
STING-Dependent Type I IFN Production Inhibits Cell-Mediated Immunity to
Infection with Listeria monocytogenes strains that enter the host cell cytosol leads to a robust cytotoxic T cell response resulting in long-lived cell-mediated immunity (CMI). Upon entry into the cytosol, L. monocytogenes secretes cyclic diadenosine monophosphate (c-di-AMP) which activates the innate immune sensor STING leading to the expression of IFN-β and co-regulated genes. In this study, we examined the role of STING in the development of protective CMI to L. monocytogenes. Mice deficient for STING or its downstream effector IRF3 restricted a secondary lethal challenge with L. monocytogenes and exhibited enhanced immunity that was MyD88-independent. Conversely, enhancing STING activation during immunization by co-administration of c-di-AMP or by infection with a L. monocytogenes mutant that secretes elevated levels of c-di-AMP resulted in decreased protective immunity that was largely dependent on the type I interferon receptor. These data suggest that L. monocytogenes activation of STING downregulates CMI by induction of type I interferon.
Published in the journal:
. PLoS Pathog 10(1): e32767. doi:10.1371/journal.ppat.1003861
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1003861
Summary
Infection with Listeria monocytogenes strains that enter the host cell cytosol leads to a robust cytotoxic T cell response resulting in long-lived cell-mediated immunity (CMI). Upon entry into the cytosol, L. monocytogenes secretes cyclic diadenosine monophosphate (c-di-AMP) which activates the innate immune sensor STING leading to the expression of IFN-β and co-regulated genes. In this study, we examined the role of STING in the development of protective CMI to L. monocytogenes. Mice deficient for STING or its downstream effector IRF3 restricted a secondary lethal challenge with L. monocytogenes and exhibited enhanced immunity that was MyD88-independent. Conversely, enhancing STING activation during immunization by co-administration of c-di-AMP or by infection with a L. monocytogenes mutant that secretes elevated levels of c-di-AMP resulted in decreased protective immunity that was largely dependent on the type I interferon receptor. These data suggest that L. monocytogenes activation of STING downregulates CMI by induction of type I interferon.
Introduction
Cell-mediated immunity (CMI) is a critical component for protection against intracellular pathogens. Upon infection, the innate immune response provides resistance and initiates the development of antigen-specific lymphocytes including cytotoxic CD8+ T cells, which ultimately kill host cells harboring pathogens [1]. The Gram-positive bacterium Listeria monocytogenes has been used for decades as a model organism to investigate the generation of CMI, as infection induces a robust effector and memory CD8+ T cell response that restricts bacterial growth following a lethal secondary challenge, resulting in long-lived sterilizing immunity [2]. Although it is generally agreed that activation of the innate immune system is critical for the initiation of adaptive immunity [3], the specific signaling pathways necessary to elicit a robust protective immune response to L. monocytogenes remain poorly understood.
L. monocytogenes is detected by multiple innate immune signaling pathways during infection [4]. Following engulfment by macrophages and dendritic cells, the bacteria reside within phagosomes where they are detected by Toll-Like Receptors (TLRs), resulting in the activation of MyD88-dependent response genes [5]. By secreting a pore-forming cytolysin, listerolysin O (LLO), L. monocytogenes escapes into the cytosol where it replicates and polymerizes actin to facilitate cell-to-cell spread [6]. L. monocytogenes is detected by several cytosolic innate immune pathways leading to a cytokine profile distinct from that of LLO-deficient bacteria, which are restricted to the phagosome [5], [7].
The primary cytosolic sensor of L. monocytogenes is STING (stimulator of interferon (IFN) genes, also known as MPYS, MITA and ERIS), an ER-localized transmembrane protein [8]. STING is activated by cyclic dinucleotides (CDNs) that are either produced by a pathogen or by an endogenous cyclic GMP-AMP synthase that is activated by DNA [9], [10]. Direct binding of CDNs to STING activates a downstream signaling cascade involving TBK1 and IRF3 [11], [12], [13]. In the case of L. monocytogenes, cyclic diadenosine monophosphate (c-di-AMP) is secreted through bacterial multi-drug efflux pumps, leading to STING activation and transcription of IFN-β and co-regulated genes [14], [15]. STING-deficient macrophages or mice are unable to produce IFN-β in response to L. monocytogenes infection indicating that STING is required for the type I IFN response to L. monocytogenes [16], [17].
Purified CDNs are immunostimulatory in vitro and in vivo. Murine and human dendritic cells exposed to cyclic diguanosine monophosphate (c-di-GMP) or c-di-AMP exhibit enhanced surface expression of costimulatory markers and T cell proliferation. Mice mount a significant antibody response following co-administration of protein antigens with c-di-GMP or c-di-AMP [13], [18], [19], [20]. CDNs also stimulate cellular immune responses. Antigen-stimulated splenocytes from mice immunized with β-ααgalactosidase in the presence of c-di-GMP or c-di-AMP proliferate and secrete cytokines [18], [19]. These data indicate that CDNs are sufficient to elicit a cell-mediated adaptive immune response.
Entry of L. monocytogenes into the host cytosol is necessary to generate secondary protective immunity, as phagosome-restricted heat-killed or LLO-deficient bacteria do not elicit functional cytotoxic T cells and long-term memory responses [21], [22], [23]. The attenuated ActA-deficient mutant strain, which escapes the phagosome but fails to polymerize actin and spread to neighboring cells, is fully immunogenic to mice [24]. Furthermore, MyD88-deficient mice, while highly susceptible to acute infection with virulent L. monocytogenes, are fully protected following secondary lethal challenge when immunized with the ActA-deficient mutant [25], [26], [27], [28]. These findings suggest that phagosomal detection of L. monocytogenes during immunization is not sufficient for the development of protective immunity.
STING activation induces an array of IRF3-dependent genes [5] as well as NF-κB and STAT6-dependent genes [29], [30]. Since LLO-deficient bacteria fail to enter the cytosol and induce STING-related genes [5], [7], we hypothesized that the detection of L. monocytogenes by STING is required for CMI. In this study, we tested whether STING signaling plays an important role in the generation of protective immunity to L. monocytogenes.
Results
The STING signaling pathway is not required for protective immunity to L. monocytogenes
In the model of protective immunity used in these studies, mice were immunized with an attenuated yet immunogenic strain of L. monocytogenes that lacks the actA and inlB genes (ActA−Lm) and challenged 30–38 days later with 2LD50 (2×105 colony forming units (CFU)) of wild-type L. monocytogenes (WT Lm). Previous studies typically immunize mice with 0.1LD50 of L. monocytogenes (1×107 CFU ActA−Lm for C57BL/6 mice) [21]. At this high immunization dose, bacterial burdens in subsequently challenged mice are below the limit of detection. In contrast, a lower immunization dose of 103 CFU (∼0.00001LD50 for C57BL/6 mice) still generated significant immunity as compared to naïve mice, but did not induce saturating immunity and thus revealed differences that might be missed using higher doses (Fig. S1A).
