
Diapause Formation and Downregulation of Insulin-Like Signaling via DAF-16/FOXO Delays Axonal Degeneration and Neuronal Loss
Axonal degeneration is a key event in the pathogenesis of neurodegenerative conditions. We show here that mec-4d triggered axonal degeneration of Caenorhabditis elegans neurons and mammalian axons share mechanistical similarities, as both are rescued by inhibition of calcium increase, mitochondrial dysfunction, and NMNAT overexpression. We then explore whether reactive oxygen species (ROS) participate in axonal degeneration and neuronal demise. C. elegans dauers have enhanced anti-ROS systems, and dauer mec-4d worms are completely protected from axonal degeneration and neuronal loss. Mechanistically, downregulation of the Insulin/IGF-1-like signaling (IIS) pathway protects neurons from degenerating in a DAF-16/FOXO–dependent manner and is related to superoxide dismutase and catalase-increased expression. Caloric restriction and systemic antioxidant treatment, which decrease oxidative damage, protect C. elegans axons from mec-4d-mediated degeneration and delay Wallerian degeneration in mice. In summary, we show that the IIS pathway is essential in maintaining neuronal homeostasis under pro-degenerative stimuli and identify ROS as a key intermediate of neuronal degeneration in vivo. Since axonal degeneration represents an early pathological event in neurodegeneration, our work identifies potential targets for therapeutic intervention in several conditions characterized by axonal loss and functional impairment.
Published in the journal:
. PLoS Genet 8(12): e32767. doi:10.1371/journal.pgen.1003141
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003141
Summary
Axonal degeneration is a key event in the pathogenesis of neurodegenerative conditions. We show here that mec-4d triggered axonal degeneration of Caenorhabditis elegans neurons and mammalian axons share mechanistical similarities, as both are rescued by inhibition of calcium increase, mitochondrial dysfunction, and NMNAT overexpression. We then explore whether reactive oxygen species (ROS) participate in axonal degeneration and neuronal demise. C. elegans dauers have enhanced anti-ROS systems, and dauer mec-4d worms are completely protected from axonal degeneration and neuronal loss. Mechanistically, downregulation of the Insulin/IGF-1-like signaling (IIS) pathway protects neurons from degenerating in a DAF-16/FOXO–dependent manner and is related to superoxide dismutase and catalase-increased expression. Caloric restriction and systemic antioxidant treatment, which decrease oxidative damage, protect C. elegans axons from mec-4d-mediated degeneration and delay Wallerian degeneration in mice. In summary, we show that the IIS pathway is essential in maintaining neuronal homeostasis under pro-degenerative stimuli and identify ROS as a key intermediate of neuronal degeneration in vivo. Since axonal degeneration represents an early pathological event in neurodegeneration, our work identifies potential targets for therapeutic intervention in several conditions characterized by axonal loss and functional impairment.
Introduction
Neuronal loss constitutes an irreversible end point of several neurodegenerative conditions triggered by diverse stimuli. Nevertheless, early neuronal dysfunction is associated with degeneration of neuronal processes, including axons and dendrites, causing a progressive loss of neuronal function [1], [2]. Axons are particularly susceptible to genetic and toxic insults and represent a key therapeutic target for neuroprotection [3], [4]. Degeneration of axons is non-apoptotic [5], [6], and depends on the activation of the mitochondrial permeability transition pore (mPTP and reference [7]), intra-axonal calcium rise, and calpain activation [8].
Much of our current knowledge regarding the mechanisms of axonal degeneration originates from studies in the Wlds mice, which show delayed axonal degeneration caused by mechanical injuries, hypoxia and other toxic stimuli [5]. The Wlds-dependent protection of axons is given by overexpression of a chimera between nuclear NMNAT1 and a non-catalytic sequence from a ubiquitin ligase [9]. Importantly, slowing down axonal degeneration by Wlds expression in mouse models of neurodegenerative conditions delays the disease progression and severity [10]–[12], pointing out to a crucial role of axonal degeneration in diseases of varied nature.
Despite the advancement in the identification of molecular players and pathways implicated in axonal degeneration, how they integrate into a degenerative process triggered by a wide variety of conditions remains inconclusive and needs further examination in vivo. Therefore it is important to characterize axonal degeneration in tractable model systems such as C. elegans, which are suitable for genetic and pharmacological interventions with functional and morphological readouts of neuronal integrity.
