
Targeted Cytotoxic Therapy Kills Persisting HIV Infected Cells During ART
Antiretroviral therapy (ART) can reduce HIV levels in plasma to undetectable levels, but rather little is known about the effects of ART outside of the peripheral blood regarding persistent virus production in tissue reservoirs. Understanding the dynamics of ART-induced reductions in viral RNA (vRNA) levels throughout the body is important for the development of strategies to eradicate infectious HIV from patients. Essential to a successful eradication therapy is a component capable of killing persisting HIV infected cells during ART. Therefore, we determined the in vivo efficacy of a targeted cytotoxic therapy to kill infected cells that persist despite long-term ART. For this purpose, we first characterized the impact of ART on HIV RNA levels in multiple organs of bone marrow-liver-thymus (BLT) humanized mice and found that antiretroviral drug penetration and activity was sufficient to reduce, but not eliminate, HIV production in each tissue tested. For targeted cytotoxic killing of these persistent vRNA+ cells, we treated BLT mice undergoing ART with an HIV-specific immunotoxin. We found that compared to ART alone, this agent profoundly depleted productively infected cells systemically. These results offer proof-of-concept that targeted cytotoxic therapies can be effective components of HIV eradication strategies.
Published in the journal:
. PLoS Pathog 10(1): e32767. doi:10.1371/journal.ppat.1003872
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1003872
Summary
Antiretroviral therapy (ART) can reduce HIV levels in plasma to undetectable levels, but rather little is known about the effects of ART outside of the peripheral blood regarding persistent virus production in tissue reservoirs. Understanding the dynamics of ART-induced reductions in viral RNA (vRNA) levels throughout the body is important for the development of strategies to eradicate infectious HIV from patients. Essential to a successful eradication therapy is a component capable of killing persisting HIV infected cells during ART. Therefore, we determined the in vivo efficacy of a targeted cytotoxic therapy to kill infected cells that persist despite long-term ART. For this purpose, we first characterized the impact of ART on HIV RNA levels in multiple organs of bone marrow-liver-thymus (BLT) humanized mice and found that antiretroviral drug penetration and activity was sufficient to reduce, but not eliminate, HIV production in each tissue tested. For targeted cytotoxic killing of these persistent vRNA+ cells, we treated BLT mice undergoing ART with an HIV-specific immunotoxin. We found that compared to ART alone, this agent profoundly depleted productively infected cells systemically. These results offer proof-of-concept that targeted cytotoxic therapies can be effective components of HIV eradication strategies.
Introduction
ART is a lifesaving and effective means to control HIV infection [1]. However, the persistent nature of this infection requires lifelong adherence to daily ART dosing [2]–[4]. This viral persistence, the cumulative costs of ART, adverse events associated with long-term ART and the constant threat of emergence of drug-resistant viral variants have led researchers to pursue HIV eradication therapies that will result in a viral rebound-free interruption of therapy [2]–[4]. Towards this goal, “kick and kill” HIV eradication strategies are being developed [5]. While interventions that can induce expression of latent HIV (e.g. histone deacetylase inhibitors and protein kinase activators) will function as the “kick” [2]–[4], it is important to note that “kill” strategies cannot rely on the induction of virus expression in latently infected cells to result in cell death [6]. Therefore, candidate “kill” agents, such as immunotoxins, are being considered for incorporation into HIV eradication protocols [7], [8]. Immunotoxins are recombinant or biochemically linked bi-functional proteins that combine the effector domain of a protein toxin with the targeting specificity of an antibody or ligand [8]–[13]. Soon after HIV was identified as the causative agent of AIDS, several immunotoxins were described as potential therapeutics for HIV [8], [10]. These interventions had effector domains from plant and bacterial protein toxins and targeting moieties against either the HIV Env glycoprotein (gp120, gp41) or cellular markers including CD4, CD25 or CD45RO [14]–[21]. The immunotoxin we chose to evaluate for in vivo efficacy herein, 3B3-PE38, combines the 3B3 scFv which targets the conserved CD4 binding site of HIV-1 gp120 with the Pseudomonas exotoxin A (PE38) effector domain [22].
The fact that tissue specific effects of ART on HIV persistence are poorly understood in patients [23] meant that there was no baseline for characterizing the systemic effects of an immunotoxin on HIV persistence during ART. Therefore, it was essential that we first fully characterize the systemic impact of ART on HIV persistence cells in vivo. To do so, we sought to analyze HIV persistence during ART in a comprehensive panel of tissues and organs over time – a study that cannot be performed in human subjects. For this reason, we used BLT humanized mice [24], the most advanced, validated, and robust small animal model available for this purpose [25], [26]. The process of bioengineering BLT mice results in systemic dissemination of human hematopoietic cells throughout the animal [24], [27]. This phenotype facilitates the simultaneous analysis of multiple tissues throughout the body. The systemic effects of HIV infection on the BLT mouse human immune system (e.g., CD4+ T cell depletion and immune activation) recapitulate what is observed in HIV-infected patients [28]–[32]. Once the tissue-specific parameters of HIV persistence during ART were established, we incorporated 3B3-PE38 into the therapeutic regimen. Systemic analyses revealed that 3B3-PE38 treatment during ART reduced the number of HIV RNA producing cells to levels significantly lower than those achieved with ART alone. This observation demonstrates that immunotoxins can play a critical role in successful HIV eradication strategies.
