organelle, the apicoplast, where it provides acetyl-CoA for fatty acid biosynthesis and possibly other acetylating reactions [4], [5]. It was shown very recently, however, that despite the absence of mitochondrial PDH, pyruvate can be metabolised by a PDH-like enzyme complex [6] and oxidised through a forward tricarboxylic acid (TCA) cycle in the erythrocytic stages of P. falciparum, being particularly important in the gametocytes, and also in the related apicomplexan parasite Toxoplasma gondii [3], [6], [7]. These findings are in contrast to previous reports that there is no link between cytosolic glucose catabolism and mitochondrial TCA metabolism [8], although this report was later retracted [9].
A striking feature of carbon metabolism in Plasmodium is their ability to fix CO2. This is utilised to generate carbamoyl phosphate and thence pyrimidines and also is incorporated into amino acids and α-ketoacids in P. lophurae, P. knowlesi and P. berghei [10]–[12]. Together with Trager's finding that P. falciparum require CO2 for in vitro growth [13], this suggests that CO2 fixation is necessary for the parasite's intra-erythrocytic survival. CO2 fixation may occur via carbamoyl phosphate synthase, phosphoenolpyruvate carboxylase (PEPC) and/or phosphoenolpyruvate carboxykinase (PEPCK), with the latter two feeding into intermediary carbon metabolism [14]–[16]. P. falciparum PEPCK is primarily expressed in gametocytes and mosquito stages [16], and is generally considered to produce phosphoenolpyruvate (PEP) and release CO2. Thus PEPC, which utilises bicarbonate, generated from CO2 by carbonic anhydrase in P. falciparum [17], to convert PEP into oxaloacetate (OAA) is a strong candidate to have a key role in P. falciparum carbon metabolism by fixing CO2. Plant and bacterial PEPCs have been well characterised [18]–[20]; the malarial enzyme has, however, been little studied. There is just one report on P. berghei PEPC activity [14], even though PEPC is absent from mammals and thus potentially offers great opportunities for exploitation by novel antimalarial intervention strategies. Thus this study aimed to confirm the operation of PEPC in erythrocytic stages of P. falciparum, to elucidate its contributions to intermediary carbon metabolism and intra-erythrocytic survival, and so determine the likelihood that it is a viable drug target.
Results
Knockout of pepc gene by homologous recombination
Initially, a disruption of the pepc gene was attempted using a single homologous recombination approach with the plasmids pHH1-Δpepc and pHH1-3′pepc. The control construct pHH1-3′pepc targeted the pepc locus and replaced the 3′ region of the gene, while the pHH1-Δpepc construct did not integrate into the correct gene locus, as shown by pulsed field gel electrophoresis (Fig. S1). These data revealed that the pepc locus is not refractory to recombination, but that a gene disruption was unsuccessful probably because the pepc gene is very important or essential for parasite survival. Parasites were then transfected with the plasmid pCC4-Δpepc, in order to target the pepc gene locus by double homologous recombination [21]. The pepc locus was not targeted when parasites were cultured in routine medium (which does not contain added malate); however, addition of 5 mM malate to the medium (malate medium) allowed the replacement of the gene with the selectable marker, blasticidin deaminase (bsd). This was verified by Southern blot analyses (Fig. 1). The D10Δpepc mutants were cloned, and two independent clones D10Δpepc-1 and D10Δpepc-2 (Fig. 1B) were used for phenotype analyses. The data for the two clones were effectively the same, and so one set of data are presented forthwith and the line referred to as D10Δpepc.
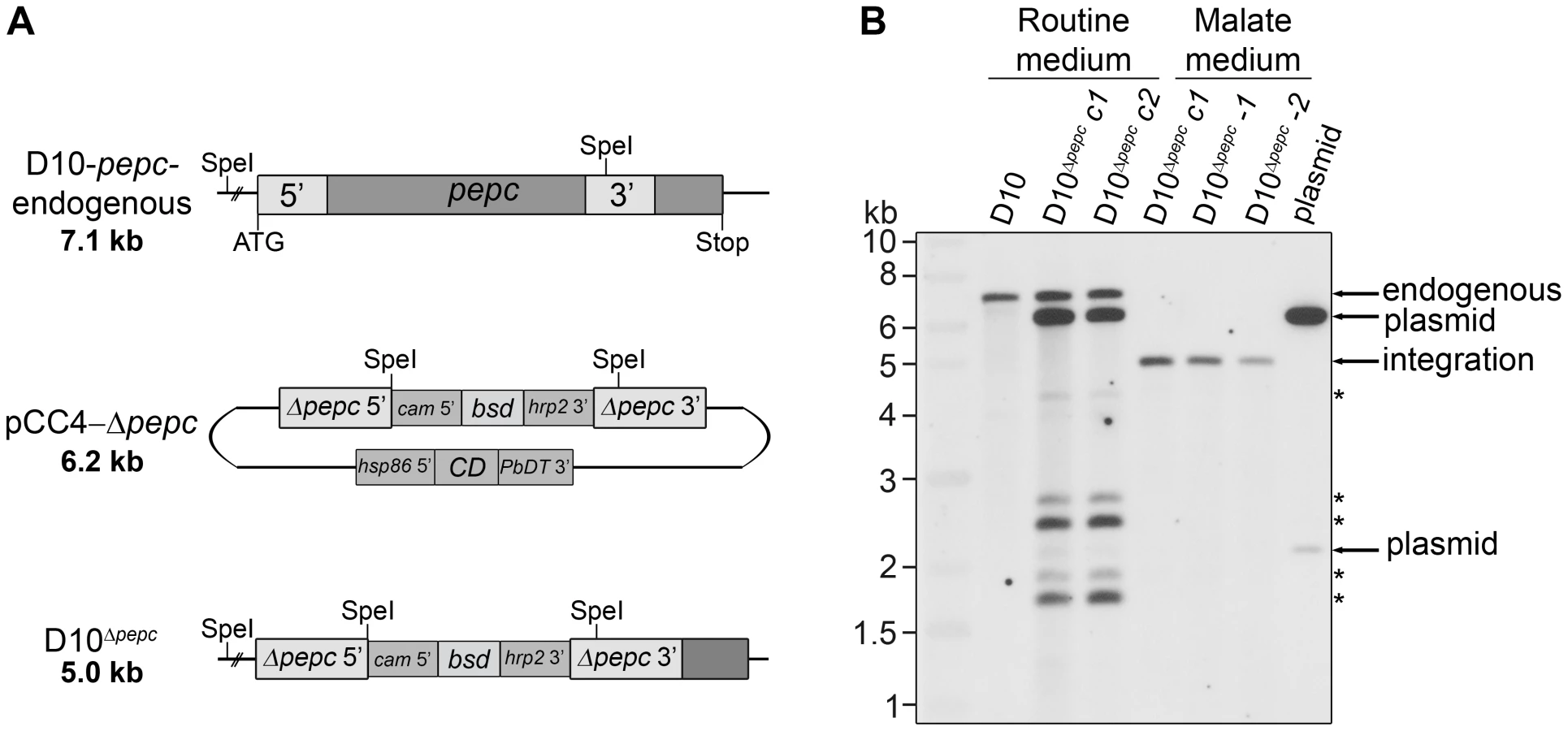
The pepc gene is important for intra-erythrocytic survival of P. falciparum
Given that the deletion of the pepc gene was achieved only when the culture medium was supplemented with 5 mM malate, the effect of withdrawing malate from the medium on the growth of D10Δpepc was analysed. Parasite growth in routine medium was followed for 14 days (Fig. 2A). After 6 days in routine medium, D10Δpepc had severely reduced growth rates and completely lost their synchronicity. Nevertheless, they continued to replicate a little, showing that in culture they are able to compensate to an extent for the loss of PEPC function. Likely mechanisms include obtaining some malate from the host erythrocyte directly or conversion from fumarate, generated as a by-product of purine salvage or itself taken up from the erythrocyte. The mutant parasites grew better in medium supplemented with malate, but the added 5 mM malate did not fully restore growth to the wild type rate (Fig. 2A). Lower concentrations of malate were less effective (Fig. 2B), whereas applying higher concentrations of malate did not improve growth further (data not shown). The beneficial effect of malate for growth of D10Δpepc seemed likely to be due to providing the mutant parasite with one of the downstream products of PEPC metabolism, and this post-PEPC metabolism is important for progression of P. falciparum through their erythrocytic cycle. In order to ascertain whether other possible downstream metabolites of PEPC could have beneficial effects, we tested several for growth stimulation of D10Δpepc. The only metabolite other than malate that significantly restored parasite growth was fumarate (Fig. 2C). This suggests that supplementation of the medium with fumarate may also have allowed experimental deletion of the pepc gene similarly to malate supplementation, although this possibility was not tested. Notably glutamine was unable to rescue growth, probably a reflection of the parasite's metabolism of this amino acid as described below.