In C. elegans, constitutively active degenerins cause degeneration of neuronal somas and axons by ultrastructural and macroscopical features reminiscent of excitotoxic neuronal death in mammals [13]. Non-apoptotic death of degenerin-expressing C. elegans neurons involves intracellular calcium rise, activation of intracellular proteases and autophagy [14]–[16], and is currently considered to occur by necrosis, also responsible for cell death in stroke, brain trauma and other human neuropathological conditions [17]. Key players in the execution of necrosis are mitochondrial dysfunction and oxidative stress [17], but the role that intracellular ROS generation plays in axonal degeneration has not been addressed in vivo.
The cell is equipped with a homeostatic network to cope with oxidative stress in physiological and pathological situations. One of the master regulators of this homeostatic response to oxidative stress is the Insulin/IGF-1-like signaling (IIS) pathway, which has been associated with the protection of diverse tissues from chemical and genetic insults [18].
Here, we show for the first time that axonal and neuronal degeneration triggered by the hyperactivated degenerin channel MEC-4d, can be prevented by diapause entry and by increasing the anti-oxidative capacity of the neuron. Mechanistically, this increase is mediated by the IIS pathway in a DAF-16/FOXO dependent manner and relies on the activity of the antioxidant enzymes superoxide dismutases (SODs) and catalases. In addition to including the axonal compartment in our analyses, we performed a functional assessment of neuronal integrity to evaluate if prevention of neurodegeneration rendered functional neurons [19], generating a powerful model for mechanistic enquiries. Neuronal function is a critical aspect to be evaluated in therapeutic applications and a readout missing from commonly used models. Importantly, we demonstrate that mec-4d-dependent degeneration can be prevented morphologically and functionally by mPTP inhibition and NMNAT overexpression, which delay axonal degeneration in mammals [7], [9]. Finally, we extend these findings to mice and demonstrate that axonal degeneration can be strongly delayed in vivo by systemic manipulation of the cellular antioxidant capacity. In summary, our work shows that prevention of neuronal degeneration and functional loss can be achieved by a systemic shift of metabolic state leading to an increase in cellular anti-oxidative defense systems.
Results
Morphological and functional time course of MEC-4d mediated degeneration of somas and axons
The use of C. elegans to systematically study axonal degeneration has not been explored so far. C. elegans touch receptor neurons (TRNs) expressing the constitutively open MEC-4d degenerin channel, constitute an ideal model for axonal degeneration as the pro-degenerating stimuli is endogenously triggered (mec-4d expression) and its degeneration and protection can be assessed morphologically and by loss of touch sensitivity. From the six TRNs (ALML, ALMR, PLML, PLMR, AVM and PVM; see Figure 1A), we investigated the AVM neuron because it arises post-embryonically, thus the degenerative process can be observed from the beginning. Additionally, the AVM by itself gives a functional response to anterior touch [19], as when AVMs arise, the other neurons of the anterior touch circuit, the ALMs, have already degenerated (data not shown). To assess the AVM neuron morphologically, we used a strain that expresses gfp under a TRN specific promoter (Pmec-17mec-17::gfp) in wild type or mec-4d(e1611) worms. AVM neurons in mec-4d mutants, appeared and extended axons that reached similar sizes to wild type AVM neurons. Thereafter, AVM somas and axons followed a stereotyped form of degeneration (Figure 1B). First, full length axons become beaded and later become truncated from the most distal end (Figure 1B). Somas become vacuolated and later disappear (Figure 1B; see Methods for a description of categories). This order of events for AVM soma and axon degeneration in mec-4d mutants is consistent with previous qualitative electron microscope analysis [13].

We performed a thorough temporal analysis of the birth and degeneration of AVM somas and axons by using gfp expression as a reporter. In wild type worms, most AVM somas and axons appear 12 hours post hatching; 24 hours after hatching, all axons have reached their full length (Figure 1C and 1D). In mec-4d mutants, 89% of somas have appeared at 12 hours, and around 48.9% axons are intact (Figure 1E and 1F). Thereafter, AVM axons degenerate as described above (Figure 1B). By 24 hours after hatching, only 7% of axons remain intact (Figure 1F), and 58% of somas appear healthy (Figure 1E). 72 hours post hatching, both somas and axons have nearly completely degenerated (Figure 1E and 1F).
We performed touch tests to mec-4d mutants at different times after hatching. The percentage of anterior-touch responsive worms at each time point correlates well with AVMs in the axon wild type (AxW) category (Figure 1G). At 12 hours after hatching, mec-4d animals do not respond because the AVB interneuron, which connects the AVM to motoneurons has not yet arisen [20].
To evaluate our genetic and pharmacological interventions on mec-4d mutants we scored at 72 hours after hatching, when AVM neurons have almost completely degenerated (Figure 1E and 1F), hence an adequate time point to observe unequivocal neuronal protection. Additional relevance comes from the fact that at 72 hours after hatching, worms have reached adulthood and have completed their life cycle.