Results
Outcomes of ART in HIV infected BLT mice
For this study, BLT mice were treated with a triple combination antiretroviral drug regimen that included the nucleotide reverse transcriptase inhibitor tenofovir disoproxil fumarate (TDF), the nucleoside reverse transcriptase inhibitor emtricitabine (FTC) and the integrase inhibitor raltegravir (RAL). This ART regimen was chosen because of its robust pharmacodynamic properties [33], superior efficacy in humans [34] and its efficacy in BLT mice [35].
In human peripheral blood [36], [37] and BLT mice (Fig. 1A), the first few weeks of ART are characterized by a rapid decline in plasma viremia (vRNA) followed by a plateau phase concomitant with a recovery of peripheral blood CD4+ T cells levels (Fig. 1B). We also observed that the blood cell-associated vDNA levels remained stable when compared to plasma vRNA levels during this treatment period (Fig. 1A) (Day −3 vs. Day42, p = 0.63, signed rank test), as seen in patients on this same ART regimen [38]. Furthermore, the robust suppression of plasma viremia by ART is consistent with the presence of each of the dosed antiretrovirals in the plasma of treated BLT mice: tenofovir (Fig. 1C), emtricitabine (Fig. 1D) and raltegravir (Fig. 1E). Such peripheral blood analyses are readily performed in both humans and BLT mice; however, the ability to simultaneously examine multiple tissues throughout the course of ART is not possible in patients. Therefore, we evaluated antiretroviral drug penetration as well as the impact of ART on vRNA production in multiple organs in BLT mice. We determined drug levels in the thymic organoid, spleen, liver, lung, terminal ileum and rectum of BLT mice. Importantly, each of the three drugs was detected in each matrix analyzed (Fig. 1C–E). To facilitate direct comparisons of drug levels between tissues and plasma, the data are presented as ng/g and ng/ml, respectively [39]. Overall, the tenofovir, FTC and RAL levels in BLT mice were ∼1.6 µM, ∼0.4 µM and ∼0.05 µM, respectively (Fig. 1C–E). Each of these values is higher than the EC50 for that drug in HIV-1 infected peripheral blood mononuclear cells (TDF: 0.005 µM; FTC: 0.01 µM; and RAL: 0.001 µM) [39]–[41]. The extensive antiretroviral drug penetration observed in BLT mouse organs during ART led us to examine the systemic impact of ART on HIV production.
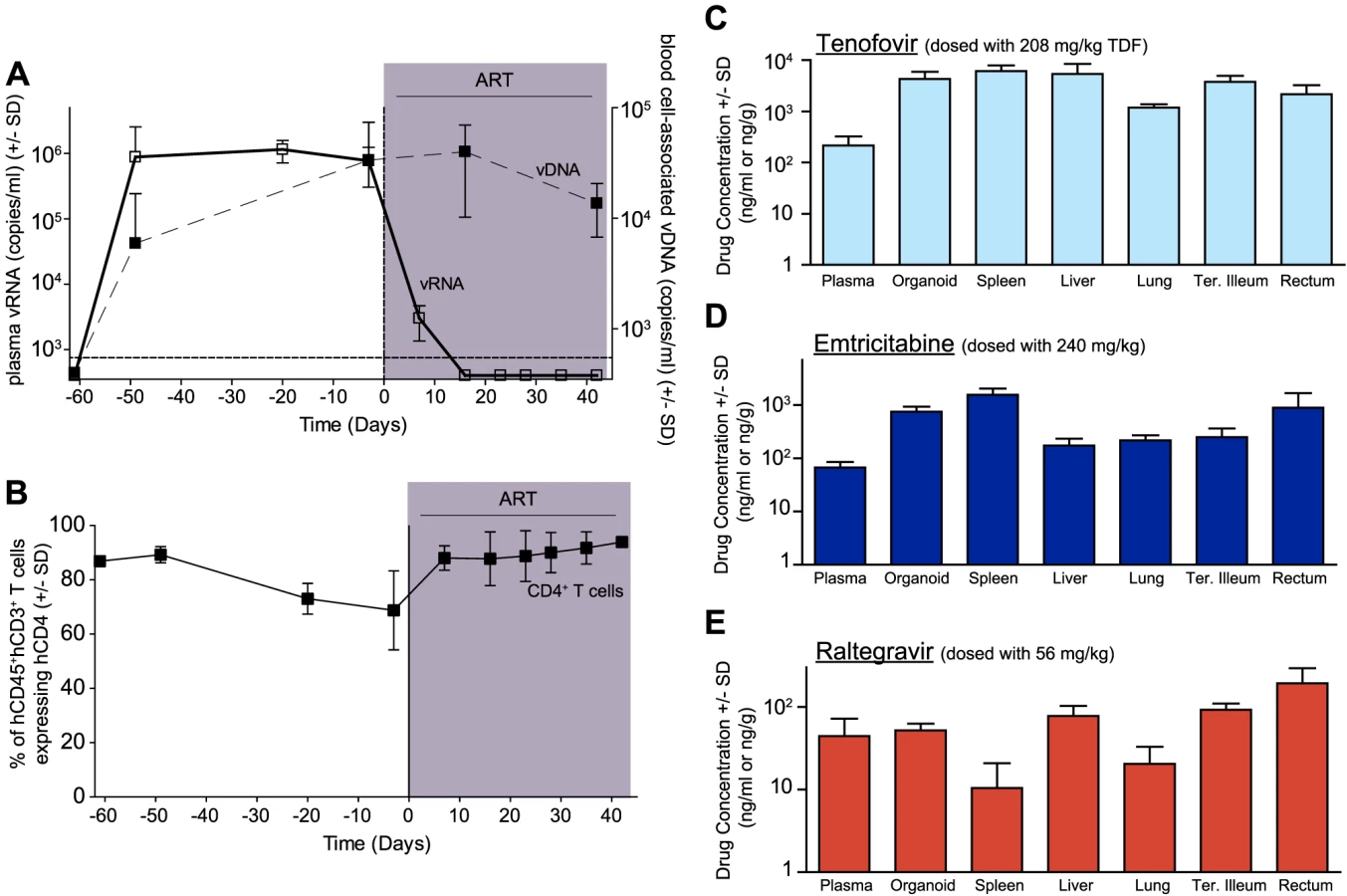
We used two different approaches to measure the systemic reduction in vRNA by ART. First, we used in situ hybridization to quantitate the number of individual cells producing vRNA in the human thymic organoid, spleen, lymph nodes, liver, lung, terminal ileum and rectum of HIV infected BLT mice receiving or not receiving ART. We found that antiretroviral penetration and activity in these tissues was sufficient to profoundly reduce the number of cells producing vRNA in all tissues (Fig. 2). However, consistent with the limited human tissue data available [42], vRNA producing cells remained detectable during therapy in all tissues analyzed (Fig. 2). We also used RT-PCR to quantitate cell-associated vRNA levels in the bone marrow, human thymic organoid, spleen, lymph nodes, liver lung, intestines and peripheral blood cells of BLT mice given ART for 0–64 days. We observed a rapid decline in cell-associated vRNA levels that plateaued by Day 28 of ART in all tissues analyzed (Fig. 3A). The longitudinal vRNA data was analyzed using two different regression models (Lowess and cubic). The observed reductions in cell-associated vRNA in each tissue tested indicate that ART penetrates these