To determine whether STING signaling is required for the generation of protective immunity to L. monocytogenes, mice lacking STING (Goldenticket, Gt) were immunized with 103 CFU of ActA−Lm expressing ovalbumin (ActA−Lm-OVA) and challenged 30–38 days later with 2LD50s of WT Lm-OVA. Surprisingly, whereas naïve STING-deficient mice had similar bacterial burdens as naïve C57BL/6 (B6) mice, immunized STING-deficient mice had approximately 1.5–2 logs fewer bacteria in spleens and livers compared to immunized B6 mice (Fig. 1A). At higher immunization doses (104 and 105 CFU), the majority of B6 and STING-deficient mice had bacterial numbers below the limit of detection in the spleen and thus no significant differences could be observed (Fig. S1B). Since cytotoxic CD8+ T cells are the major mediator of L. monocytogenes clearance following secondary challenge [2], [31], the number of OVA-specific CD8+ T cells were measured by staining splenocytes with a MHC class I restricted OVA tetramer (Kb/OVA257–264). STING-deficient mice had significantly higher total numbers of OVA-specific CD8+ T cells compared to B6 mice (Fig. 1B). These data suggested that STING-deficient mice exhibited enhanced immunity.

STING stimulation leads to the activation of the transcription factor, IRF3 [12]. In addition, IRF7 contributes to IFN-α production in response to L. monocytogenes in vivo [32]. To determine the role of these downstream effectors of STING signaling, mice lacking IRF3 (IRF3−/−) or IRF3 and IRF7 (IRF3/7−/−) were examined for protective immunity. Compared to B6 mice, IRF3-deficient mice had less bacteria in the livers and IRF3/7-deficient mice had less bacteria in the spleens and livers (Fig. 1A). Both groups had significantly higher numbers of OVA-specific CD8+ T cells than B6 mice (Fig. 1B), suggesting that these transcription factors contribute to the STING-mediated decrease in immunity.
To evaluate whether T cells from mice lacking STING signaling possessed effector functions, splenocytes from ActA−Lm-OVA-immunized mice were stimulated with either the MHC class I-restricted peptide OVA257–264, or the MHC class II-restricted peptide LLO190–201 from L. monocytogenes and measured for IFN-γ, TNF-α and IL-2 production by intracellular cytokine staining and flow cytometry. STING-deficient mice had significantly higher numbers of polyfunctional IFN-γ-, TNF-α - and IL-2 - producing CD8+ and CD4+ T cells compared to B6 mice (Fig. 1C). IRF3/7-deficient but not IRF3-deficient mice also had higher numbers of IFN-γ - and TNF-α-producing CD8+ T cells. These data suggested that in the absence of STING or its downstream transcription factors IRF3 and IRF7, CD8+ T cell expansion, cytokine production and bacterial clearance was enhanced.
In the absence of MyD88, STING plays a role in the innate but not the adaptive immune response
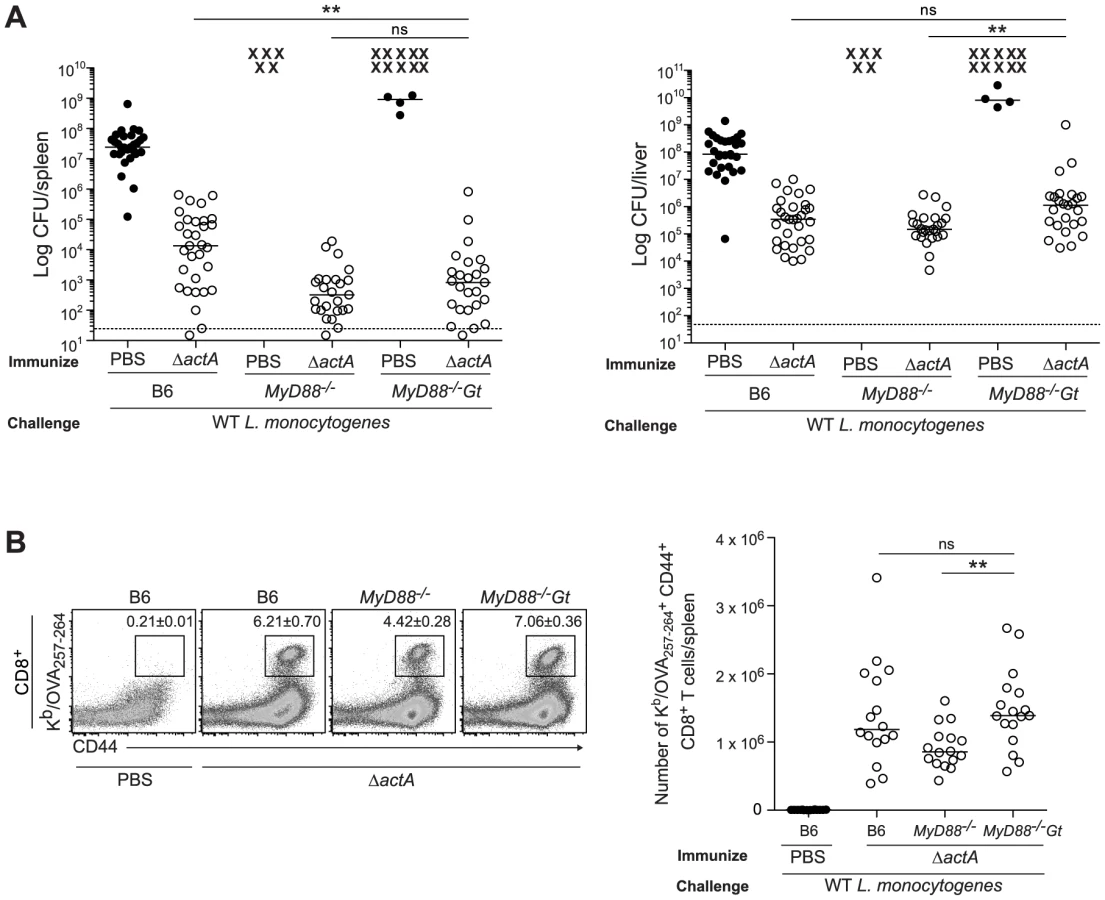
L. monocytogenes activates TLRs which signal via the adaptor MyD88 [4]. It is possible that both MyD88 - and STING-dependent response genes play redundant roles in generating protective immunity to L. monocytogenes. To test this hypothesis, we bred mice lacking both MyD88 and STING (MyD88−/−Gt). Bone marrow-derived macrophages (BMMs) from MyD88/STING-deficient mice infected with WT Lm had low or non-detectable expression of the cytokines IFN-β, IL-12 p40, TNF-α and IL-6 (Fig. S2A).