In summary, we have introduced two novel elements in the model of degeneration triggered by mec-4d: we included the axonal compartment and performed functional analyses of neuronal integrity, which allows a behavioral evaluation of treatments that delay or prevent neuronal degeneration.
Diapause entry prevents neuronal degeneration
Upon harsh conditions, C. elegans larvae enter a diapause state called dauer, from where they resume development once the environment becomes favorable [21]. Among other stress resistant changes, dauers have enhanced anti-oxidative defense systems [22]–[25] due to downregulation of the IIS pathway, which leads to the activation of transcription factors DAF-16/FOXO [26] and SKN-1/Nrf-2 [27]. DAF-16 targets genes like sod-3 and ctl-1 [28], while SKN-1/Nrf-2 mobilizes the phase 2 detoxification response [29], [30].
To examine whether neuronal degeneration is hampered by diapause entry, mec-4d worms were induced to form dauer by starvation and kept in diapause for a week or a month. Strikingly, there was an almost complete prevention of somatic and axonal degeneration by morphological criteria in mec-4d dauer, independently of the diapause length (Figure 2A–2C). Furthermore, AVM neurons of one-month old mec-4d dauers responded to the anterior touch (Figure 2D). In one-month old wild type dauers, 21.9% were anterior and posteriorly touch insensitive (Figure 2D). Considering this percent of insensitive wild type dauers, there is a precise correlation between morphologically wild type axons of mec-4d AVMs and their ability to respond to touch. Noticeably, this protection is stronger than most other genetic or pharmacological treatment reported previously for the mec-4d stressor, and additionally includes a functional readout. Dauer mec-4(u253) loss of function mutants were largely insensitive to touch (2 out of 90 responded to two of ten touches), demonstrating that touch response during dauer is MEC-4 dependent.

We then examined if the protection conferred by dauer was reversed upon exit from this state. For this purpose, dauer worms were placed on food and AVM integrity was scored every 24 hours for 120 hours. Degeneration of somas and axons during dauer exit resumes in a dauer length-independent fashion (Figure 2E–2H).
To rule out that AVM protection during dauer was due to a reduction of mec-4d expression in diapause, we followed MEC-4::GFP expression by means of the uIs58 transgene (Pmec-4mec-4::gfp) in dauer and non-dauer wild type animals. The MEC-4 channel is patterned in regularly spaced puncta along the TRN axons (Figure 2I and reference [31]), allowing the quantification of both intensity by gfp expression and the interpuncta distance, which reflects the distribution of the mechanosensory channel. The same criteria was used to assess whether in mec-4d dauers the expression of MEC-4 was impaired or diminished by the mec-4d (e1611) dominant mutation. Dauer entry did not affect MEC-4 expression nor did the mec-4d (e1611) mutation, as GFP intensity was similar in all three conditions (Figure 2I and 2J). Importantly, the interpuncta distance was also unchanged in both wild type and mec-4d animals compared to non-dauer wild type worms (Figure 2I and 2K), confirming that the distribution of the mechanosensory channel is not affected neither by the mec-4d mutation nor by dauer entry. In addition, both wild type and mec-4d dauers respond to anterior gentle touch (Figure 2D), strongly suggesting that MEC-4 is expressed and functional.