tissues sufficiently to significantly reduce viremia in a related manner in each tissue as illustrated in the plots depicting the individual Lowess curves graphed together (Fig. 3B). We found that a Lowess curve generated with cell-associated vRNA data for all tissues together (Fig. 3C) is very similar to the reduction in plasma viremia observed specifically in human peripheral blood soon after ART initiation [36]. We also noted significant (p<0.001) differences in each tissue, including peripheral blood, when we compared cell-associated vRNA levels from mice in an untreated state versus those from treated mice with vRNA levels within the plateau stage (Fig. 4). These similarities in vRNA reduction observed between human peripheral blood and all of the examined tissues from BLT mice during the first months following ART initiation suggest that blood is a reasonable surrogate for the impact of ART throughout the body.



Impact of HIV-specific immunotoxin on residual vRNA production during ART
Since neither cytopathic effects of HIV expression or antiviral immune responses are sufficient to deplete cells expressing virus [6], these cells represent potential targets for cytotoxic HIV-specific immunotherapies such as the immunotoxin 3B3-PE38. This immunotoxin has been shown to be active against HIV-1-infected primary CD4+ T cells, macrophages and thymocytes [43], [44]. Knowing that the effect of ART on the vRNA production reached a plateau by Day 28 of treatment led us to ask whether targeted killing of the persisting HIV producing cells during this plateau phase would augment the vRNA decline during ART [8]. As with most anti-HIV agents, monotherapy with immunotoxins is not clinically viable [8]; therefore, we quantitated the systemic impact of 3B3-PE38 on HIV persistence during ART (7 IP doses on alternate days beginning on Day 28 of ART). Compared to ART alone, ART plus 3B3-PE38 reduced the cell-associated vRNA by 3.2 logs (>1,000-fold) in the bone marrow (Fig. 5A). Complementing ART with 3B3-PE38 also led to a reduction in the persisting cell-associated vRNA levels in the human thymic organoid, spleen, lymph nodes, liver lung, intestines and peripheral blood cells (range: 0.4 to 1.5 logs) (Fig. 5A). When all tissues were analyzed together, ART plus 3B3-PE38 reduced cell-associated vRNA levels in all organs combined by an additional 0.8 logs compared to ART alone (p<0.001) (Fig. 5B).

Following the initiation of ART in humans, there is a multi-phasic decay in viremia that reflects the rate of turnover of productively infected cells [36], [45]. Most models of viral dynamics assume that the drugs completely block new infections of susceptible cells, with the observed decay reflecting the decay rate of cells infected prior to the initiation of ART [36], [46]. Because this decay is constantly progressing, it is likely that the number of vRNA producing cells would decline over time during ART in the absence of immunotoxin treatment. However, given that we observed an overall 0.8 log reduction in cell-associated vRNA levels beyond the effect of ART alone (Fig. 5B), we used in situ hybridization to examine whether there was an associated reduction in the number of cells producing vRNA. We found that complementation of ART with 3B3-PE38 led to a 1.8 log reduction in the numbers of vRNA producing cells throughout the body (p = 0.007) (Fig. 5C). This profound reduction in the number of vRNA producing cells provides a mechanistic explanation for the rapid reduction in tissue cell-associated vRNA levels relative to the ART only experimental group.