Analysis of the pepc gene product
The predicted protein encoded by the pepc gene of P. falciparum has clear similarity with PEPCs of other organisms but rather limited identity. Amino acid identities between P. falciparum PEPC and plant enzymes are in the range of 30% (those of Zea mays and Arabidopsis thaliana are 29% and 31% identical, respectively) whereas identity with Escherichia coli PEPC is 25%. An extension at the N-terminus of the P. falciparum protein, a 29 amino acid central insertion and a long insertion towards the C-terminus (see alignment Fig. S2) are the prime reasons for this low degree of sequence identity and also the larger mass of the parasite protein (134 kDa compared with 100 kDa of bacterial PEPC and 116 kDa of plant PEPC) [18]. Nevertheless, the parasite protein does contain residues known to be crucial to the activities of other PEPCs. The C-terminal catalytic peptide at position 1143 to 1148 (equivalent to 966 to 970 of maize PEPC) (see Fig. S2) is completely conserved in P. falciparum PEPC as are residues R685, which is involved in PEP binding (equivalent to R647 in maize PEPC), and H193 (equivalent to H177 in maize PEPC), which acts as catalytic base in the active site of the protein [18]. In addition, the two loops probably involved in regulation of PEPC catalytic activity (loop I and loop II in Fig. S2) are also highly conserved in P. falciparum PEPC. Loop I comprising residues 678GRGGXXG685RGG (XX = SV in P. falciparum PEPC; = TV in maize PEPC) includes the active site residue R685 and allows flexibility of this residue during the catalytic cycle. It is suggested that in the maize enzyme when in its inactive state the equivalent arginine residue is ‘attracted away’ from the active site, whereas in the active state R685 is attracted to the active cavity through interaction with the C-terminal glycine. Loop II is located at the catalytic cavity and forms a bridge above the β-barrel; it is thought to be involved in binding of HCO3− and may have a role in covering the active site cavity to protect catalytic intermediates from unwanted reactions with surrounding water [18]. Moreover, a homology model of P. falciparum PEPC that was generated using ITASSER (Fig. 3) is consistent with it having a good degree of structural similarity with E. coli PEPC. This shows that the core structure and the location of catalytically and regulatory amino acids in P. falciparum PEPC are structurally in good agreement with the equivalent in the PEPC of E. coli, with the β-barrel that generates the active site cavity being structurally conserved and similarly surrounded by α-helices. The homology model also shows that the N-terminus of P. falciparum PEPC forms a non-homologous loop (residues Met1 to Cys46 in Fig. 3). The two other insertion regions also show clear differences in structure from the E. coli PEPC, both being presented as surface-located loops (Fig. 3).

A full phylogenetic analysis of PEPCs was reported last year [22] and this suggests that P. falciparum PEPC is aligned with the C4 plant PEPCs as it possesses the signature amino acids K373 and S818; these are equivalent to K353 and S780 in the maize PEPC sequence (Fig. S2), are invariably found in C4 plant PEPCs and are thought to influence kinetic parameters and inhibition/activation features of the enzymes. In contrast, these residues in C3 plant PEPCs are arginine and alanine, respectively [23]. Scrutiny of other amino acid residues important for the protein's catalytic activity and binding of allosteric activators or inhibitors [18], [23], [24] are consistent with this conclusion. Interestingly, however, P. falciparum PEPC does not possess the N-terminal serine residue present in plant PEPC proteins that is phosphorylated in order to regulate PEPC activity of C4 plants [19]. However, analyses of the phosphoproteome of P. falciparum suggests that S1109 is phosphorylated during blood stage development of the parasites [25] which implies that P. falciparum PEPC activity may be regulated through phosphorylation of this residue.
The conservation of all of these features in the P. falciparum PEPC suggests that the gene encodes a functional PEPC protein. Unfortunately detailed biochemical and structural analyses of Plasmodium PEPC recombinant protein have so far been ruled out by our lack of success in attempts to express parasite protein in enzymatically active form; despite very extensive attempts using different expression systems and modified gene constructs (with the long insertions deleted), we have been unable to generate active recombinant enzyme that could be studied in a meaningful way. The native enzyme has been purified from P. berghei and displayed a molecular size of 280 kDa [14], implying that it forms a homodimer. Other larger PEPC species were also found after sucrose gradient centrifugation, suggesting that similar to PEPC isolated from other organisms the parasite enzyme may also form a tetramer [18]. Such analyses of the PEPC of P. falciparum have not been reported, obtaining sufficient pure enzyme has been problematic.
Sensitivity of D10Δpepc to metabolic inhibitors
To probe the mechanism facilitating the survival of D10Δpepc, its susceptibility to metabolic inhibitors was quantified. L-cycloserine, a competitive inhibitor of pyridoxal phosphate-dependent enzymes such as aspartate aminotransferase (AAT) [26], showed a clear differential effect on the parasite lines. D10Δpepc in routine medium was more sensitive (IC50 of 30±1 µM compared to 100±6 µM for D10, p<0.05; Fig. 4A), an effect partially reversed when D10Δpepc was cultured in malate supplemented medium (IC50 of 70±3 µM). We analysed by western blotting the expression levels of both AAT and malate dehydrogenase (MDH), enzymes important for metabolism of OAA generated by PEPC, and showed that they were not significantly affected by the deletion of the pepc gene regardless of the presence or absence of malate in the culture medium (Fig. S3). These data are entirely consistent with D10Δpepc depending on the activity of aminotransferases such as AAT and being more sensitive to the competitive inhibitor as result of lower OAA levels in the absence of PEPC.