To examine whether the loss of both the MyD88 and STING signaling pathways affect bacterial clearance during an acute infection, B6, MyD88 - and MyD88/STING-deficient mice were infected with WT Lm. MyD88/STING-deficient mice had similar bacterial burdens as MyD88-deficient mice early after infection, however by day 3, had significantly higher CFU in the spleen and the liver (Fig. S2B). Although mice lacking MyD88 cannot survive infection with WT Lm, MyD88/STING-deficient mice died earlier than MyD88-deficient mice (data not shown). These data indicated that in the absence of MyD88, STING contributes to L. monocytogenes clearance during an acute response.
To determine whether the loss of MyD88 and STING affects initiation of the adaptive response to L. monocytogenes, the upregulation of costimulatory molecules on splenic dendritic cells from immunized B6, STING-, MyD88 - and MyD88/STING-deficient mice was measured. Surface expression of CD86 and CD40 was decreased in MyD88 - and STING-deficient mice and further reduced in the MyD88/STING-deficient mice, suggesting an additive effect of these two pathways (Fig. 2A). Upregulation of the activation marker CD69 on CD8+ and CD4+ T cells was also decreased in both MyD88 - and STING-deficient mice and ablated in MyD88/STING-deficient mice (Fig. 2B). Furthermore, IL-6, TNF-α and MCP-1 in the serum were mostly dependent on MyD88 but further reduced in the MyD88/STING-deficient mice. IL-12 p70 was non-detectable in either MyD88 - or MyD88/STING-deficient mice (Fig. 2C). These data indicated that mice lacking MyD88 and STING signaling had significantly reduced dendritic and T cell activation and cytokine production in vivo in response to L. monocytogenes immunization.

Mice lacking both MyD88 and STING were tested for the ability to develop protective immunity. Similar to B6 and MyD88-deficient mice, MyD88/STING-deficient mice were protected following secondary lethal challenge and showed no signs of disease unlike naïve mice which were either moribund or dead at the time of sacrifice. MyD88/STING-deficient mice had a small increase in the number of bacteria in the livers compared to MyD88-deficient mice (Fig. 3A). However, MyD88/STING-deficient mice had significantly higher number of OVA-specific CD8+ T cells compared to MyD88-deficient mice (Fig. 3B), suggesting that the higher bacterial loads in the liver was likely due to loss of innate rather than adaptive immune responses. These data indicated that regardless of the presence or absence of MyD88, protective immunity to L. monocytogenes is enhanced in the absence of STING.
Enhanced STING activation blocks the development of adaptive immunity
Mice immunized with LLO-deficient L. monocytogenes fail to develop protective immunity [21], [22]. Since LLO-deficient bacteria do not activate STING, we hypothesized that c-di-AMP-mediated STING activation might be sufficient to restore immunity in LLO−Lm-immunized mice. In support of previous reports, we found that BMDCs stimulated with c-di-AMP secreted cytokines and upregulated costimulatory markers in vitro (Fig. S3). Furthermore, mice administered c-di-AMP intravenously exhibited increased surface expression of CD86 and CD40 on splenic dendritic cells and of the activation marker CD69 on splenic CD8+ and CD4+ T cells in a STING-dependent manner (Fig. 4A and 4B), indicating that c-di-AMP can induce an inflammatory response in our model.

To test our hypothesis, B6 mice were immunized with LLO−Lm-OVA in the presence of c-di-AMP. While ActA−Lm-OVA-immunized mice restricted bacterial growth following challenge, the co-administration of c-di-AMP with LLO−Lm-OVA did not rescue protective immunity (Fig. 4C). Instead, the presence of c-di-AMP significantly reduced immunity in ActA−Lm-OVA-immunized mice suggesting that STING signaling inhibits protective immunity to L. monocytogenes infection.
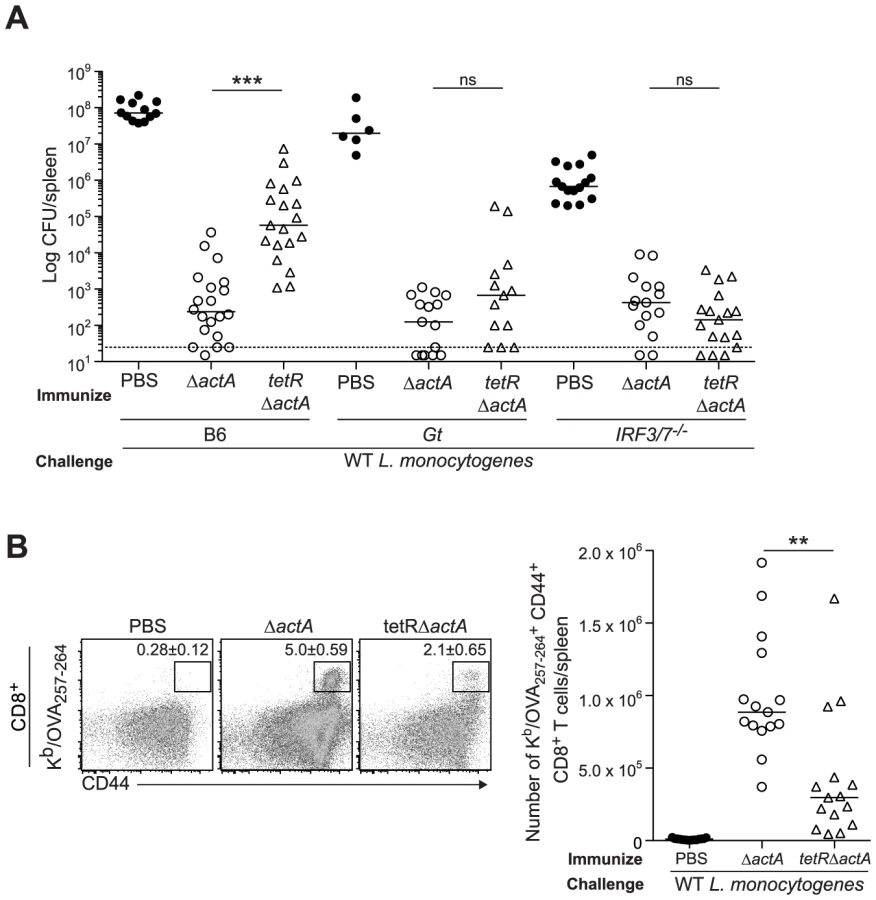
To further examine the effect of c-di-AMP on immunity to L. monocytogenes, B6, STING - and IRF3/7-deficient mice were immunized with ActA−Lm-OVA in the presence or absence of c-di-AMP. Following challenge, B6 mice immunized in the presence of c-di-AMP had significantly higher bacterial numbers and had fewer numbers of total and OVA-specific CD8+ T cells in the spleen than mice immunized with ActA−Lm-OVA alone (Fig. 4D, 4E and S4A). STING-deficient mice were protected and had a robust CD8+ T cell expansion confirming that c-di-AMP-mediated inhibition of immunity was STING-dependent. IRF3/7-deficient mice were significantly protected as compared to naïve mice and had a population of OVA-specific CD8+ T cells, suggesting that c-di-AMP-mediated inhibition is partially due to IRF3 and IRF7 (Fig. 4D and 4E). Interestingly, IRF3/7-deficient mice were less protected compared to mice immunized with ActA−Lm-OVA alone, indicating that STING-dependent, IRF3/7-independent signaling also plays a role in loss of protective immunity.