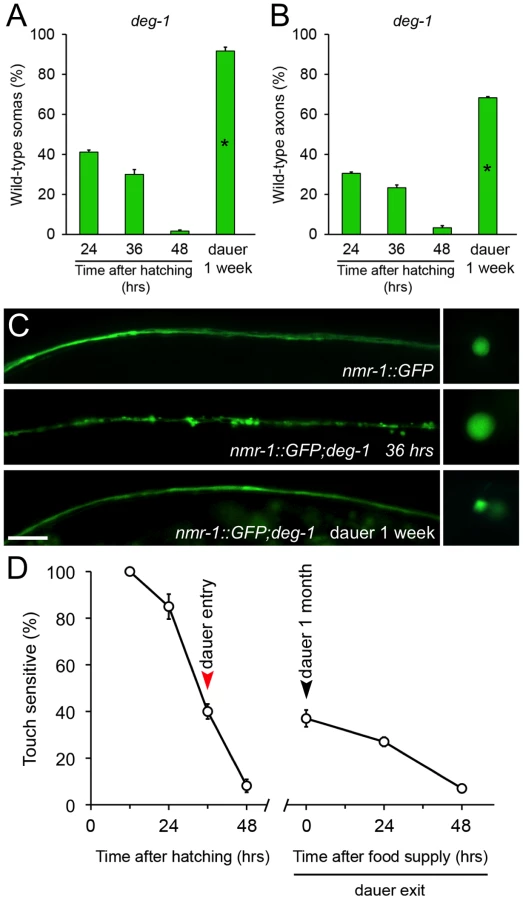
To study whether this phenomenon was extensive to other cells and pro-degenerative stimuli, we tested whether dauer entry prevented degeneration of PVC neurons caused by the deg-1(u38) mutation. Degeneration of the PVC renders worms insensitive to posterior touch [32]. To observe degeneration of somas and axons caused by the deg-1(u38) dominant mutation, we created a strain that expresses gfp under the nmr-1 promoter (Pnmr-1nmr-1::gfp, reference [33]) in a deg-1 (u38) mutant background. At the time corresponding to dauer entry, 30% of PVC somas and 23% of axons were wild type (Figure 3A and 3B). We induced deg-1(u38) animals to form dauer for one month and analyzed the morphology of their PVC neurons and performed a posterior touch test. Impressively, 91% of somas and 68% of axons remained wild type in dauer, and the protection of touch sensitivity was also maintained (Figure 3A–3D), and decayed as worms exited this stage by food reposition (Figure 3D). These results demonstrates that protection conferred by dauer to degenerating neurons is independent of cell type and stimuli, and persists as long as animals remain in diapause.
IIS operates cell autonomously to delay neuronal degeneration
The C. elegans insulin receptor DAF-2 negatively controls DAF-16/FOXO and SKN-1/Nrf-2 activity, and daf-2 deficient worms share with dauers similar transcription signatures leading to an increase in expression of antioxidant enzymes (Figure 4A and references [22], [27]). We wanted to dissect out the DAF-2 dependent components of dauer that protects mec-4d neurons from degeneration. To this end, we generated a strain with a temperature sensitive mutation in daf-2(e1370ts), together with mec-4d(e1611) and a fluorescent marker in TRNs (Pmec-17mec-17::gfp). At 25°C, DAF-2 is not functional in daf-2(e1370ts) mutants and nuclear translocation of DAF-16 and SKN-1 activates their target genes [30], [34]. At 25°C, mec-4d AVMs appear and degenerate and by 72 hours almost no axons or somas remain (Figure 4B and 4C). In contrast, raising daf-2(e1370ts);mec-4d(e1611) at 25°C from the time of hatching confers an almost complete protection to both axons and somas similar to the provided in dauer (Figure 4B and 4C).

Insulin pathway in C. elegans mediates life span extension, dauer formation and stress resistance by systemic and cell autonomous mechanisms [35]–[38]. We wanted to know if cell autonomous downregulation of DAF-2 in TRN prevents mec-4d-dependent TRN degeneration. To this end, we created a systemic RNAi defective strain with TRN-enhanced RNAi also carrying the mec-4d(e1611) mutation and a fluorescent marker in touch neurons (Pmec-17mec-17::gfp, WCH6; see Methods). We fed daf-2 and control unc-22(RNAi) to newly hatched nematodes and scored AVMs 72 hours later. As expected, unc-22(RNAi) treated animals shown no twitching phenotype, since systemic RNAi is impaired in this strain. Impressively, TRN-autonomous daf-2(RNAi) prevented AVM degeneration, rendering about 69% somas and 33% axons in a wild type category, compared to 8% and 3% in control RNAi (Figure 4D and 4E). Taken together our data suggest that repression of the IIS pathway leads to prevention of neuronal demise triggered by mec-4d and that downregulation of the IIS only in TRN is sufficient to provide this protection.
DAF-16 contributes to inhibition of neuronal degeneration associated with reduced IIS signaling
To define the contribution of DAF-16 and SKN-1 in the prevention of mec-4d-dependent neuronal degeneration by DAF-2 downregulation, we performed double RNAi of daf-2 together with daf-16 or skn-1 TRN-autonomously. Combinatorial RNAi has been shown extensively to be an efficient tool to target two genes concomitantly [39]. daf-2/skn-1(RNAi) resembled the protection that daf-2(RNAi) alone conferred to axons and somas while daf-2/daf-16(RNAi) reverted this protection (Figure 5A and 5B). This result suggests that elevation in DAF-16, but not SKN-1 targets in TRN is responsible for neuronal protection after DAF-2 downregulation. No effects in degeneration of somas or axons triggered by mec-4d were observed when skn-1 or daf-16(RNAi) were performed alone (Figure 5A and 5B).