AAT generates aspartate, which is a precursor for pyrimidine synthesis. Therefore the parasites' sensitivity to DSM190, an inhibitor of the key enzyme of pyrimidine biosynthesis dihydroorotate dehydrogenase (DHOD) [27], was determined and it was found that D10 and D10Δpepc were equally susceptible to the inhibitor, regardless of the presence of malate (Fig. 4B), suggesting that DHOD function is not affected in D10Δpepc. In the mitochondrion, malate is converted to OAA by malate∶quinone oxidoreducatase (MQO) which transfers electrons to the mitochondrial electron transport chain (mtETC) [28] and so we hypothesised that mtETC could be compromised by the absence of PEPC. To address whether this was the case, the sensitivities to atovaquone, an inhibitor acting against cytochrome bc1 complex in the mtETC [29], [30], were determined for D10 and D10Δpepc in routine or malate medium. Atovaquone inhibited the parasite lines with similar IC50 values between 0.7 and 1.2 nM (Fig. 4C). We also compared the mitochondrial membrane potential of D10 and D10Δpepc parasites by monitoring the uptake of the fluorophore MitoTracker. The data acquired indicate that the electrochemical gradient across the inner mitochondrial membrane is not affected by the loss of PEPC function (Fig. S4). To confirm the validity of this approach, the mitochondrial membrane potential was collapsed with valinomycin; this resulted in an even distribution of the fluorophore throughout the cytosol of the parasite.
Metabolomic analysis of D10Δpepc
The data above are consistent with PEPC generating OAA through CO2 fixation with subsequent conversion into malate and aspartate, and that these pathways are important for parasite development. That both malate and fumarate restore the growth phenotype implies that they feed into the same pathways. Intermediary metabolism is, however, a complicated matrix of interactions and the exogenously supplied malate and fumarate could provide their beneficial effects via conversion to a number of metabolites including OAA itself. So we decided that an analysis of key metabolites in this part of metabolism would inform in more detail on the contributions of PEPC. Thus a targeted metabolomics analysis using liquid chromatography-mass spectrometry (LC-MS) with the stable-isotope labelled nutrients 13C-U-D-glucose, 13C-bicarbonate and 13C,15N-U-glutamine was conducted.
One difficulty in performing these labelling experiments was that the growth defect of the mutant parasites restricted the experiments that could be done. Whereas wild type D10 parasites can be synchronised and maintained for the 28-hour incubation period with the stable isotope at up to 10% parasitaemia, the D10Δpepc mutant grown without added malate for 9 days only reached a parasitaemia of 6%, maximally. In addition, these parasites could not be concentrated efficiently using MACS columns in the same way as the D10 parasites. The explanation for this is unclear, but perhaps could reflect some disruption of haemozoin synthesis in the mutant parasites. To overcome these difficulties, we carried out direct comparisons of D10Δpepc - and D10-infected RBC at 6% parasitaemia and, to ensure that the data from these experiments were robust, we also analysed D10 parasites incubated under the same conditions but subsequently concentrated to 90% parasitaemia via the MACS column methodology [31].
Incubation of parasites with 13C-U-D-glucose led, as expected, to an efficient incorporation of the 13C-labelled carbon skeleton into glycolytic intermediates and the end product lactate, both intracellularly and in the spent medium (Fig. 5, Table S1). The pentose phosphate pathway (PPP) metabolites ribulose 5-phosphate/ribose 5-phosphate and sedoheptulose 7-phosphate were also heavily labelled and apparently more abundant in D10Δpepc compared with D10 (Figs. 5B, 5C), providing evidence that the PPP is operational. In addition, levels of glycerol 3-phosphate were also greatly increased in D10Δpepc and strongly labelled (Figs. 5B, 5C). The proportion of incorporation of 13C label into most of these metabolites was not apparently different in D10Δpepc compared with D10, the only clear difference observed was reduced incorporation of 13C label into sedoheptulose 7-phosphate. Analysis of metabolites in the spent media of the samples (Table S1) indicated a large reduction of lactate production in D10Δpepc, reflecting a similar reduction in glucose utilisation, signifying that the mutant parasites have a lower metabolic capacity than D10. To confirm this and quantify the relative generation of metabolic end-products and utilisation of substrates fully (mass spectrometric analysis is only semi-quantitative and is usually not used to quantify metabolite concentrations, although it can give an indication), we determined the concentrations of various metabolites in the spent media of the different parasite lines using enzyme-based assays. The results (Table 1) show that both the consumption of glucose and release of lactate are reduced to approximately half in the D10Δpepc, and that lactate apparently accounted for approximately 75% and 70% of the glucose consumed by D10 and D10Δpepc, respectively. The slightly lower relative generation of lactate by D10Δpepc is consistent with some glucose being diverted into pathways other than only glycolysis, such as the PPP and glycerol 3-phosphate production (see Fig. 5).


D10 parasites also incorporated 13C label from glucose into intracellular 13C-3-labelled malate, fumarate and aspartate, the first two also being released into the medium, which can be explained by 13C-3-labelled PEP being utilised by PEPC to fix CO2 and form OAA (Fig. 5B). Most importantly, this was confirmed by the finding that these metabolites were not labelled in D10Δpepc, validating the activity of PEPC in generating them. The presence of 13C in these metabolites was relatively greater in the samples extracted from D10-infected RBC concentrated to 90% compared to those with 6% parasitaemia, whereas no 13C label was detected in the RBC control (Fig. 5B), thus substantiating that the incorporation was parasite-specific. The total levels of malate, fumarate and aspartate were lower in D10Δpepc compared to D10-infected RBC, in contrast to glycerol 3-phosphate and the PPP metabolites (Fig. 5C). The release of malate and fumarate into the spent medium by D10 parasites, although detected by the mass spectrometric analysis, was less than we could measure using our enzyme-based assays (5 and 33 µM, respectively) (Table 1), emphasising that these metabolites are important in intermediary carbon metabolism in the parasite rather than being metabolic end-products (they could account for <0.5% and <2% of the glucose used, maximally).
Interestingly, we also detected 13C-2 - and 13C-5 - labelled citrate in D10, strongly supporting the presence of a canonical TCA metabolism operating in the erythrocytic stages of P. falciparum at least as far as citrate. Citrate was not 13C-labelled in D10Δpepc, thus demonstrating that PEPC is a key node in intermediary carbon metabolism and forms a crucial link between glycolysis and mitochondrial TCA metabolism.
These findings using 13C-U-D-glucose were further validated by the analyses of parasites incubated with 13C-HCO3− (Fig. 6, Tables S3 and S4). The single 13C carbon present in bicarbonate was detected in malate, fumarate and aspartate in D10, with generally higher levels of incorporation detected in the samples extracted from infected RBC with 90% parasitaemia. Importantly, the incorporation of 13C label was completely absent in D10Δpepc-infected RBC (the 13C label present in these samples was at a similar level to that in the RBC control and attributed to natural occurrence of 13C-1 isotopomers), confirming that CO2 fixation into these metabolites is indeed attributable to PEPC. Overall these data provide clear evidence that PEPC functions in CO2 fixation and has an anaplerotic role.

We also investigated the catabolism of 13C 15N-U-glutamine by the parasites. The data obtained (Fig. S5 and Tables S5 and S6,) showed that there was a good flux from glutamine, via glutamate, to α-ketoglutarate and subsequently succinate, which were present as 13C-5 - and 13C-4-labelled molecules, respectively, in high proportion. The amount of 13C-4-labelled succinate in the D10Δpepc parasites was less than in D10, reflecting the lower catabolic rate generally in the mutant and also the lower consumption of glutamine (Table 1). There was, however, very little 13C label detected in the malate and fumarate pools. This is consistent with the conversion of succinate to these metabolites being rather low as also described by others recently [6], although the dilution effect of unlabelled forms of these metabolites in the host erythrocytes is also a factor. This may also explain why added glutamine was unable to restore growth of D10Δpepc, unlike malate and fumarate (Fig. 2), although the mitochondrial location of the metabolites generated from glutamine may also be an important factor.