Discussion
Despite the lifesaving benefits of ART in HIV patients, relatively little is known regarding the organ specific impact of this therapy on HIV production and persistence [23]. Improving our knowledge of the systemic effects of ART is a critical step in the development of successful HIV eradication therapies. To address this need, we characterized the impact of ART by analyzing both drug penetration and HIV production in different tissues throughout BLT humanized mice. We found that ART penetration into multiple organ systems is sufficient to significantly reduce the number of cells producing HIV, as well as cell-associated vRNA levels, throughout the body. However, HIV persists during ART in every organ analyzed.
HIV-infected cells persisting during ART represent suitable targets for cytotoxic HIV-specific immunotherapies such as the 3B3-PE38 immunotoxin. Since such therapies target HIV proteins expressed by infected cells, their efficacy requires active virus transcription. Therefore there is no predicted impact of immunotoxin treatment on the size of the transcriptionally silent latent HIV reservoir. For this reason, the HIV latent reservoir was not quantitated in this study and our conclusions are based on the quantitation of cell-associated vRNA and vRNA-producing cell numbers. Specifically, we demonstrate that 3B3-PE38 kills vRNA producing cells throughout the body such that the reduction of vRNA levels during combined therapy is more rapid than with ART only (Fig. 6). A recent report described the ability of new broadly neutralizing monoclonal antibodies to suppress HIV-1 rebound after termination of ART [47] which led us to consider the possibility that the observed activity of the immunotoxin was due in part to neutralization by the 3B3 scFv moiety. In the Horwitz, et al. study, the amount of IgG (10.5 mg/injection) administered was over 2000 fold higher than our doses of 3B3-PE38 (0.005 mg/injection). Thus, the circulating 3B3-PE38 levels in our study could not reach the levels required for neutralization by 3B3 [48]. It is therefore unlikely under our experimental conditions that neutralization by the 3B3 scFv moiety of the immunotoxin accounts for the significant 3B3-PE38-mediated reduction we observed in tissue cell-associated vRNA levels.

Comparing and contrasting data from BLT mice with data from patients and NHP is essential to understanding the predictive nature of our model. The most notable difference between our study and those in patients and NHP is the duration of ART. Our study examined the impact of ART in BLT mice over ∼2 months, while patient and macaque studies have examined cell-associated vRNA levels during several years of ART [49], [50]. These differences in ART duration prevent direct comparisons of the impact of long-term ART on persistent vRNA production within these experimental platforms. Despite this constraint, we can compare data across all three experimental platforms for consistency. For example, we found that our data showing the continued presence vRNA in BLT mouse peripheral blood and tissue cells during ART are consistent with the continued presence of vRNA within patient peripheral blood cells, ileal biopsy cells and rectal biopsy cells from patients treated for a median of 12.5 months with ART [50] and within macaque cells from throughout the body after 26 weeks on ART [49]. In addition to future studies incorporating longer treatment windows in BLT mice, it will be important to determine whether the vRNA plateau phase reached after 28 days is directly comparable to that seen in humans on long term ART. For this purpose, incorporation of an additional drug into the ART regimen could be tested. If the plateau state in BLT mice is comparable to the human situation during long-term ART, then future studies using more sensitive assay for HIV-1 RNA in conjunction with ART intensification should not detect any additional reduction in the steady state level of viremia during this timeframe.
The results presented here provide proof of concept for targeted cytotoxic therapy as a successful complement to ART for the depletion of persisting HIV infected cells. In addition to the 3B3-PE38, alternative immunotoxins with different effector domains and targeting moieties are available [8]–[10]. This is important because: (i) treatments with different cytotoxic interventions during ART will likely be required to completely eradicate HIV producing cells due to the immunogenicity of the immunotoxin molecules [8]–[13], [51], [52], the need to target the diverse cell types that are productively infected with HIV [2]–[4], and (iii) the likely selection of viral mutants resistant to any single immunotoxin [53]. Beyond immunotoxins, alternative targeted cytotoxic therapy strategies developed for cancer treatment could also be adapted to generate comprehensive anti-HIV cytotoxic therapy regimens [54], [55]. Possibilities include: aptamer—toxin conjugates; radioimmunotherapies; antibody—cytotoxic drug conjugates; targeted cytolytic viruses; targeted delivery of a cytotoxic peptides; and adoptive immunotherapy with ex vivo expanded natural or genetically modified T cells [54], [55]. Importantly, the model system developed herein to quantitate the effects of 3B3-PE38 can be used to determine the in vivo efficacy of any of these other strategies alone or in combinations.