Next, we evaluated a L. monocytogenes mutant, tetR::Tn917, that secretes 20-fold more c-di-AMP than WT L. monocytogenes [14], [15]. B6, STING - and IRF3/7-deficient mice were immunized with either ActA−Lm-OVA or the tetR::Tn917 mutant in the ActA−Lm-OVA background (tetRActA−Lm-OVA). B6 mice immunized with tetRActA−Lm-OVA had significantly higher bacterial numbers in the spleen compared to ActA−Lm-OVA-immunized mice whereas STING - and IRF3/7-deficient mice exhibited no significant difference (Fig. 5A). Furthermore, B6 mice immunized with tetRActA−Lm-OVA had a smaller population of total and OVA-specific CD8+ T cells compared to ActA−Lm-OVA-immunized mice (Fig. 5B and S4B). These data supported our finding that enhanced STING signaling lead to a reduction in protective immunity.
Enhanced STING activation inhibits CD8+ T cell priming
To determine whether enhanced STING signaling reduces T cell priming, OVA-specific CD8+ T cells were measured at the peak of the primary response from mice immunized with ActA−Lm-OVA in the presence or absence of c-di-AMP or tetRActA−Lm-OVA. At 7 days post immunization, mice immunized in the presence of c-di-AMP had significantly fewer OVA-specific CD8+ T cells compared to mice immunized with ActA−Lm-OVA alone. Mice immunized with the tetRActA−Lm-OVA mutant also had a small decrease in antigen-specific cells (Fig. 6A). Furthermore, the small population of OVA-specific CD8+ T cells that were present in mice immunized with elevated STING activation had significantly higher surface expression of the naïve T cell maker CD62L, suggesting that enhanced STING signaling elicited fewer antigen-specific effector T cells (Fig. 6B). In addition, peptide-stimulated CD8+ and CD4+ splenocytes from ActA−Lm-OVA-immunized mice in the presence of c-di-AMP or tetRActA−Lm-OVA-immunized mice produced fewer cytokines than those from ActA−Lm-OVA-immunized mice (Fig. 6C). These data indicate that mice immunized in the presence of enhanced STING signaling exhibited reduced T cell priming.

STING-mediated loss of immunity is dependent on type I IFNs
To determine the role of type I IFNs in STING-mediated CMI, IFN-αβR-deficient mice were tested for protective immunity. IFN-αβR-deficient mice restricted bacterial growth better than B6 mice (Fig. 7A and 7B), indicating that like STING - and IRF3/7-deficient mice, IFN-αβR-deficient mice also hyper-immunize. Although IFN-αβR-deficient mice immunized in the presence of c-di-AMP had higher bacterial numbers compared to mice immunized with ActA−Lm-OVA alone, these mice were significantly more protected than naïve mice, indicating a role for both type I IFN-dependent and independent mechanisms of suppression (Fig. 7A). IFN-αβR-deficient mice immunized with tetRActA−Lm-OVA were completely protected (Fig. 7B). These data indicated that the c-di-AMP-mediated inhibition of protective immunity is largely dependent on type I IFNs. Interestingly, we found that mice immunized in the presence of the synthetic double-stranded RNA, polyinosinic∶polycytidylic acid (poly(I∶C)), a STING-independent agonist of TLR3 and IRF3, also lost the ability to restrict bacterial growth following challenge (Fig S5), suggesting that type I IFN-mediated inhibition of immunity is unlikely STING specific.

We next determined whether inhibition of T cell priming by STING-dependent type I IFNs acted directly on lymphocytes. CD8+ T cells lacking the IFN-αβR undergo clonal expansion in response to primary L. monocytogenes infection so an adoptive transfer model could be used [33], [34]. B6, STING-, or IFN-αβR-deficient mice were injected with WT and IFN-αβR-deficient OT-I splenocytes and subsequently immunized with ActA−Lm-OVA in the presence or absence of c-di-AMP. At 7 days, WT and IFN-αβR-deficient OT-I cells expanded in ActA−Lm-OVA-immunized B6 mice, whereas in the presence of c-di-AMP, both WT and IFN-αβR-deficient OT-I cells had significantly reduced populations indicating that type I IFNs were not directly blocking T cell priming (Fig. 8A and 8B). Inhibition of T cell expansion by c-di-AMP was rescued in IFN-αβR - and STING-deficient mice further indicating that type I IFN-mediated suppression of immunity is not T cell intrinsic.

Discussion
The results of this study show that the STING signaling pathway is not required to elicit CMI to L. monocytogenes. In fact, the absence of STING or IRF3 and IRF7 led to a higher number of antigen-specific CD8+ T cells and increased levels of protective immunity. Mice lacking both MyD88 and STING were also protected upon secondary challenge, indicating that not only is STING dispensable for the generation of a protective response to L. monocytogenes, it does not act in a redundant fashion with the TLR-MyD88 signaling pathway. Conversely, when STING activity was enhanced either by administering c-di-AMP during immunization or using a bacterial mutant that secretes elevated levels of c-di-AMP, mice failed to immunize and had decreased numbers of antigen-specific T cells following reinfection. This suppressive effect was largely due to the induction of type I IFNs since IFN-αβR-deficient mice immunized with either ActA−Lm-OVA in the presence of c-di-AMP or the tetR mutant were protected. Collectively, these findings suggest that L. monocytogenes-induced STING activation reduces the host adaptive immune response by induction of type I IFN.