These data for glutamine metabolism together with those presented in Figs. 5 and 6 for metabolism of glucose and bicarbonate suggest that there is a full forward TCA cycle operating in the parasites. However, there appear to be three separate segments of the TCA pathway which are apparently fed by separate ‘entry points’ (malate, pyruvate and α-ketoglutarate, respectively). The fluxes through the reactions from malate and pyruvate to citrate as well as that from α-ketoglutarate to succinate seem to be greater than the flux through the other segments of the cycle. This could be due to citrate and succinate being removed from the cycle and fed into other metabolic activities, indeed citrate exiting the mitochondrion could be part of the parasite's malate shuttle as discussed below.
Given the complex matrix of interconnections between different areas of metabolism, it was conceivable that the deletion of pepc would impact upon parts of metabolism in addition to redox homeostasis and the TCA cycle and that these effects could be contributing to the growth defect. Purine and pyrimidine metabolism were candidate areas. Thus we analysed the mass spectrometry data to find whether or not the levels of several metabolites in this metabolic area are changed in the mutant parasites. The results (Table S7, Fig. 5C) suggest that there are relatively minor changes to carbamoyl aspartate, orotate, dihydroorotate and hypoxanthine, indicating that these pathways are not greatly affected by the gene deletion.
PEPC as a possible drug target
The lack of PEPC in mammals and the importance of this enzyme to P. falciparum, as this study confirms, makes the parasite enzyme a possible drug target of interest. Plant and bacterial PEPCs are inhibited by 3,3-dichloro-2-(dihydroxyphosphinoylmethyl) propenoate (DCDP), an analogue of PEP. DCDP has a strong inhibitory effect on photosynthetic activity of plants at 1 mM and inhibits PEPC enzyme activity in the µM range [32]. The lack of recombinant protein has negated testing the effect of this PEP analog on the activity of P. falciparum PEPC, but we have assessed the efficacy of the inhibitor against parasite growth in vitro. The IC50 value for the inhibition of D10 was found to be 10.6±0.9 mM (n = 5); given this high concentration required to achieve a lethal effect on wild type parasites we did not test this compound against the mutant line. This poor efficacy of DCDP is likely to be due to its charged nature and thus to its inability to penetrate the parasite membranes.
Discussion
Early studies with various Plasmodium species showed that bicarbonate incorporation into intermediary metabolites potentially involved TCA activity, suggesting a link between CO2 fixation and TCA anaplerosis in these parasites [10]–[12]. However, subsequent analyses revealed that Plasmodium species lack typical anaplerotic enzymes such as pyruvate carboxylase and malic enzyme (providing OAA and malate, respectively, to the TCA cycle in other organisms), and also mitochondrial aspartate∶glutamate antiporter and aminotransferase [33]–[35], all integral to the malate shuttle in mammalian cells. Moreover, it was reported and apparently accepted dogma that there was no link between cytosolic glucose catabolism and mitochondrial TCA metabolism [8], [36], although the original paper has been recently retracted [9]. Therefore, to reconcile these reports we postulated and tested the hypothesis that PEPC, a CO2 fixing enzyme usually found in plants and bacteria, performs anaplerosis in erythrocytic P. falciparum by maintaining a mitochondrial malate shuttle and thereby performs an important metabolic function essential for the growth of the parasite.
Our demonstration that the generation of D10Δpepc null mutants was only possible in the presence of mM concentrations of malate provides strong evidence that PEPC is indeed a critical enzyme for providing malate via OAA for important metabolic reactions. The severe growth defect of D10Δpepc in the absence of malate and the partial rescue upon malate re-supplementation support this conclusion. D10Δpepc survival in the absence of added malate or fumarate is presumably mediated through salvage of these metabolites from the host erythrocyte, although clearly the availability is insufficient to rescue the severely limited growth of D10Δpepc in culture (Fig. 2A). These data suggest that PEPC could be essential for in vivo growth and viability of the parasites and so potentially it is a valid drug target. Efforts have been made to target PEPC in plants for development of a novel herbicide [32], [37] and our results suggest that such inhibitors are worth considering for their antimalarial potential. We have tested a commonly known PEPC inhibitor for its ability to inhibit P. falciparum D10 viability and indeed this was possible, although only at very high concentrations. This poor efficacy is likely to be a reflection of the charged nature of the compound and it would be interesting, therefore, to assess in future studies the efficacy against P. falciparum of uncharged PEP analogues similar to those previously described [37], [38].
The availability of D10Δpepc has allowed us to investigate in detail the contributions that the enzyme makes to intermediary metabolism in the erythrocytic stages of the parasite. Using only samples at 6% parasitaemia means that a large component of the metabolites detected were from the erythrocytes themselves, which effectively dilutes out labelling of the same metabolites present in the parasites. Thus, we validated the data that we obtained with low parasitaemia cultures by concentrating labelled D10-infected RBC using the MACS procedure [31] and this reassuringly provided similar data. More detailed analysis of intermediary metabolism will require development of methodology allowing the analysis of the D10Δpepc null mutants at higher parasitaemia.
The targeted metabolomics analysis of the parasite-infected RBC, uninfected RBC and spent culture medium after incubation with 13C-U-D-glucose and 13C-bicarbonate has provided definitive evidence that PEPC produces OAA for the generation of malate, fumarate and aspartate and feeds into mitochondrial TCA metabolism. The proportion of the metabolites malate, fumarate and aspartate 13C-labelled in the D10-infected RBC was maximally <20%, even in the samples obtained from D10-infected RBC concentrated to ∼90% parasitaemia. This contrasts to the glycolytic and PPP intermediates and lactate that were extensively labelled from 13C-U-D-glucose. There are several contributory factors for this. Other substrates could feed into the PEPC pathway, such as fumarate from purine salvage using aspartate derived from haemoglobin digestion, and also OAA generated by PEPC being converted to aspartate via AAT – the idea of the latter being important is supported by the differential inhibition of D10 and D10Δpepc by L-cycloserine. The involvement of fumarate in post-PEPC metabolism is further supported by its ability to partially rescue the growth defect of D10Δpepc (Fig. 2), which also suggests that fumarate might be salvaged from the host cell, and is in line with the finding that radiolabelled fumarate is converted into malate, aspartate and OAA in isolated parasites [39]. Similarly, exogenous malate clearly can be used and is probably salvaged from the host erythrocyte, which could account for the survival of D10Δpepc in the absence of malate supplementation. Malate and fumarate are known to be interconverted in erythrocytes [40], [41], thus either of them may be the main metabolite salvaged, and it is probable that they are also interconverted by fumarase in the mitochondrion as this is generally a reversible reaction. In addition, glutamine is another substrate feeding into mitochondrial TCA metabolism, as shown here and previously by MacRae and colleagues and Cobbold and colleagues for P. falciparum and also T. gondii [3], [6], [7], leading to generation of (unlabelled) fumarate and malate. Importantly, however, the relatively high amounts of these metabolites in the RBC themselves are all unlabelled (Fig. 5, Table S1) and effectively dilute the labelled proportion of these metabolites detected in infected RBC, even in the concentrated samples of D10-infected RBC (Table S2). The detection of 13C - malate and 13C-fumarate in the spent medium of the 13C -U-glucose and 13C-bicarbonate experiments with D10 parasites, and the absence of this from the spent medium of D10Δpepc and RBC, also strongly supports the proposed activity of PEPC (Tables S1, S3). The explanation for the release of these metabolites requires further analysis, although our quantitative data on metabolites in the spent media show clearly not only that D10Δpepc has a catabolic deficiency reflecting its growth defect but also that in both D10 and D10Δpepc the major route of glucose catabolism is to lactate and there is very little release of succinate, malate or fumarate (Table 1). Overall, the data suggest that, as expected, there is a complicated interplay in intermediary metabolism between the parasite and its host cell.