In summary, we determined that the systemic penetration of antiretroviral drugs into different organs throughout the body was sufficient to reduce cell-associated vRNA levels ART alone was unable to completely eliminate vRNA expressing cells in tissues. This observation provided a quantitative framework for the systemic in vivo efficacy evaluation of interventions to destroy HIV producing cells during therapy. Within this framework we were able to demonstrate that persistent HIV producing cells systemically present during ART were significantly depleted by an Env-targeting immunotoxin. Such targeted cytotoxic interventions may prove to be critically important components of an effective HIV eradication strategy.
Materials and Methods
Ethics statement
All animal experiments were conducted following NIH guidelines for housing and care of laboratory animals and in accordance with The University of North Carolina at Chapel Hill (UNC Chapel Hill) regulations after review and approval by the UNC Chapel Hill Institutional Animal Care and Use Committee (permit number 12-171).
Generation of BLT humanized mice
BLT mice were prepared essentially as previously described [27]. Briefly, thy/liv implanted NOD/SCID-gamma chain null mice (NSG; The Jackson Laboratories, Stock #5557) were transplanted with autologous human fetal liver CD34+ cells (Advanced Bioscience Resources) and monitored for human reconstitution in peripheral blood by flow cytometry [24], [27], [32], [35]. All BLT mice (n = 40) used for these experiments were characterized for human immune system reconstitution prior to HIV infection. Their peripheral blood contained an average of 57% (+/−15 SD) human CD45+ cells of which 58% (+/−22 SD) were human T cells. Of the human T cells, 81% (+/−6 SD) were human CD4+ T cells.
Analysis of HIV infection of BLT mice
Infection of BLT mice with HIV-1JRCSF was monitored in peripheral blood by determining plasma levels of vRNA as described (level of detection = 750 copies per mL of plasma; 40 µl mouse plasma sample size) using one-step reverse transcriptase real-time PCR [ABI custom TaqMan Assays-by-Design] [35], [56]–[58]. Tissues were harvested and cells isolated as we have previously described for RNA isolation or flow cytometric analysis [24], [32]. Flow cytometry data were collected using a BD FACSCanto cytometer and analyzed using BD FACSDiva software (v. 6.1.3).
For HIV RNA in situ hybridization (ISH), tissues were fixed overnight in 4% paraformaldehyde at 4°C and then transferred to 70% ethanol. Tissues were then embedded in paraffin for sectioning. After deparaffinization with xylene and rehydration through graded ethanols, tissue sections were treated with HCl, triethanolamine, digitonin and 4 µg/mL Proteinase K as previously described [59]. After acetylation with acetic anhydride and dehydration, tissue sections were hybridized at 45°C overnight with a 35S labeled antisense riboprobe and 0.5 mM aurintricarboxylic acid in the hybridization mix. After extensive washes and ribonuclease treatment, tissue sections were dehydrated, coated in Ilford K5 emulsion diluted with glycerol and ammonium acetate, exposed at 4°C for 7–14 days, and developed and fixed per manufacturer's instructions. They were stained with Hematoxylin, dehydrated, and mounted with Permount. Non-infected control tissues and sense riboprobe controls were analyzed when appropriate. Photographic images using epifluorescence were taken with a digital camera and the tiffs were analyzed for the area of the sections and the area occupied by silver grains using Photoshop with Fovea Pro. Section weights were estimated from their 5-micron thickness and their area. These methods have been extensively reviewed [59].
Treatment of HIV infection of BLT mice
ART in infected BLT mice consisted of three antiretrovirals administered daily via intraperitoneal injection: tenofovir disoproxil fumarate (TDF; 208 mg/kg), emtricitabine (FTC; 240 mg/kg body weight) and raltegravir (RAL; 56 mg/kg) [35]. The gp120-targeting immunotoxin 3B3(Fv)-PE38 [22] (here referred to as 3B3-PE38) was administered intraperitoneally every other day beginning on Day 28 of ART for a total of 7 doses: the first 4 were 1 µg/25 g and the last 3 were 5 µg/25 g.
Analytical methods for drug quantification
Plasma and snap frozen tissue samples from BLT mice receiving daily ART were analyzed for drug concentrations. Quantification of FTC, tenofovir (TFV) and RAL plasma concentrations was performed by protein precipitation and LC-MS/MS analysis. 10 µl of each stored plasma sample was mixed with 75 µL of methanol containing the isotopically-labeled internal standards (13C TFV and 13C 15N FTC). Following vortexing and centrifugation, the supernatant was removed and evaporated to dryness. The extracts were reconstituted with 1 mM ammonium phosphate prior to LC-MS/MS analysis. All compounds were eluted from a Phenomenex Synergi Polar-RP (4.6×50 mm, 4 µm particle size) analytical column. An API-5000 triple quadrupole mass spectrometer (AB Sciex, Foster City, CA) was used to detect the analytes. Data were collected using AB Sciex Analyst Chromatography Software (Analyst version 1.6.1). The dynamic range of this assay was 1–1000 ng/mL for each compound using a 1/concentration2 weighted linear regression. Calibrators and quality control samples were within 15% of the nominal value for all compounds. Plasma samples with concentrations above the calibration range were diluted into the calibration range with blank plasma prior to reanalysis.