To rule out that the effect observed in daf-2/skn-1(RNAi) was due to inefficient skn-1(RNAi), we fed bacteria expressing skn-1 dsRNA to mec-4d worms to reveal the embryonic lethal phenotype that has been reported for this gene (www.wormbase.org). More than 90% of mec-4d embryos on skn-1(RNAi) did not hatch, confirming the expected phenotype. Additionally, it is unlikely that the lack of effect of daf-2/skn-1(RNAi) on neuronal protection is due to an artifact of double RNAi inefficiency since daf-16 combined with daf-2(RNAi) successfully reverted the daf-2(RNAi) protection.
Key enzymes that control oxidative stress downstream of DAF-2 are superoxide dismutases and catalases [40], which are compartmentalized within the cell to distinct organelles. To directly test whether antioxidant endogenous defenses are needed for the protection conferred by downregulation of the IIS pathway, we performed combinatorial TRN-autonomous RNAi of daf-2 with sod-2, sod-4, ctl-1 or ctl-2. Protection of AVM somas and axons by daf-2(RNAi) was reverted by sod-2, sod-4, ctl-1 and ctl-2(RNAi) in somas and axons (Figure 5C and 5D). These results strongly argue that protection of neuronal degeneration by downregulation of DAF-2 relies on the activity of antioxidant enzymes, supporting a role for ROS in neuronal degeneration triggered by MEC-4d.
Antioxidants prevent neuronal degeneration
Heightened antioxidant capacity of worms by activation of DAF-16/FOXO after daf-2 downregulation, dramatically prevented somatic and axonal degeneration of AVM touch neurons in mec-4d mutants. Importantly, TRNs in mec-4d animals were positive for a fluorogenic ROS-dependent dye (Figure 5E), demonstrating a heightened oxidative stress state in TRNs during the degenerative process. We then tested whether the ROS scavengers trolox, a vitamin E derivative, and ascorbic acid (AA) acutely protect the AVM from mec-4d dependent degeneration. Trolox and AA treatment from the time of hatching greatly protected AVM somas and axons from degeneration at 72 hours (Figure 5F and 5G). Protection of neuronal somas was almost complete, from 8% of worms with wild type somas in controls (no treatment) to 88% and 55% in trolox and AA-treated worms, respectively (Figure 5F). Axons were also protected, with 6% of worms with intact axons in controls, rising to 34% and 22% under trolox and AA treatment, respectively (Figure 5G).
From these results, we conclude that elevated ROS is a key intermediate pathway for neuronal degeneration triggered by MEC-4d stimuli, and that degeneration of both somas and axons can be prevented by systemic antioxidant treatment.
Dietary restriction prevents neuronal degeneration in C. elegans
Animals fed with a caloric restricted diet have reduced oxidative stress levels, and this is proposed as one of the underlying causes for life span extension and delay in age-associated conditions [41]. Hence, we wondered if dietary restriction also impacted degeneration of the AVM in mec-4d worms. We designed an intermittent fasting protocol during dauer exit as mec-4d dauers have protected AVMs and are developmentally synchronized. To this end, mec-4d dauers (Figure 2A–2D) were exited from diapause by a 3 hour food period followed by 9 hours without food (see Methods). This schedule was repeated until 72 hours and AVM integrity was compared to dauers exiting in an ad-libitum food regime. Importantly, nematodes under intermittent fasting developed normally and were fertile when they reached adulthood (data not shown). Dietary restriction conferred a strong protection of 69% to somas and 35% to axons, compared to 7% and 13% in the ad libitum regime, respectively (Figure 6A–6C). This demonstrates, that dietary restriction, a treatment that lowers oxidative stress, protects the TRN from degenerating.

MEC-4d and mammalian axonal degeneration are mechanistically similar
Ideally, the knowledge we gather from a simple model organism as C. elegans should be translated into a mammalian animal model. To this end, we tested if mec-4d triggered neuronal degeneration proceeds by similar mechanisms to degeneration of mammalian neurons. As reported only for somas [14], supplementing EGTA to mec-4d mutants from the time of hatching also significantly reduces degeneration of AVM axons at 72 hours (Figure 7A–7C), as in mammals [8]. Second, it has been shown that axonal degeneration in mammalian neurons requires mPTP activation [7], which is pharmacologically inhibited by cyclosporin A (CsA). CsA treatment strongly reduces both soma and axon demise (Figure 7A and 7B). Finally, delay of axonal degeneration by NMNAT overexpression after diverse stimuli constitutes a molecular signature of non-apoptotic neuronal degeneration in several animal models, including mice, rats, flies and zebrafish [9], [42]–[46]. We asked if overexpression of C. elegans NMNAT protected against mec-4d-triggered neurodegeneration. To this end, we expressed F26H9.4, one of the two C. elegans NMNAT genes, fused to gfp under the TRN specific mec-18 promoter. As expression of the F26H9.4 protein is restricted to the cytosol (Figure 7D), like NMNAT-2 in mammalian cells, and is similar in sequence (data not shown), we called it NMAT-2. Overexpression of NMAT-2::GFP in TRN strongly protected AVM somas and axons against mec-4d mediated degeneration at 72 hours post-hatching (Figure 7E and 7F). Importantly, AVM function was also rescued by NMAT-2 overexpression when the touch response was evaluated (Figure 7G). These results demonstrate that C. elegans neurons degenerate by similar mechanisms to mammalian neurons giving a strong base for extending our findings to a mouse model of neuronal degeneration.