During asexual erythrocytic life, P. falciparum generates ATP through glycolysis and primarily yields lactate as metabolic end-product [36] and our data confirm this (Fig. 5; Table S1). Importantly, our study corroborates the recent finding [3], [6] that an oxidative TCA pathway is also operational. The presence of the 13C-2-labelled citrate suggests that acetyl-CoA is used in the production of citrate in the mitochondrion, despite the lack of a mitochondrial PDH [4] which in most eukaryotes catalyses acetyl-CoA production in mitochondria, and supports the recent suggestion that instead branched chain α-ketoacid dehydrogenase acts to effect the conversion [3], [6]. The additional occurrence of 13C-5-labelled citrate, which is absent in D10Δpepc, clearly supports malate being an additional entry point for mitochondrial TCA metabolism. This entry of malate into mitochondrial metabolism also allows for the transfer of reducing equivalents into the mtETC via MQO (Fig. 5).
The labelling pattern obtained with D10 and D10Δpepc-infected RBC with 13C-U-D-glucose, 13C-bicarbonate and 13C,15N-U-glutamine has allowed us to construct a scheme of the likely metabolic pathways operating in intermediary metabolism of asexual erythrocytic stages of P. falciparum and to postulate how the D10Δpepc mutants have adapted to facilitate survival in vitro. We propose (summarised in Fig. 7) that in wild type parasites the OAA resulting from PEPC activity is converted to malate that enters the mitochondrion where it is converted to OAA through the action of MQO. The presence of a cytosolic MDH is well established, a malate/ketoglutarate antiporter has been reported and the presence of MQO is confirmed [28], [42], [43]. The OAA generated probably can leave the mitochondrion, via a transporter postulated previously [39], [43], to complete an abbreviated, parasite-specific, malate shuttle. An alternative possibility postulated recently [6] is that it is citrate that exits the mitochondrion and this is subsequently converted to OAA and acetyl-CoA by citrate synthase II in a citrate lyase-like reaction, but there is no experimental evidence available to date to support this suggestion. Whichever of these happens, the shuttle leads to oxidation of cytosolic NADH, pivotal in maintaining cytosolic redox balance. The importance of this shuttle itself rather than the TCA cycle per se is emphasised by the finding that chemical ablation of the TCA cycle using fluoroacetate, which inhibits the aconitase step and thus is subsequent to OAA/citrate formation in the malate shuttle, had no inhibitory effect upon growth of erythrocytic stages of the parasite [3]. An additional factor is that the electrons transferred into the mitochondrion through malate are passed into the ubiquinone pool, which helps maintain mtETC. As the loss of PEPC resulting in no flux into malate from glucose does not affect the mtETC electrochemical gradient (Fig. S4) nor, apparently, the activity of complex III or DHOD (Figs. 4,5C, Table S7), it seems that the reduced contribution of MQO to the mtETC in these erythrocytic stages of D10Δpepc can be compensated for by other inputs. Our findings that there are elevated levels of both DHAP/glyceraldehyde 3-phosphate and glycerol 3-phosphate in the mutants and that they are as strongly labelled with 13C in the mutants as in D10, despite a reduced rate of glucose consumption, could mean that there is an increased flux from glycolytic triose phosphates to glycerol 3-phosphate. This is then potentially converted back to triose phosphate via the mitochondrial glycerol 3-phosphate dehydrogenase [44] feeding electrons into mtETC and so increasing flux, thereby compensating for the reduced flux through MQO. This diversion of glycolytic flux would also result in oxidation of NADH (in the conversion of triose phosphate to glycerol 3-phosphate) and so help to compensate for the lack of this occurring in the MDH step. Caution is required, however, in interpreting the data so far, as the elevated levels of trioses in the mutant parasites could reflect lower rates of their use rather than elevated flux to them. Studies in recent years on the mtETC in erythrocytic P. falciparum have indicated that it is more complex than had been understood previously [29], [30] and we suggest that the pepc KO line that we have generated and characterised could be an important tool with which to dissect this further.

Another result of the lack of malate generation from glucose and the consequent reduction in malate feeding into TCA metabolism would be a reduced flux through isocitrate dehydrogenase (IDH), considered a major source of the generation of NADPH [45]. The higher levels of ribulose 5-P/ribose 5-P in D10Δpepc, together with the heavy labelling of the metabolite despite the lower glucose utilisation, could reflect an increased flux through oxidative PPP, which would result in elevated generation of NADPH, compensating for the lower IDH flux. The finding that sedoheptulose 7-P although apparently elevated in abundance is less heavily labelled by 13C in D10Δpepc than in D10 itself could be due to the flux from ribulose 5-P to sedoheptulose 7-P (transketolase activity) not being similarly increased in D10Δpepc as is the flux through the oxidative PPP and also that sedoheptulose 7-P is utilised less, neither of which would necessarily impact upon redox homeostasis. Clearly, however, more analyses will be required to fully elucidate the adaptive changes of the mutant parasites.
Thus the modifications to metabolism that we have found in the D10Δpepc mutant have provided a better understanding and new insights into the crucial function of PEPC of P. falciparum. The OAA generated by PEPC feeds into pathways central to maintaining cytosolic redox balance (NADH oxidation by MDH) and the mtETC (via MQO), and generating NADPH (via IDH) for antioxidant and biosynthetic reactions. Together these are major contributions to parasite intermediary and redox metabolism, and our findings suggest that interfering with this could have major repercussions for parasite survival in vivo, and consequently PEPC inhibitors should be investigated as possible novel antimalarial agents.
Materials and Methods
Materials
WR99210 was a kind gift from Jacobus Pharmaceuticals (USA). 5-Fluorocytosine (5-FC) was obtained from The Royal Infirmary Pharmacy, Glasgow. Blasticidin was purchased from Invitrogen (UK). Plasmids pHH1 and pCC4 were kind gifts from Professor A. F. Cowman (The Walter and Eliza Hall Institute, Melbourne, Australia). Human blood was obtained from the Glasgow Blood Transfusion Services. Specific antibodies, used in western blot experiments, were generated in rabbits using recombinant expressed protein (Eurogentec, Belgium). Secondary antibodies were purchased from Promega. Anti-MDH and anti-AAT antibodies were a kind gift from Professor H. Balaram (Jawaharlal Nehru Centre for Advanced Scientific Research, Bangalore, India). DSM190 was a kind gift from Professor M.A. Phillips (Department of Pharmacology, University of Texas Southwestern Medical Center, Dallas, USA). [8-3H]-hypoxanthine (specific activity: 10–30 Ci/mmol) was from American Radiolabeled Chemicals. Other chemicals were purchased from Sigma UK unless otherwise stated.