The quantitation of FTC, TFV and RAL in tissues started with the homogenization of each tissue in a 70% acetonitrile + 30% 1 mM ammonium phosphate solution. A portion of the resulting homogenate was extracted by protein precipitation with acetonitrile containing isotopically-labeled internal standards (13C TFV, and 13C 15N FTC). Following vortexing and centrifugation, the supernatant was removed and evaporated to dryness. The extracts were reconstituted with 1 mM ammonium phosphate prior to LC-MS/MS analysis. TFV and FTC were eluted from a Waters Atlantis T3 (100×2.1 mm, 3 µm particle size) analytical column. RAL was eluted from a Phenomenex Synergi Polar-RP (50×4.6 mm, 4 µm particle size) analytical column. An API - 5000 triple quadrupole mass spectrometer was used to detect all analytes. Data were collected using AB Sciex Analyst Chromatography Software. The dynamic range of this assay was 0.3–300 ng/mL of homogenate for each compound using a 1/concentration2 weighted linear regression. Based upon the mass of the tissue extracted, the concentrations were converted from ng/mL homogenate to ng/g tissue. Calibrators and quality control samples were within 15% of the nominal value all compounds.
Statistics
All statistical tests performed using an alpha level of 0.05. Exact log rank tests were utilized to generate p values for Fig. 2 (R v2.14.1). Mann-Whitney comparisons were utilized to generate the p values presented in Figs. 4 & 5A (Prism v4). Wilcoxon rank-sum statistics with repeated measures corrections were utilized to generate p values presented in Fig. 5B (R). Exact log rank tests with repeated measures corrections were utilized to generate p values for Fig. 5C (R). For Figs. 3 & 6, the Lowess and cubic regression models were fit using R. The linear heterogeneity models that allowed for the residual variance to change over time were computed in SAS/STAT software. Graphs were generated in Prism v4 with the exception of the fitted Lowess and cubic regression models which were generated in R.
Zdroje
1. NakagawaF, MayM, PhillipsA (2013) Life expectancy living with HIV: recent estimates and future implications. Curr Opin Infect Dis 26 : 17–25.
2. RichmanDD, MargolisDM, DelaneyM, GreeneWC, HazudaD, et al. (2009) The challenge of finding a cure for HIV infection. Science 323 : 1304–1307.
3. DeeksSG, AutranB, BerkhoutB, BenkiraneM, CairnsS, et al. (2012) Towards an HIV cure: a global scientific strategy. Nat Rev Immunol 12 : 607–614.
4. DurandCM, BlanksonJN, SilicianoRF (2012) Developing strategies for HIV-1 eradication. Trends Immunol 33 : 554–562.
5. DeeksSG (2012) HIV: Shock and kill. Nature 487 : 439–440.
6. ShanL, DengK, ShroffNS, DurandCM, RabiSA, et al. (2012) Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 36 : 491–501.
7. HamerDH (2004) Can HIV be Cured? Mechanisms of HIV persistence and strategies to combat it. Curr HIV Res 2 : 99–111.
8. BergerEA, PastanI (2010) Immunotoxin complementation of HAART to deplete persisting HIV-infected cell reservoirs. PLoS Pathog 6: e1000803.
9. ThrushGR, LarkLR, ClinchyBC, VitettaES (1996) Immunotoxins: an update. Annu Rev Immunol 14 : 49–71.
10. PincusSH (1996) Therapeutic potential of anti-HIV immunotoxins. Antiviral Res 33 : 1–9.
11. Van OijenMG, PreijersFW (1998) Rationale for the use of immunotoxins in the treatment of HIV-infected humans. J Drug Target 5 : 75–91.
12. LiuS, SchubertRL, BuggeTH, LepplaSH (2003) Anthrax toxin: structures, functions and tumour targeting. Expert Opin Biol Ther 3 : 843–853.
13. EngertA, SausvilleEA, VitettaE (1998) The emerging role of ricin A-chain immunotoxins in leukemia and lymphoma. Curr Top Microbiol Immunol 234 : 13–33.
14. TillMA, Zolla-PaznerS, GornyMK, PattonJS, UhrJW, et al. (1989) Human immunodeficiency virus-infected T cells and monocytes are killed by monoclonal human anti-gp41 antibodies coupled to ricin A chain. Proc Natl Acad Sci U S A 86 : 1987–1991.
15. PincusSH, WehrlyK, ChesebroB (1989) Treatment of HIV tissue culture infection with monoclonal antibody-ricin A chain conjugates. J Immunol 142 : 3070–3075.
16. TillMA, GhetieV, GregoryT, PatzerEJ, PorterJP, et al. (1988) HIV-infected cells are killed by rCD4-ricin A chain. Science 242 : 1166–1168.
17. ChaudharyVK, MizukamiT, FuerstTR, FitzGeraldDJ, MossB, et al. (1988) Selective killing of HIV-infected cells by recombinant human CD4-Pseudomonas exotoxin hybrid protein. Nature 335 : 369–372.
18. BergerEA, ClouseKA, ChaudharyVK, ChakrabartiS, FitzGeraldDJ, et al. (1989) CD4-Pseudomonas exotoxin hybrid protein blocks the spread of human immunodeficiency virus infection in vitro and is active against cells expressing the envelope glycoproteins from diverse primate immunodeficiency retroviruses. Proc Natl Acad Sci U S A 86 : 9539–9543.