The mechanism of type I IFN-mediated inhibition of T cell priming remains unclear. We found that both WT and IFN-αβR-deficient CD8+ T cells had significantly reduced expansion in the presence of c-di-AMP, suggesting that c-di-AMP-mediated suppression of lymphocyte priming is T cell extrinsic. One possibility is that the uptake of type I IFN-induced apoptotic cells by macrophages results in the release of the immunosuppressive cytokine IL-10 [35]. However, mice administered c-di-AMP did not have detectable levels of IL-10 in the serum. Furthermore, similar to B6 mice, IL-10-deficient mice immunized in the presence of c-di-AMP were unable to elicit OVA-specific CD8+ T cells following challenge (data not shown). Thus, IL-10 is not the downstream effector of STING/type I IFN-mediated suppression of adaptive immunity in our model of protective immunity. Other mechanisms including indoleamine 2,3-dioxygenase (IDO)-mediated T cell suppression, which is upregulated by type I IFN [36], may play a role in c-di-AMP-mediated inhibition of immunity. Indeed, Huang et al. found that splenic DCs from mice treated with DNA nanoparticles suppress ex vivo T cell proliferation in a STING - and IDO-dependent manner [37].
While spleens from immunized STING-deficient mice contained fewer bacteria than B6 mice, bacterial clearance by immunized IRF3/7 - and IFN-αβR-deficient mice was even higher. One possibility is that there are STING-independent sources of type I IFNs in response to L. monocytogenes. Supporting this hypothesis, we observed that MyD88/STING-deficient macrophages produced low levels of IFN-β eight hours post infection. Recent studies found that RIG-I-deficient cells had reduced IFN-β secretion in response to L. monocytogenes infection suggesting that RNA from L. monocytogenes can be detected by the host [38], [39]. Supporting this hypothesis, we found that immunized mice lacking MAVS, the signaling adaptor for RIG-I, were more protected than control mice upon reinfection (data not shown). Thus, RIG-I-dependent, STING-independent induction of type I IFNs via IRF3 and IRF7 may also contribute to the inhibition of protective immunity. Another possibility is that since naïve IRF3/7 - and IFN-αβR-deficient mice exhibit heightened bacterial clearance compared to both naïve B6 and STING-deficient mice, innate immune factors are also likely restricting reinfection. In fact, IFN-αβR-deficient mice have higher neutrophil recruitment in response to L. monocytogenes infection [40]. Thus, careful examination of each mouse strain will be necessary to determine the extent of the contribution of innate versus adaptive immune mechanisms in protective immunity.
Type I IFN-mediated inhibition of immunity is unlikely specific to STING activation. Mice immunized in the presence of the poly(I∶C), which induces type I IFNs independently of STING, also lost the ability to restrict bacterial growth following challenge. Several studies have shown that administering poly(I∶C) prior to a protein antigen inhibits clonal expansion of antigen-specific CD8+ T cells, supporting our findings that systemic type I IFN-induced inflammation reduces T cell priming [41], [42].
The importance of type I IFNs during bacterial infection is less understood than for viruses [43]. For example, IFN-β is the highest upregulated gene following L. monocytogenes cytosolic invasion, yet mice lacking the IFN-αβR are more resistant to acute infection, suggesting that type I IFNs may promote pathogenesis [35], [44], [45]. However, strains of L. monocytogenes that secrete elevated levels of c-di-AMP and induce higher levels of type I IFNs, are not hypervirulent [46], but induce considerably less T cell immunity as shown in this study. Thus it is possible that secretion of c-di-AMP and consequent type I IFN production may play a role in L. monocytogenes pathogenesis by suppressing the development of adaptive immunity. Although L. monocytogenes generally causes acute infections, recent studies have found that type I IFNs promote chronic infections with LCMV by suppressing cell-mediated mechanisms of viral control [47], [48]. In the case of Mycobacterium tuberculosis, mice lacking IRF3 are more resistant to infection with M. tuberculosis suggesting that IRF3 activation is detrimental to host clearance [49]. In humans, IFN-α treatment leads to higher incidences of TB reactivation [50]. Furthermore, active TB patients exhibit an increase in type I IFN-inducible transcripts in the blood, which correlated with disease severity [51]. Therefore, type I IFNs may exacerbate or maintain secondary or long-term chronic infections. Interestingly, human STING often contains polymorphisms that makes it resistant to bacterial but not host derived CDNs [52].
STING-mediated suppression of protective immunity was not solely due to type I IFNs. Although protective immunity in tetRActA−Lm-OVA-immunized mice was rescued in the absence of IRF3/7 and the IFN-αβR, mice immunized in the presence of c-di-AMP exhibited type I IFN-independent suppression. Since STING activates NF-κB as well as IRF3, it is possible that NF-κB-dependent inflammation also plays a role in restricting immunity. We believe that administering c-di-AMP activates STING more robustly compared to infection with the tetRActA−Lm strain and thus inhibition of immunity by type I IFN-independent inflammation would become more apparent.
The results of this and other studies suggest an inverse relationship between the extent of inflammation and the development of adaptive immunity. For example, IL-12-deficient mice immunized with L. monocytogenes develop higher numbers of CD8+ memory T cells and are more resistant to reinfection [53]. In previous studies, we found that a L. monocytogenes strain engineered to activate the inflammasome, and consequently induce high levels of IL-1β secretion, was a poor inducer of adaptive immunity [54]. Furthermore, co-administration of heat-killed or LLO-deficient L. monocytogenes blocked immunity to WT bacteria in a MyD88-dependent manner [55]. Thus, activation of three distinct signaling pathways, STING, MyD88, and caspase-1 all resulted in the inhibition of the development of adaptive immunity. Therefore, there appears to be a dichotomy between innate immune pathways that are necessary for survival (for example, MyD88 for L. monocytogenes and type I IFN for viruses), and those that lead to adaptive immunity. In fact, our data and work from others suggest that lack of inflammation represents an ideal environment for the generation of memory T cells [31], [56]. Indeed, mice deficient for both MyD88 and STING are fully immunized by L. monocytogenes even though there was a significant reduction of dendritic and T cell activation and cytokine production following immunization.