Generation of P. falciparum pepc knockout construct
The plasmid pCC4 [21] contains two homologous regions, to both the 5′ and 3′ end of the target gene, and was used to generate the pCC4-Δpepc knockout construct. The primers used to amplify the 5′fragment (1–498 bp of the pepc ORF) and the 3′ fragment (2440–2939 bp of the pepc ORF) are shown in Table S8. The sequence of pCC4-Δpepc was verified (Eurofins MWG Operon).
Parasite culture, transfection, determination of IC50 values, parasite growth rate, and metabolite concentrations in spent media
P. falciparum D10 (Papua New Guinea) was cultured according to Trager and Jensen [13] in RPMI 1640 (Invitrogen, UK) containing 11 mM glucose, 0.5% (w/v) Albumax II (Invitrogen, UK), 200 µM hypoxanthine, 20 µg/ml gentamycin (PAA, UK) (designated routine medium) in human erythrocytes between 0.5% and 5% haematocrit. Parasite cultures were maintained under an atmosphere of reduced oxygen (1% oxygen, 3% CO2 and 96% nitrogen). Parasites were synchronised using sorbitol [46] and freed from erythrocytes using saponin [47]. Parasitaemia was determined using Giemsa-stained thin smears. Transfection of into P. falciparum erythrocytic stages was performed as described previously [48]. Transfectants were selected with 2.5 µg/ml blasticidin and they were maintained in either routine medium (which does not contain added malate) or routine medium supplemented with 5 mM malate (malate medium). Before cloning by limiting dilution according to Kirkman et al. [49], pCC4-Δpepc transfectants were subjected to negative selection with 1 µM 5-FC.
Growth rates of D10 and D10Δpepc were determined as described by Günther et al. [50]. For the rescue experiments, D10Δpepc were cultured in routine medium for 9 days, synchronised, diluted to 1% parasitaemia and cultured for 5 days in routine medium or routine medium supplemented with increasing concentrations of malate (0.5 mM to 5 mM), 5 mM aspartate, 5 mM fumarate, 5 mM succinate, 5 mM glycerol, 5 mM glutamine or 0.5 mM citrate. D10 were grown in the same media as controls. The cultures were diluted 1∶5 on day 3 and the parasitaemia of 1000 RBC was determined. The effects of L-cycloserine, DSM190 and atovaquone on the viability of the parasites were determined by measuring the incorporation of [3H]-hypoxanthine in the presence of increasing drug concentrations according to Desjardins et al. [51]. D10 and D10Δpepc were maintained in either routine (D10Δpepc for 9 days) or malate medium and synchronised two days prior to the experiment. IC50 values were calculated by nonlinear regression of the sigmoidal dose-response equation using GraFit version 7 (Erithacus, UK).
The concentrations of glucose, lacatate, glutamine, succinate, malate and fumarate in the spent media from RBC, D10 and D10Δpepc cultures were assayed using the following kits: glucose (Megazyme D-Glucose-HK), lactate (Megazyme D-/L-Lactic Acid (D-/L-Lactate) (Rapid)), glutamine (Megazyme L-Glutamine/Ammonia (Rapid)), malate (Megazyme L-Malic Acid), succinate (Megazyme Succinic Acid) and fumarate (Sigma Fumarate Assay Kit). All assays were carried out as per manufacturer's recommendation.
Targeted metabolomics using 13C-metabolites
Targeted metabolomics was performed with D10 and D10Δpepc cultured in routine medium for 9 days. After synchronisation, the RBC number of D10 and D10Δpepc infected cultures was determined using a Scepter 2.0 Handheld Automated Cell Counter (Millipore, UK) and adjusted to 6% parasitaemia and 1% haematocrit with culture medium containing 13C-labelled metabolites. Uninfected RBC of the same batch were used as control at the same haematocrit. Either glucose, bicarbonate or glutamine were replaced with 11 mM 13C-U - D-glucose (99%, CK Gas Products Ltd., UK), 0.2% 13C sodium bicarbonate (99%, CK Gas Products, UK) or 2 mM 13C, 15N-U-glutamine (99%, CK Gas Products, UK), respectively, and cultures were incubated for 28 h post-synchronisation. Spent medium was collected and metabolites were extracted in HPLC-grade chloroform∶methanol (1∶3 v/v). Metabolism of parasitized RBC was quenched to 4°C using a dry ice/70% ethanol bath [52]. Infected RBC were washed in ice-cold PBS and 3.5×108 cells were used for metabolite extraction using HPLC-grade chloroform∶methanol∶water (1∶3∶1 v/v/v). Samples were sonicated using a sonicating waterbath and then metabolites extracted for 1 h at 4°C under shaking conditions. The extracted samples were centrifuged at 16,000 g for 15 min at 4°C and the supernatants were transferred into glass vials and stored at −80°C until LC-MS analysis. Late stage D10 cultures obtained 28 hours post-sorbitol synchronisation were also enriched by magnetic purification using the Vario MACS magnetic system and LS columns (Miltenyi Biotec) [31], [53], [54]. This procedure resulted in an accumulation of infected RBC to 90% and metabolites of 3×107 infected RBC were analysed.
Liquid chromatography-mass spectrometry analyses
All samples were analysed using a liquid chromatography (LC) mass spectrometry system (MS) (Ultimate 3000 LC system, Dionex, UK, connected to an Exactive mass spectrometer, Thermo Scientific, Germany). The LC system was controlled using Chromeleon (Dionex, UK), and the MS was controlled by the software Xcalibur (Thermo Scientific, Germany), which allowed recording data for both positive and negative ionisation mode. Separation of analytes was achieved via a ZIC-pHILIC chromatography column (150 mm×4.6 mm×5 µm; Sequant, Uemå, Sweden) by a two solvent system consisting of solvent A: 20 mM ammonium carbonate and solvent B: acetonitrile. The chromatography conditions are summarised in Table S9. Both positive and negative mode spectra were acquired using 3 microscans over a scan range of 75.0–1200.0 m/z at a resolution of 50,000 (FWHM at 500 m/z) with an automatic gain control (AGC) target of 1×106 and a maximum inject time of 250 milliseconds. Specific instrument settings are supplied in the supporting information.
Data analysis
Data processing initially involved centroiding and converting vendor-specific raw LC-MS files into the mzXML open format. Chromatographic peaks in these files were extracted using the detection algorithm from XCMS [55] and stored in corresponding PeakML files [56]. Subsequently, PeakML files representing replicates were aligned and combined using mzMatch.R [57] after filtering out all peaks that were not reproducibly detected. The combined PeakML files were subjected to additional noise filtering, gap-filling and metabolite identification steps, using authentic metabolic standards for all metabolites of interest to ensure reliable identification. The proportion of each metabolite labelled with the stable isotopes was determined (see Figure S6) together with information on the overall abundance of metabolites present in the respective parasite extract samples or the culture medium at the time of harvesting of the parasites. The PeakML file obtained after filtering and identification was scanned for labelled metabolites using the PeakML.Isotope.TargettedIsotopes function of mzMatch-ISO [58]. Further details are provided in the supporting information.