Systemic antioxidant treatment and intermittent fasting delay axonal degeneration in mice
We investigated if systemic antioxidant treatment (AOX) has an effect in Wallerian degeneration in mice, a well established model of axonal degeneration triggered by axotomy, and delayed by NMNAT overexpression [5]. Wild-type mice (C57BL/6J strain) were supplemented with ascorbic acid in the drinking water for 10 days before a nerve injury was performed in the sciatic nerve. After 3, 5 and 7 days, the degree of axonal degeneration distal to the injury site was quantified. In non-treated mice, axons disconnected from their somas degenerate to a large extent in 3 days as seen by the almost complete disappearance of neurofilament markers (Figure 8A and 8D). AOX treatment strongly protected axons from degeneration over 3, 5 and even 7 days (Figure 8B and 8D). Ultrastructurally, control sciatic nerves display mixed populations of conserved myelinated and non-myelinated axons. Three days after damage, distal nerves exhibit degenerating axons and collapsed myelin sheaths (Figure 8E). Consistent with the immunofluorescence results, AOX treatment robustly protects axons from axotomy-dependent degeneration when examined at the EM level (Figure 8F).

We then assessed if dietary restriction replicate in mice the protective effects obtained in mec-4d worms. To this end, mice were subjected to intermittent fasting by means of ad libitum feeding every other day for 10 days. Subsequently, sciatic nerves were injured and analyzed 3, 5 and 7 days later as described above. Impressively, dietary restriction in mice delays axonal degeneration at 3, 5 and 7 days after axotomy as seen both by immunofluorescence (Figure 8C and 8D) and ultrastructural analyses (Figure 8G). Ultrastructurally, the reported increase in axonal mitochondria diameter at 3 days post-axotomy (Figure 8H and reference [7]) was prevented by both AOX and dietary restriction (Figure 8H). In contralateral, non-damaged nerves, neither AOX nor DR treatments affect axonal parameters as density or ultrastructural characteristics (Figure 8B–8D, 8F and 8G).
Due to the robust protection over injury-induced axonal degeneration conferred by AOX, we wanted to establish the dynamics of oxidative stress after nerve damage. To this end, we measured lipid peroxidation, a widely used reporter of oxidative modifications [47]. Injured sciatic nerves kept ex vivo showed a gradual increase in lipid peroxidation over time (Figure 8I), and at 3 days post injury, lipoperoxide levels increased about 20-fold compared to non-injured sciatic nerves (Figure 8I). Importantly, this increase in lipid peroxidation was significantly prevented by AOX treatment 3 days after nerve damage (Figure 8I). In situ observation of oxidative stress using a ROS-sensitive dye shows a clear appearance of positive signal in sciatic nerve axons 24 hours after nerve damage (Figure 8J), consistent with the increase in lipid peroxidation (Figure 8I).
Taken together, our data demonstrate that in mice, as in C. elegans, axonal degeneration depends on the anti-oxidative cellular capacity, which can be enhanced directly by antioxidant treatment or indirectly by dietary restriction.
Discussion
The Insulin/IGF-1-like signaling (IIS) pathway is implicated in a number of cellular processes, including aging and stress resistance; its known role in the regulation of antioxidative mechanisms has been associated with the protection of diverse tissues from chemical and genetic insults [18]. In this work, we show for the first time that downregulation of the IIS pathway in vivo prevents degeneration of both neuronal somas and axons in a cell autonomous fashion. We then show in mice that systemic treatments that increase the antioxidative cellular state protect axons from Wallerian degeneration, a process that has been associated with the progression of several neurodegenerative conditions.