Considering that the innate immune response is believed to be required for the initiation of adaptive immunity, we were surprised that MyD88/STING-deficient mice immunized with L. monocytogenes were protected after reinfection. This raises the question, which innate immune signaling pathways contribute to the initiation of T cell priming to L. monocytogenes? Previous work from our group found that NOD2 detects cytosolic L. monocytogenes [5]. However, immunized MyD88/NOD1/2-deficient mice clear bacteria upon secondary lethal challenge (data not shown), suggesting that protective immunity is not due to redundancy between MyD88 and NOD-like signaling pathways. Future studies to identify which innate immune detection pathways are required for L. monocytogenes-mediated CMI would provide a greater understanding of how pathogens and adjuvants elicit protective immunity, knowledge that can be used for the development and improvement of vaccines.
Materials and Methods
Ethics statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All protocols were reviewed and approved by the Animal Care and Use Committee at the University of California, Berkeley (MAUP# R235-0813B).
Mouse strains
C57BL/6 mice were purchased from The Jackson Laboratory. Goldenticket (Gt) mice were generated from an ENU mutagenesis screen. Gt mice contain a single nucleotide mutation in STING resulting in the absence of the STING protein [17]. All mice were in the C57BL/6 genetic background. Gt, MyD88−/−, MyD88−/−Gt, IRF3−/−, IRF3/7−/− and IFNAR1−/− mice were bred in our facilities. GFP+/+ OT-I+/+ RAG2−/− and IFNAR1−/− OT-I+/+ Ly5.1+/+ RAG2−/− mice were generously provided by Ellen Robey.
Bacterial strains and culture
All L. monocytogenes strains were in the 10403S background. ActA−Lm-OVA (ΔactAΔinlB) (DP-L6014) [57], WT Lm-OVA (DP-L6018) and LLO−Lm-OVA (Δhly) (DP-L6017) [21] L. monocytogenes were previously described. For tetRActA−Lm-OVA (DP-L6015), the tetR::Tn917 transposon [14] was transduced into ActA−Lm-OVA. L. monocytogenes were grown in brain heart infusion (BHI) media at 30°C overnight without shaking to stationary phase. For in vitro infections, L. monocytogenes was washed 3× in PBS. For in vivo infections, L. monocytogenes was diluted in BHI at 37°C shaking for ∼2 hours until they reached an OD600 0.4–0.6.
In vivo infections
Eight to 12 week old sex-matched mice were infected intravenously with 103 CFU (unless otherwise indicated) of L. monocytogenes diluted in phosphate buffered saline (PBS) in a total volume of 200 µl. For acute infections and primary immunization studies, mice were sacrificed at 1, 2 and 3 or 7 days post infection, respectively. For challenge studies, mice immunized 30–38 days prior were infected with 2×105 CFU of WT Lm-OVA. Where indicated, mice were administered either 50 µg or 100 µg of c-di-AMP or 50 µg of poly(I∶C) (InvivoGen) with the bacterial inoculum. Three days later, spleens and livers were homogenized in 0.1% IGEPAL CA-630 (Sigma) and plated on LB-strep plates to enumerate CFU. For analysis of CD8+ T cell responses, spleens were divided and weighed. For splenic dendritic and T cell activation, mice were immunized with 105 CFU of ActA−Lm-OVA. A higher dose was used to allow for the easy detection of activated cells.
For OT-I cells, splenocytes from GFP+/+ OT-I+/+ RAG2−/− and IFNAR1−/− OT-I+/+ Ly5.1+/+ RAG2−/− mice were isolated and washed 3× with PBS. Percent of CD8+ OT-I cells was determined by flow cytometry. 2×104 of each cell type was injected intravenously into each mouse (4×104 total cells/mouse) 1 day prior to immunization with L. monocytogenes.
C-di-AMP was generated by Josh Woodward as previously described [58]. Purified c-di-AMP was resuspended in tissue culture grade PBS. LPS was removed from the prepared c-di-AMP using Detoxi-gel endotoxin removing gel (Pierce) according to the manufacturers instructions. Endotoxin content was measured using the Toxinsensor Chromogenic LAL Endotoxin assay kit (Genescript). The nucleotide solution was passed through the Detoxi-gel until endotoxin levels were <0.0125 EU/ml. Nucleotide was then diluted to 500 µg/ml.
Dendritic cell and T cell analysis
For analysis of T cell responses, spleen halves were dissociated and filtered through a 70 µm cell strainer. Red blood cells were lysed with Red Blood Cell Lysing Buffer (Sigma). To determine OVA-specific cells, splenocytes were stained with anti-mouse CD8, CD44, CD62L and a Kb/OVA257–264 tetramer. Representative FACs plots are gated on CD8+ cells and values show the median percentage of Kb/OVA257–264 tetramer+ CD44+ within the CD8+ cell population ± SEM. For peptide stimulation assays, splenocytes were stimulated for 5 hours with 2 µM OVA257–264 or LLO190–201 peptide in the presence of GolgiPlug (BD Biosciences). Cells were surface stained with anti-mouse CD8 and CD4, fixed and permeabilized using Cytofix/Cytoperm (BD Biosciences), and stained for intracellular anti-mouse IFN-γ, TNF-α and IL-2. For splenic dendritic cells and T cells, splenocytes were stained with anti-mouse CD11b, CD11c, CD86 and CD40 or anti-mouse CD8, CD4 and CD69, respectively. Flurophore-conjugated antibodies were purchased from eBioscience. Samples were acquired using an LSRII flow cytometer (BD Biosciences) and analyzed using FlowJo software (Tree Star).
Quantitative PCR
BMMs were generated as previously described [59]. In a 6-well plate, 2×106 BMMs were either infected with WT Lm at a multiplicity of infection (MOI) of 2 bacteria per cell or stimulated with 10 µM c-di-AMP. At 30 minutes post infection, gentamicin was added for a final concentration of 50 µg/ml. At 4 and 8 hours post infection, cells were harvested and RNA was purified using the RNAqueous kit (Ambion). RNA was then DNase treated, processed and analyzed as previously described [5].
Bone marrow-derived dendritic cells
Bone marrow from femurs was plated in media containing 20 ng/ml recombinant murine GMCSF (ProSpec) at a density of 5×105 cells/ml in a 24-well plate. At days 2, 4 and 5 media was replaced with fresh media containing 20 ng/ml GMCSF and cells were harvested on day 6. BMDCs were plated at 3×105 cells/well in 48-well plates. Cells were incubated with either 10 µM c-di-AMP, 100 ng/ml lipopolysaccharide (LPS), or 20 µg/ml poly(I∶C) (InvivoGen). After 24 hours, supernatant was assayed for IFN-β using ISRE-L929 cells as previously described [14], or for MCP-1, IL-12p40 (BD OptEIA kit, BD Biosciences) and IL-6 (eBioscience) by ELISA.