Genomic DNA and Southern blotting
Genomic DNA was isolated from saponin isolated parasites using the QIAamp DNA Mini Kit (Qiagen, UK). 1–3 µg of genomic DNA, digested with SpeI, were separated on a 0.8% agarose gel and blotted onto positively charged nylon membrane (GE Healthcare, UK). The blots were pre-hybridized and probed at 55°C. Probes were labelled with thermostable alkaline phosphatase using the AlkPhos Direct Labeling and Detection System (VWR, UK) and successive washes were performed according to the manufacturer's instructions at 55°C. The blots were exposed to hypersensitive autoradiography film (VWR, UK) for 1–16 h.
Homology modelling of P. falciparum PEPC
Homology modelling of P. falciparum PEPC (PfPEPC) was performed using the I-TASSER server for protein structure and function prediction (http://zhanglab.ccmb.med.umich.edu/I-TASSER/) [59], [60]. The top three templates used for structure prediction of PfPEPC were the PEPC from E. coli (EcPEPC) (PDB entry 1JQN, [61]), PEPC from maize (ZmPEPC) (PDB entry 1JQO, [61]) and the archaeal-type PEPC (PDB entry 3ODM, [62]). Root mean square deviation (RMSD) values showed PfPEPC to be most similar to EcPEPC (0.56 Å), followed by ZmPEPC (2.49 Å) and archaeal-PEPC (3.80 Å). Based on these templates, five I-TASSER models were obtained for PfPEPC with C-scores ranging from −0.86 to −1.23. The structural alignment of the best model, that is, the model with the highest C-score (−0.86), with the top structural analogue, that is EcPEPC (0.56 Å), was generated using Magic Fit in Swiss-PDB Viewer 4.1.0 (http://www.expasy.org/spdbv/) [63]. This is shown in Fig. 3.
Statistical analyses
See supporting information for details.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. WHO, (2012) World Malaria Report 2012, World Health Organisation, Geneva, Switzerland, 59.
2. HaySI, GuerraCA, GethingPW, PatilAP, TatemAJ, et al. (2009) A world malaria map: Plasmodium falciparum endemicity in 2007. PLoS Med 6: e1000048.
3. MacRaeJI, DixonMW, DearnleyMK, ChuaHH, ChambersJM, et al. (2013) Mitochondrial metabolism of sexual and asexual blood stages of the malaria parasite Plasmodium falciparum. BMC Biol 11 : 67.
4. FothBJ, StimmlerLM, HandmanE, CrabbBS, HodderAN, et al. (2005) The malaria parasite Plasmodium falciparum has only one pyruvate dehydrogenase complex, which is located in the apicoplast. Mol Microbiol 55 : 39–53.
5. PeiY, TarunAS, VaughanAM, HermanRW, SolimanJM, et al. (2010) Plasmodium pyruvate dehydrogenase activity is only essential for the parasite's progression from liver infection to blood infection. Mol Microbiol 75 : 957–971.
6. CobboldSA, VaughanAM, LewisIA, PainterHJ, CamargoN, et al. (2013) Kinetic flux profiling elucidates two independent acetyl-CoA biosynthetic pathways in Plasmodium falciparum. J Biol Chem Oct 25 [Epub ahead of print].
7. MacRaeJI, SheinerL, NahidA, TonkinC, StriepenB, et al. (2012) Mitochondrial metabolism of glucose and glutamine is required for intracellular growth of Toxoplasma gondii. Cell Host Microbe 12 : 682–692.
8. OlszewskiKL, MatherMW, MorriseyJM, GarciaBA, VaidyaAB, et al. (2010) Branched tricarboxylic acid metabolism in Plasmodium falciparum. Nature 466 : 774–778.
9. OlszewskiKL, MatherMW, MorriseyJM, GarciaBA, VaidyaAB, et al. (2013) Retraction: Branched tricarboxylic acid metabolism in Plasmodium falciparum. Nature 497 : 652.
10. ShermanIW, TingIP (1966) Carbon dioxide fixation in malaria (Plasmodium lophurae). Nature 212 : 1387–1388.
11. ShermanIW, TingIP (1968) Carbon dioxide fixation in malaria. II. Plasmodium knowlesi (monkey malaria). Comp Biochem Physiol 24 : 639–642.
12. NagarajanK (1968) Metabolism of Plasmodium berghei. 3. Carbon dioxide fixation and role of pyruvate and dicarboxylic acids. Exp Parasitol 22 : 33–42.
13. TragerW, JensenJB (1976) Human malaria parasites in continuous culture. Science 193 : 673–675.
14. McDanielHG, SiuPM (1972) Purification and characterization of phosphoenolpyruvate carboxylase from Plasmodium berghei. J Bacteriol 109 : 385–390.
15. BlumJJ, GinsburgH (1984) Absence of alpha-ketoglutarate dehydrogenase activity and presence of CO2-fixing activity in Plasmodium falciparum grown in vitro in human erythrocytes. J Protozool 31 : 167–169.
16. HaywardRE (2000) Plasmodium falciparum phosphoenolpyruvate carboxykinase is developmentally regulated in gametocytes. Mol Biochem Parasitol 107 : 227–240.
17. KrungkraiSR, SuraveratumN, RochanakijS, KrungkraiJ (2001) Characterisation of carbonic anhydrase in Plasmodium falciparum. Int J Parasitol 31 : 661–668.
18. IzuiK, MatsumuraH, FurumotoT, KaiY (2004) Phosphoenolpyruvate carboxylase: a new era of structural biology. Annu Rev Plant Biol 55 : 69–84.
19. O'LearyB, ParkJ, PlaxtonWC (2011) The remarkable diversity of plant PEPC (phosphoenolpyruvate carboxylase): recent insights into the physiological functions and post-translational controls of non-photosynthetic PEPCs. Biochem J 436 : 15–34.
20. West-EberhardMJ, SmithJA, WinterK (2011) Plant science. Photosynthesis, reorganized. Science 332 : 311–312.
21. MaierAG, BraksJA, WatersAP, CowmanAF (2006) Negative selection using yeast cytosine deaminase/uracil phosphoribosyl transferase in Plasmodium falciparum for targeted gene deletion by double crossover recombination. Mol Biochem Parasitol 150 : 118–121.
22. PengY, CaiJ, WangW, SuB (2012) Multiple inter-kingdom horizontal gene transfers in the evolution of the phosphoenolpyruvate carboxylase gene family. PLoS One 7: e51159.
23. SvenssonP, BläsingOE, WesthoffP (2003) Evolution of C4 phosphoenolpyruvate carboxylase. Arch Biochem Biophys 414 : 180–188.
24. WesthoffP, GowikU (2004) Evolution of c4 phosphoenolpyruvate carboxylase. Genes and proteins: a case study with the genus Flaveria. Ann Bot 93 : 13–23.
25. TreeckM, SandersJL, EliasJE, BoothroydJC (2011) The phosphoproteomes of Plasmodium falciparum and Toxoplasma gondii reveal unusual adaptations within and beyond the parasites' boundaries. Cell Host Microbe 10 : 410–419.
26. JanskiAM, CornellNW (1981) Inhibition by cycloserine of mitochondrial and cytosolic aspartate aminotransferase in isolated rat hepatocytes. Biochem J 194 : 1027–1030.
27. GujjarR, El MazouniF, WhiteKL, WhiteJ, CreasonS, et al. (2011) Lead optimization of aryl and aralkyl amine-based triazolopyrimidine inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with antimalarial activity in mice. J Med Chem 54 : 3935–3949.
28. VaidyaAB, MatherMW (2009) Mitochondrial evolution and functions in malaria parasites. Annu Rev Microbiol 63 : 249–267.