19. BellKD, RamiloO, VitettaES (1993) Combined use of an immunotoxin and cyclosporine to prevent both activated and quiescent peripheral blood T cells from producing type 1 human immunodeficiency virus. Proc Natl Acad Sci U S A 90 : 1411–1415.
20. McCoigC, Van DykeG, ChouCS, PickerLJ, RamiloO, et al. (1999) An anti-CD45RO immunotoxin eliminates T cells latently infected with HIV-1 in vitro. Proc Natl Acad Sci U S A 96 : 11482–11485.
21. PincusSH, McClureJ (1993) Soluble CD4 enhances the efficacy of immunotoxins directed against gp41 of the human immunodeficiency virus. Proc Natl Acad Sci U S A 90 : 332–336.
22. BeraTK, KennedyPE, BergerEA, BarbasCF3rd, PastanI (1998) Specific killing of HIV-infected lymphocytes by a recombinant immunotoxin directed against the HIV-1 envelope glycoprotein. Mol Med 4 : 384–391.
23. CoryTJ, SchackerTW, StevensonM, FletcherCV (2013) Overcoming pharmacologic sanctuaries. Curr Opin HIV AIDS 8 : 190–195.
24. MelkusMW, EstesJD, Padgett-ThomasA, GatlinJ, DentonPW, et al. (2006) Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat Med 12 : 1316–1322.
25. DentonPW, GarciaJV (2011) Humanized mouse models of HIV infection. AIDS Rev 13 : 135–148.
26. DentonPW, GarciaJV (2012) Mucosal HIV-1 transmission and prevention strategies in BLT humanized mice. Trends Microbiol 20 : 268–274.
27. DentonPW, NochiT, LimA, KriskoJF, Martinez-TorresF, et al. (2012) IL-2 receptor gamma-chain molecule is critical for intestinal T-cell reconstitution in humanized mice. Mucosal Immunol 5 : 555–566.
28. LongBR, StoddartCA (2012) Alpha interferon and HIV infection cause activation of human T cells in NSG-BLT mice. J Virol 86 : 3327–3336.
29. BrainardDM, SeungE, FrahmN, CariappaA, BaileyCC, et al. (2009) Induction of robust cellular and humoral virus-specific adaptive immune responses in human immunodeficiency virus-infected humanized BLT mice. J Virol 83 : 7305–7321.
30. ZouW, DentonPW, WatkinsRL, KriskoJF, NochiT, et al. (2012) Nef functions in BLT mice to enhance HIV-1 replication and deplete CD4+CD8+ thymocytes. Retrovirology 9 : 44.
31. SunZ, DentonPW, EstesJD, OthienoFA, WeiBL, et al. (2007) Intrarectal transmission, systemic infection, and CD4+ T cell depletion in humanized mice infected with HIV-1. J Exp Med 204 : 705–714.
32. DentonPW, EstesJD, SunZ, OthienoFA, WeiBL, et al. (2008) Antiretroviral pre-exposure prophylaxis prevents vaginal transmission of HIV-1 in humanized BLT mice. PLoS Med 5: e16.
33. JilekBL, ZarrM, SampahME, RabiSA, BullenCK, et al. (2012) A quantitative basis for antiretroviral therapy for HIV-1 infection. Nat Med 18 : 446–451.
34. RockstrohJK, DejesusE, LennoxJL, YazdanpanahY, SaagMS, et al. (2013) Durable Efficacy and Safety of Raltegravir versus Efavirenz When Combined With Tenofovir/Emtricitabine In Treatment-Naive HIV-1 Infected Patients: Final Five-Year Results From STARTMRK. J Acquir Immune Defic Syndr 63 (1) 77–85.
35. DentonPW, OlesenR, ChoudharySK, ArchinNM, WahlA, et al. (2012) Generation of HIV latency in BLT humanized mice. J Virol 86 : 630–634.
36. PalmerS, MaldarelliF, WiegandA, BernsteinB, HannaGJ, et al. (2008) Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc Natl Acad Sci U S A 105 : 3879–3884.
37. AutranB, CarcelainG, LiTS, BlancC, MathezD, et al. (1997) Positive effects of combined antiretroviral therapy on CD4+ T cell homeostasis and function in advanced HIV disease. Science 277 : 112–116.
38. MurrayJM, McBrideK, BoeseckeC, BaileyM, AminJ, et al. (2012) Integrated HIV DNA accumulates prior to treatment while episomal HIV DNA records ongoing transmission afterwards. AIDS 26 : 543–550.
39. RobbinsBL, SrinivasRV, KimC, BischofbergerN, FridlandA (1998) Anti-human immunodeficiency virus activity and cellular metabolism of a potential prodrug of the acyclic nucleoside phosphonate 9-R-(2-phosphonomethoxypropyl)adenine (PMPA), Bis(isopropyloxymethylcarbonyl)PMPA. Antimicrob Agents Chemother 42 : 612–617.
40. SchinaziRF, McMillanA, CannonD, MathisR, LloydRM, et al. (1992) Selective inhibition of human immunodeficiency viruses by racemates and enantiomers of cis-5-fluoro-1-[2-(hydroxymethyl)-1,3-oxathiolan-5-yl]cytosine. Antimicrob Agents Chemother 36 : 2423–2431.