Touch receptor neuron degenerating by the persistent activity of the MEC-4d degenerin channel is an excellent model to study mechanisms of neuronal degeneration. We improved the model by including detailed morphological description of different neuronal compartments, including somas and axons, and a functional readout of neuronal integrity. To date there is no in vivo electrophysiological data about MEC-4d functionality in the touch receptor neurons. We hypothesize that MEC-4d channels, despite exhibiting a persistent sodium current, can be further activated. A deeper exploration on MEC-4d properties could be performed by in vivo electrophysiological recording of TRNs in mec-4d mutants as in O'Hagan et al. [48] under conditions that prevent axonal degeneration.
Degeneration by mec-4d expression is mechanistically overlapping to mammalian neuronal degeneration, as its depends on extracellular Ca2+ ions [14], is delayed by CsA treatment [7], and is protected by overexpression of NMAT-2, the C. elegans homologue of mammalian NMNAT-2 (Figure 7D–7G).
Even though in the C. elegans model of neuronal demise presented here the relationship between soma and axonal degeneration is not distinguished, several evidences suggest that the pro-degenerative stimuli act directly in the axonal compartment, including the expression of MEC-4d in the axon (Figure 2I), their early degeneration compared to somas (Figure 1E–1F) and the axonal protection conferred by NMAT-2 overexpression (Figure 7F), a manipulation known to directly affect axon degeneration in other systems.
Downregulation of the IIS pathway and dauer formation prevent neuronal degeneration
Lack of nutrients signals the downregulation of the IIS pathway in C. elegans and the formation of the dauer larvae, a diapause state that can last for up to four months [18], [49], [50]. Entry into dauer prevented neuronal degeneration as long as worms were kept in diapause. Exit from dauer resumes the degeneration program of TRNs. Protection from neuronal degeneration during dauer was not exclusive of the mec-4d triggered death: nematodes with the deg-1 mutation, which kills the PVC cells, also were protected from neuronal demise during diapause. This speaks of a broad mechanism of neuronal protection in the dauer state, which represents a novel characteristic of this diapause condition.
Downregulation of the IIS pathway by daf-2 mutation protected TRN from degeneration to a great extent, comparable to the dauer-dependent protection, suggesting that the mechanism responsible for neuronal protection in dauer is related to this pathway. By using neuronal-specific RNAi we demonstrated that protective mechanisms of the IIS pathway are activated in a neuronal autonomous fashion (Figure 4D–4E).
Downstream of DAF-2, the transcription factors DAF-16/FOXO and SKN-1/Nrf2 activate different mechanisms of oxidative stress surveillance. DAF-16 targets SODs and catalases while SKN-1 activates the detoxification phase II genes such as gcs-1 [30]. Our results demonstrate that AVM protection from mec-4d induced degeneration is at least in part due to the transcriptional activation of DAF-16 downstream of DAF-2 in a neuronal autonomous fashion (Figure 5A–5B). Consistently, the protection granted by DAF-2 downregulation requires the expression of the antioxidant enzymes SODs and catalases (Figure 5C–5D). It is likely that reduction of cellular oxidative stress by activation of DAF-16/FOXO represents a neuroprotective mechanism in part by its ability to inhibit axonal degeneration, preventing the consequent demise of the neuronal soma. At the same time, the expression of organelle specific anti-ROS systems (i.e. SODs and catalases) as a result of DAF-16 activation may simultaneously prevent soma and axonal demise.
Systemic antioxidant treatment and intermittent fasting delay neurodegeneration in worms and mice
There is a good correlation between factors that promote longevity and those that we have found here to prevent neuronal degeneration [51]. Likely these are processes that rely on mechanisms that preserve the cellular integrity through lowering, for example, oxidative stress.
Caloric restriction (CR) decreases oxidative stress in several tissues [41] and in mammals, reduces insulin levels with a concomitant nuclear translocation of FOXO and activation of its target genes [52]. Several studies have shown that caloric restriction increases life expectancy and is beneficial for healthy aging of the nervous system [52], [53]. CR also reduces brain damage in models of focal cerebral ischemia [54] and Parkinson's disease [55].
In our work, both adult worms and mice subject to CR showed delayed axonal degeneration. CR and reduced IIS are believed to exert their effects by a decrease in cellular oxidative damage [41]. CR causes a decrease in energy expenditure, ROS production and oxidative damage, while downregulation of the IIS increases cellular antioxidant defenses, which elevates resistance to oxidative stress [56]. CR has the advantage of being an intervention that is readily applicable to humans [57]–[59]. Importantly, these acute treatments can be implemented once organisms reach their fertile age with very short or even no pretreatment in the case of C. elegans and mice, and rendering impressive results both by genetic pro-degenerative triggers (worms, degenerin activation) as well as mechanical damage (mice, nerve damage).