29. EsserL, QuinnB, LiYF, ZhangM, ElberryM, et al. (2004) Crystallographic studies of quinol oxidation site inhibitors: a modified classification of inhibitors for the cytochrome bc(1) complex. J Mol Biol 341 : 281–302.
30. BartonV, FisherN, BiaginiGA, WardSA, O'NeillPM (2010) Inhibiting Plasmodium cytochrome bc1: a complex issue. Curr Opin Chem Biol 14 : 440–446.
31. RibautC, BerryA, ChevalleyS, ReybierK, MorlaisI, et al. (2008) Concentration and purification by magnetic separation of the erythrocytic stages of all human Plasmodium species. Malar J 7 : 45.
32. JenkinsCL (1989) Effects of the phosphoenolpyruvate carboxylase inhibitor 3,3-dichloro-2-(dihydroxyphosphinoylmethyl)propenoate on photosynthesis: C(4) selectivity and studies on C(4) photosynthesis. Plant Physiol 89 : 1231–1237.
33. GardnerMJ, HallN, FungE, WhiteO, BerrimanM, et al. (2002) Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419 : 498–511.
34. GibalaMJ, YoungME, TaegtmeyerH (2000) Anaplerosis of the citric acid cycle: role in energy metabolism of heart and skeletal muscle. Acta Physiol Scand 168 : 657–665.
35. JitrapakdeeS, Vidal-PuigA, WallaceJC (2006) Anaplerotic roles of pyruvate carboxylase in mammalian tissues. Cell Mol Life Sci 63 : 843–854.
36. OlszewskiKL, LlinásM (2011) Central carbon metabolism of Plasmodium parasites. Mol Biochem Parasitol 175 : 95–103.
37. García-AllesLF, ErniB (2002) Synthesis of phosphoenol pyruvate (PEP) analogues and evaluation as inhibitors of PEP-utilizing enzymes. Eur J Biochem 269 : 3226–3236.
38. IzuiK, MatsudaY, KameshitaI, KatsukiH, WoodsAE (1983) Phosphoenolpyruvate carboxylase of Escherichia coli. Inhibition by various analogs and homologs of phosphoenolpyruvate. J Biochem 94 : 1789–1795.
39. BulusuV, JayaramanV, BalaramH (2011) Metabolic fate of fumarate, a side product of the purine salvage pathway in the intraerythrocytic stages of Plasmodium falciparum. J Biol Chem 286 : 9236–9245.
40. TanakaKR, ValentineWN (1961) Fumarase activity of human leukocytes and erythrocytes. Blood 17 : 328–333.
41. SimpsonRJ, BrindleKM, CampbellID (1982) Spin ECHO proton NMR studies of the metabolism of malate and fumarate in human erythrocytes. Dependence on free NAD levels. Biochim Biophys Acta 721 : 191–200.
42. TripathiAK, DesaiPV, PradhanA, KhanSI, AveryMA, et al. (2004) An alpha-proteobacterial type malate dehydrogenase may complement LDH function in Plasmodium falciparum. Cloning and biochemical characterization of the enzyme. Eur J Biochem 271 : 3488–3502.
43. NozawaA, FujimotoR, MatsuokaH, TsuboiT, TozawaY (2011) Cell-free synthesis, reconstitution, and characterization of a mitochondrial dicarboxylate-tricarboxylate carrier of Plasmodium falciparum. Biochem Biophys Res Commun 414 : 612–617.
44. van DoorenGG, StimmlerLM, McFaddenGI (2006) Metabolic maps and functions of the Plasmodium mitochondrion. FEMS Microbiol Rev 30 : 596–630.
45. WrengerC, MüllerS (2003) Isocitrate dehydrogenase of Plasmodium falciparum. Eur J Biochem 270 : 1775–1783.
46. LambrosC, VanderbergJP (1979) Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol 65 : 418–420.
47. UmlasJ, FallonJN (1971) New thick-film technique for malaria diagnosis. Use of saponin stromatolytic solution for lysis. Am J Trop Med Hyg 20 : 527–529.
48. WuY, SifriCD, LeiHH, SuXZ, WellemsTE (1995) Transfection of Plasmodium falciparum within human red blood cells. Proc Natl Acad Sci USA 92 : 973–977.
49. KirkmanLA, SuXZ, WellemsTE (1996) Plasmodium falciparum: isolation of large numbers of parasite clones from infected blood samples. Exp Parasitol 83 : 147–149.
50. GüntherS, WallaceL, PatzewitzEM, McMillanPJ, StormJ, et al. (2007) Apicoplast lipoic acid protein ligase B is not essential for Plasmodium falciparum. PLoS Pathog 3: e189.
51. DesjardinsRE, CanfieldCJ, HaynesJD, ChulayJD (1979) Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob Agents Chemother 16 : 710–718.
52. De SouzaDP, SaundersEC, McConvilleMJ, LikićVA (2006) Progressive peak clustering in GC-MS metabolomic experiments applied to Leishmania parasites. Bioinformatics 22 : 1391–1396.
53. TrangDT, HuyNT, KariuT, TajimaK, KameiK (2004) One-step concentration of malarial parasite-infected red blood cells and removal of contaminating white blood cells. Malar J 3 : 7.
54. AhnSY, ShinMY, KimYA, YooJA, KwakDH, et al. (2008) Magnetic separation: a highly effective method for synchronization of cultured erythrocytic Plasmodium falciparum. Parasitol Res 102 : 1195–1200.
55. TautenhahnR, BöttcherC, NeumannS (2008) Highly sensitive feature detection for high resolution LC/MS. BMC Bioinformatics 9 : 504.
56. ScheltemaRA, JankevicsA, JansenRC, SwertzMA, BreitlingR (2011) PeakML/mzMatch: a file format, Java library, R library, and tool-chain for mass spectrometry data analysis. Anal Chem 83 : 2786–2793.
57. JankevicsA, MerloME, De VriesM, VonkRJ, TakanoE, et al. (2012) Separating the wheat from the chaff: a prioritisation pipeline for the analysis of metabolomics datasets. Metabolomics 8 : 29–36.
58. ChokkathukalamA, JankevicsA, CreekDJ, AchcarF, BarrettMP, et al. (2013) mzMatch-ISO: an R tool for the annotation and relative quantification of isotope-labelled mass spectrometry data. Bioinformatics Oxford England 29 : 281–283.
59. ZhangY (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 23 9 : 40.
60. RoyA, KucukuralA, ZhangY (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5 : 725–738.
61. MatsumuraH, XieY, ShirakataS, InoueT, YoshinagaT, et al. (2002) Crystal structures of C4 form maize and quaternary complex of E. coli phosphoenolpyruvate carboxylases. Structure 10 : 1721–1730.
62. DharmarajanL, KraszewskiJL, MukhopadhyayB, DuntenPW (2011) Structure of an archaeal-type phosphoenolpyruvate carboxylase sensitive to inhibition by aspartate. Proteins 79 : 1820–1829.
63. GuexN, PeitschMC (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18 : 2714–2723.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 1
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Lyme Disease: Call for a “Manhattan Project” to Combat the Epidemic
- Origin, Migration Routes and Worldwide Population Genetic Structure of the Wheat Yellow Rust Pathogen f.sp.
- IFNγ/IL-10 Co-producing Cells Dominate the CD4 Response to Malaria in Highly Exposed Children
- Human and Plant Fungal Pathogens: The Role of Secondary Metabolites