41. ScopellitiF, PollicitaM, Ceccherini-SilbersteinF, Di SantoF, SurdoM, et al. (2011) Comparative antiviral activity of integrase inhibitors in human monocyte-derived macrophages and lymphocytes. Antiviral Res 92 : 255–261.
42. LafeuilladeA, KhiriH, ChadapaudS, HittingerG, HalfonP (2001) Persistence of HIV-1 resistance in lymph node mononuclear cell RNA despite effective HAART. AIDS 15 : 1965–1969.
43. KennedyPE, BeraTK, WangQC, GalloM, WagnerW, et al. (2006) Anti-HIV-1 immunotoxin 3B3(Fv)-PE38: enhanced potency against clinical isolates in human PBMCs and macrophages, and negligible hepatotoxicity in macaques. J Leukoc Biol 80 : 1175–1182.
44. GoldsteinH, Pettoello-MantovaniM, BeraTK, PastanIH, BergerEA (2000) Chimeric toxins targeted to the human immunodeficiency virus type 1 envelope glycoprotein augment the in vivo activity of combination antiretroviral therapy in thy/liv-SCID-Hu mice. J Infect Dis 181 : 921–926.
45. BlanksonJN, FinziD, PiersonTC, SabundayoBP, ChadwickK, et al. (2000) Biphasic decay of latently infected CD4+ T cells in acute human immunodeficiency virus type 1 infection. J Infect Dis 182 : 1636–1642.
46. SedaghatAR, DinosoJB, ShenL, WilkeCO, SilicianoRF (2008) Decay dynamics of HIV-1 depend on the inhibited stages of the viral life cycle. Proc Natl Acad Sci U S A 105 : 4832–4837.
47. HorwitzJA, Halper-StrombergA, MouquetH, GitlinAD, TretiakovaA, et al. (2013) HIV-1 suppression and durable control by combining single broadly neutralizing antibodies and antiretroviral drugs in humanized mice. Proc Natl Acad Sci U S A 110 : 16538–16543.
48. BarbasCF3rd, HuD, DunlopN, SawyerL, CababaD, et al. (1994) In vitro evolution of a neutralizing human antibody to human immunodeficiency virus type 1 to enhance affinity and broaden strain cross-reactivity. Proc Natl Acad Sci U S A 91 : 3809–3813.
49. NorthTW, HigginsJ, DeereJD, HayesTL, VillalobosA, et al. (2010) Viral sanctuaries during highly active antiretroviral therapy in a nonhuman primate model for AIDS. J Virol 84 : 2913–2922.
50. YuklSA, ShergillAK, HoT, KillianM, GirlingV, et al. (2013) The distribution of HIV DNA and RNA in cell subsets differs in gut and blood of HIV-positive patients on ART: implications for viral persistence. J Infect Dis 208 : 1212–1220.
51. WooJH, FrankelA, NevilleDM (2010) Cytotoxic therapy with diphtheria toxin fusion proteins. Drugs of the Future 35 : 823–831.
52. WeldonJE, PastanI (2011) A guide to taming a toxin–recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. FEBS J 278 : 4683–4700.
53. FangH, PincusSH (1995) Unique insertion sequence and pattern of CD4 expression in variants selected with immunotoxins from human immunodeficiency virus type 1-infected T cells. J Virol 69 : 75–81.
54. BergerEA (2011) Targeted cytotoxic therapy: adapting a rapidly progressing anticancer paradigm for depletion of persistent HIV-infected cell reservoirs. Curr Opin HIV AIDS 6 : 80–85.
55. MitsuyasuR (2013) Curing HIV: lessons from cancer therapy. Curr Opin HIV AIDS 8 : 224–229.
56. DentonPW, OthienoF, Martinez-TorresF, ZouW, KriskoJF, et al. (2011) One percent tenofovir applied topically to humanized BLT mice and used according to the CAPRISA 004 experimental design demonstrates partial protection from vaginal HIV infection, validating the BLT model for evaluation of new microbicide candidates. J Virol 85 : 7582–7593.
57. WahlA, SwansonMD, NochiT, OlesenR, DentonPW, et al. (2012) Human breast milk and antiretrovirals dramatically reduce oral HIV-1 transmission in BLT humanized mice. PLoS Pathog 8: e1002732.
58. KriskoJF, Martinez-TorresF, FosterJL, GarciaJV (2013) HIV Restriction by APOBEC3 in Humanized Mice. PLoS Pathog 9: e1003242.
59. HaaseAT, HenryK, ZupancicM, SedgewickG, FaustRA, et al. (1996) Quantitative image analysis of HIV-1 infection in lymphoid tissue. Science 274 : 985–989.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 1
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Lyme Disease: Call for a “Manhattan Project” to Combat the Epidemic
- Origin, Migration Routes and Worldwide Population Genetic Structure of the Wheat Yellow Rust Pathogen f.sp.
- IFNγ/IL-10 Co-producing Cells Dominate the CD4 Response to Malaria in Highly Exposed Children
- Human and Plant Fungal Pathogens: The Role of Secondary Metabolites