Serum cytokines
Serum cytokines were measured using the CBA Mouse Inflammation Kit (BD Biosciences) and analyzed on the LSRII flow cytometer.
Statistical analysis
A two-tailed, Mann-Whitney U test was used to analyze the significance of differences in the means between groups. Significance is indicated as * p<0.05, ** p<0.005, *** p<0.0005 or ns = not significant.
Supporting Information
Zdroje
1. HartyJT, TvinnereimAR, WhiteDW (2000) CD8+ T cell effector mechanisms in resistance to infection. Annual review of immunology 18 : 275–308.
2. PamerEG (2004) Immune responses to Listeria monocytogenes. Nat Rev Immunol 4 : 812–823.
3. SchentenD, MedzhitovR (2011) The control of adaptive immune responses by the innate immune system. Advances in immunology 109 : 87–124.
4. WitteCE, ArcherKA, RaeCS, SauerJD, WoodwardJJ, et al. (2012) Innate immune pathways triggered by Listeria monocytogenes and their role in the induction of cell-mediated immunity. Advances in immunology 113 : 135–156.
5. LeberJH, CrimminsGT, RaghavanS, Meyer-MorseNP, CoxJS, et al. (2008) Distinct TLR - and NLR-mediated transcriptional responses to an intracellular pathogen. PLoS Pathog 4: e6.
6. CossartP, Toledo-AranaA (2008) Listeria monocytogenes, a unique model in infection biology: an overview. Microbes and infection/Institut Pasteur 10 : 1041–1050.
7. O'RiordanM, YiCH, GonzalesR, LeeKD, PortnoyDA (2002) Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. Proc Natl Acad Sci U S A 99 : 13861–13866.
8. BarberGN (2011) Innate immune DNA sensing pathways: STING, AIMII and the regulation of interferon production and inflammatory responses. Current opinion in immunology 23 : 10–20.
9. BarkerJR, KoestlerBJ, CarpenterVK, BurdetteDL, WatersCM, et al. (2013) STING-dependent recognition of cyclic di-AMP mediates type I interferon responses during Chlamydia trachomatis infection. mBio 4: e00018–00013.
10. ZhangX, ShiH, WuJ, SunL, ChenC, et al. (2013) Cyclic GMP-AMP Containing Mixed Phosphodiester Linkages Is An Endogenous High-Affinity Ligand for STING. Molecular cell 51 : 226–235.
11. BurdetteDL, MonroeKM, Sotelo-TrohaK, IwigJS, EckertB, et al. (2011) STING is a direct innate immune sensor of cyclic di-GMP. Nature 478 : 515–518.
12. IshikawaH, MaZ, BarberGN (2009) STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461 : 788–792.
13. McWhirterSM, BarbalatR, MonroeKM, FontanaMF, HyodoM, et al. (2009) A host type I interferon response is induced by cytosolic sensing of the bacterial second messenger cyclic-di-GMP. J Exp Med 206 : 1899–1911.
14. CrimminsGT, HerskovitsAA, RehderK, SivickKE, LauerP, et al. (2008) Listeria monocytogenes multidrug resistance transporters activate a cytosolic surveillance pathway of innate immunity. Proc Natl Acad Sci U S A 105 : 10191–10196.
15. WoodwardJJ, IavaroneAT, PortnoyDA (2010) c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328 : 1703–1705.
16. JinL, HillKK, FilakH, MoganJ, KnowlesH, et al. (2011) MPYS is required for IFN response factor 3 activation and type I IFN production in the response of cultured phagocytes to bacterial second messengers cyclic-di-AMP and cyclic-di-GMP. Journal of immunology 187 : 2595–2601.
17. SauerJD, Sotelo-TrohaK, von MoltkeJ, MonroeKM, RaeCS, et al. (2011) The N-Ethyl-N-Nitrosourea-Induced Goldenticket Mouse Mutant Reveals an Essential Function of Sting in the In Vivo Interferon Response to Listeria monocytogenes and Cyclic Dinucleotides. Infect Immun 79 : 688–694.
18. EbensenT, LibanovaR, SchulzeK, YevsaT, MorrM, et al. (2011) Bis-(3′,5′)-cyclic dimeric adenosine monophosphate: Strong Th1/Th2/Th17 promoting mucosal adjuvant. Vaccine 29 : 5210–5220.
19. EbensenT, SchulzeK, RieseP, LinkC, MorrM, et al. (2007) The bacterial second messenger cyclic diGMP exhibits potent adjuvant properties. Vaccine 25 : 1464–1469.
20. KaraolisDK, MeansTK, YangD, TakahashiM, YoshimuraT, et al. (2007) Bacterial c-di-GMP is an immunostimulatory molecule. J Immunol 178 : 2171–2181.
21. BahjatKS, LiuW, LemmensEE, SchoenbergerSP, PortnoyDA, et al. (2006) Cytosolic entry controls CD8+-T-cell potency during bacterial infection. Infect Immun 74 : 6387–6397.
22. BercheP, GaillardJL, SansonettiPJ (1987) Intracellular growth of Listeria monocytogenes as a prerequisite for in vivo induction of T cell-mediated immunity. J Immunol 138 : 2266–2271.
23. von KoenigCH, FingerH, HofH (1982) Failure of killed Listeria monocytogenes vaccine to produce protective immunity. Nature 297 : 233–234.
24. HartyJT, BevanMJ (1995) Specific immunity to Listeria monocytogenes in the absence of IFN gamma. Immunity 3 : 109–117.
25. EdelsonBT, UnanueER (2002) MyD88-dependent but Toll-like receptor 2-independent innate immunity to Listeria: no role for either in macrophage listericidal activity. Journal of immunology 169 : 3869–3875.
26. KursarM, MittruckerHW, KochM, KohlerA, HermaM, et al. (2004) Protective T cell response against intracellular pathogens in the absence of Toll-like receptor signaling via myeloid differentiation factor 88. International immunology 16 : 415–421.
27. SekiE, TsutsuiH, TsujiNM, HayashiN, AdachiK, et al. (2002) Critical roles of myeloid differentiation factor 88-dependent proinflammatory cytokine release in early phase clearance of Listeria monocytogenes in mice. Journal of immunology 169 : 3863–3868.
28. WaySS, KollmannTR, HajjarAM, WilsonCB (2003) Cutting edge: protective cell-mediated immunity to Listeria monocytogenes in the absence of myeloid differentiation factor 88. J Immunol 171 : 533–537.
29. ChenH, SunH, YouF, SunW, ZhouX, et al. (2011) Activation of STAT6 by STING is critical for antiviral innate immunity. Cell 147 : 436–446.
30. IshikawaH, BarberGN (2008) STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455 : 674–678.
31. CondottaSA, RicherMJ, BadovinacVP, HartyJT (2012) Probing CD8 T cell responses with Listeria monocytogenes infection. Advances in immunology 113 : 51–80.
32. StockingerS, KastnerR, KernbauerE, PilzA, WestermayerS, et al. (2009) Characterization of the interferon-producing cell in mice infected with Listeria monocytogenes. PLoS pathogens 5: e1000355.
33. KepplerSJ, RosenitsK, KoeglT, VucikujaS, AicheleP (2012) Signal 3 cytokines as modulators of primary immune responses during infections: the interplay of type I IFN and IL-12 in CD8 T cell responses. PLoS One 7: e40865.
34. ThompsonLJ, KolumamGA, ThomasS, Murali-KrishnaK (2006) Innate inflammatory signals induced by various pathogens differentially dictate the IFN-I dependence of CD8 T cells for clonal expansion and memory formation. J Immunol 177 : 1746–1754.
35. CarreroJA, CalderonB, UnanueER (2004) Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J Exp Med 200 : 535–540.
36. HuangL, LemosHP, LiL, LiM, ChandlerPR, et al. (2012) Engineering DNA nanoparticles as immunomodulatory reagents that activate regulatory T cells. Journal of immunology 188 : 4913–4920.
37. HuangL, LiL, LemosH, ChandlerPR, PacholczykG, et al. (2013) Cutting Edge: DNA Sensing via the STING Adaptor in Myeloid Dendritic Cells Induces Potent Tolerogenic Responses. Journal of immunology 191 : 3509–13.
38. AbdullahZ, SchleeM, RothS, MraheilMA, BarchetW, et al. (2012) RIG-I detects infection with live Listeria by sensing secreted bacterial nucleic acids. The EMBO journal 31 : 4153–4164.
39. HagmannCA, HerznerAM, AbdullahZ, ZillingerT, JakobsC, et al. (2013) RIG-I Detects Triphosphorylated RNA of Listeria monocytogenes during Infection in Non-Immune Cells. PLoS One 8: e62872.
40. Brzoza-LewisKL, HothJJ, HiltboldEM (2012) Type I interferon signaling regulates the composition of inflammatory infiltrates upon infection with Listeria monocytogenes. Cellular immunology 273 : 41–51.
41. MarshallHD, UrbanSL, WelshRM (2011) Virus-induced transient immune suppression and the inhibition of T cell proliferation by type I interferon. Journal of virology 85 : 5929–5939.
42. NgoiSM, St RoseMC, MenoretAM, SmithDE, ToveyMG, et al. (2012) Presensitizing with a Toll-like receptor 3 ligand impairs CD8 T-cell effector differentiation and IL-33 responsiveness. Proceedings of the National Academy of Sciences of the United States of America 109 : 10486–10491.
43. MonroeKM, McWhirterSM, VanceRE (2010) Induction of type I interferons by bacteria. Cellular microbiology 12 : 881–890.
44. AuerbuchV, BrockstedtDG, Meyer-MorseN, O'RiordanM, PortnoyDA (2004) Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med 200 : 527–533.
45. O'ConnellRM, SahaSK, VaidyaSA, BruhnKW, MirandaGA, et al. (2004) Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med 200 : 437–445.
46. SchwartzKT, CarletonJD, QuillinSJ, RollinsSD, PortnoyDA, et al. (2012) Hyperinduction of host beta interferon by a Listeria monocytogenes strain naturally overexpressing the multidrug efflux pump MdrT. Infection and immunity 80 : 1537–1545.
47. WilsonEB, YamadaDH, ElsaesserH, HerskovitzJ, DengJ, et al. (2013) Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 340 : 202–207.
48. TeijaroJR, NgC, LeeAM, SullivanBM, SheehanKC, et al. (2013) Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340 : 207–211.
49. ManzanilloPS, ShilohMU, PortnoyDA, CoxJS (2012) Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell host & microbe 11 : 469–480.
50. TelescaC, AngelicoM, PiccoloP, NosottiL, MorroneA, et al. (2007) Interferon-alpha treatment of hepatitis D induces tuberculosis exacerbation in an immigrant. The Journal of infection 54: e223–226.
51. BerryMP, GrahamCM, McNabFW, XuZ, BlochSA, et al. (2010) An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466 : 973–977.
52. DinerEJ, BurdetteDL, WilsonSC, MonroeKM, KellenbergerCA, et al. (2013) The Innate Immune DNA Sensor cGAS Produces a Noncanonical Cyclic Dinucleotide that Activates Human STING. Cell reports 3 : 1355–1361.
53. PearceEL, ShenH (2007) Generation of CD8 T cell memory is regulated by IL-12. Journal of immunology 179 : 2074–2081.
54. SauerJD, PereyreS, ArcherKA, BurkeTP, HansonB, et al. (2011) Listeria monocytogenes engineered to activate the Nlrc4 inflammasome are severely attenuated and are poor inducers of protective immunity. Proceedings of the National Academy of Sciences of the United States of America 108 : 12419–12424.
55. BahjatKS, Meyer-MorseN, LemmensEE, ShugartJA, DubenskyTW, et al. (2009) Suppression of cell-mediated immunity following recognition of phagosome-confined bacteria. PLoS pathogens 5: e1000568.
56. BadovinacVP, PorterBB, HartyJT (2004) CD8+ T cell contraction is controlled by early inflammation. Nature immunology 5 : 809–817.
57. BrockstedtDG, GiedlinMA, LeongML, BahjatKS, GaoY, et al. (2004) Listeria-based cancer vaccines that segregate immunogenicity from toxicity. Proc Natl Acad Sci U S A 101 : 13832–13837.
58. WitteCE, WhiteleyAT, BurkeTP, SauerJD, PortnoyDA, et al. (2013) Cyclic di-AMP Is Critical for Listeria monocytogenes Growth, Cell Wall Homeostasis, and Establishment of Infection. mBio 4: e00282–13.
59. PortnoyDA, JacksPS, HinrichsDJ (1988) Role of hemolysin for the intracellular growth of Listeria monocytogenes. J Exp Med 167 : 1459–1471.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 1
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- Lyme Disease: Call for a “Manhattan Project” to Combat the Epidemic
- Origin, Migration Routes and Worldwide Population Genetic Structure of the Wheat Yellow Rust Pathogen f.sp.
- IFNγ/IL-10 Co-producing Cells Dominate the CD4 Response to Malaria in Highly Exposed Children
- Human and Plant Fungal Pathogens: The Role of Secondary Metabolites