Although the mechanisms by which CR increases resistance of cells to age-related pathology are not firmly established, two not mutually exclusive possibilities have been suggested: a reduction in mitochondrial oxyradical production by lowering metabolic flux [60] and activation of cellular antioxidant mechanisms [61]–[64] by an hormetic cellular response after a mild-stress associated to intermittent induction of mitochondrial metabolism and sub-lethal ROS production [65].
Integrative model for ROS–dependent neuronal degeneration
Degeneration of TRN somas by mec-4d expression has been studied as a model of necrosis [66]. Calcium increase, activation of intracellular proteases (including calpains and cathepsins) and autophagy, are intermediary steps leading to vacuolation of TRN somas, which are later engulfed by neighboring cells in a ced-2, 5 and 10-regulated process [67]. Our results demonstrate a critical role for ROS in this process, which can be controlled by manipulating the IIS pathway. This, together with its nmat-2 dependence, better defines this necroptotic death mechanism of neuronal somas. We have further demonstrated that neuronal degeneration in worms and mice also depends on ROS production, which can be counteracted by genetic or pharmacological means, as well as reduction in the metabolic burden to the organism by CR.
Based on our results and published data, we propose a model in which opening of the MEC-4d channel results in an increase in intracellular calcium, which together with the endoplasmic reticulum calcium contribution [14], leads to mitochondrial dysfunction, ATP depletion and ROS generation, which enters into positive feedback loops, finally leading to activation of proteases in somas and axons. Importantly, the presence of axonal beading in mec-4d worms suggests that axonal transport defects might represent an early event triggering degeneration, in agreement with recent data [68]. In both dauer and daf-2 mutants, anti-ROS systems are elevated, decreasing mitochondrial dysfunction and blocking neuronal degeneration (Figure 9).

Accumulative stress with age just as a consequence of living may resemble the acute effect caused by damage to neurons. Factors such as DAF-16/FOXO integrate and orchestrate a response to oxidative stress provoked by either processes being physiological or pathological. Since neurons are mostly irreplaceable, the nervous system is particularly susceptible to aging and injury and may profit from an intrinsic ability to detoxify itself that can be exploited for therapeutic purposes.
Methods
C. elegans growth and maintenance
C. elegans wild type (N2), mutants (daf-2(e1370)III, deg-1(e38)X, mec-4d(e1611)X) and transgenic strains [TU2773 {uIs31(Pmec-17mec-17::gfp); mec-4d(e1611)X}, TU3755 {uIs58(Pmec-4mec-4::gfp)}, VM484 {akIs3(Pnmr-1nmr-1::gfp)}] were grown as previously described [69]. For time course experiments we synchronized worms by collecting the newly hatched L1 two hours after washing off a plate with mixed stage worms. Groups of 30 to 50 worms were gently collected in M9 using a mouth pipette and placed in new plates with food to be examined at the appropriate times. All experiments, unless noted were done at 20°C.
Expression constructs and transformation
A 1 kb fragment 5′ from the start of translation of the F26H9.4 (nmat-2) gene was amplified from genomic DNA using the following primers AAAAGGATCCATGAAACGAGTCGCTCTTCTTGC and TTTTGGATCCATTTTCTGATACAGATTATTCTCCC, that introduced 5′ and 3′ BamHI sites. The resulting PCR fragment was cloned between the mec-18 promoter and yfp coding sequence in TU#739 [70] to create WCH#19. We generated transgenic nematodes by microinjection [71] of 10 ng/µl of WCH#19, 30 ng/µl of pCW2.1 (a ceh-22::gfp plasmid [72]) and 60 ng/µl of pBSK (Stratagene) as filling DNA.
Transgenic strains generated
WCH1 contains wchEx1, an extrachromosomal array containing Pmec-18nmat-2::yfp; Pceh-22gfp expressed in wild-type worms. wchEx1 was crossed into mec-4d(e1611)X to create WCH2. WCH4 contains uIs31(Pmec-17mec-17::gfp) in a daf-2(e1370ts)III; mec-4d(e1611) double mutant. WCH6 contains uIs71(Pmec-18sid-1;Pmyo-2mcherry) and uIs31(Pmec-17mec-17::gfp) arrays in a sid-1(pk3321)V, mec-4d(e1611)X double mutant. uIs58(Pmec-4mec-4::gfp) was crossed into mec-4d(e1611)X animals to create WCH32, and akIs31(Pnmr-1nmr-1::gfp) was crossed into deg-1(u38)X mutants to create WCH33.
Criteria for touch neuron integrity
To assess touch neuron morphology, worms were immobilized by placing them on a drop of 20 mM sodium azide directly on a 2% agarose pad. All worms were observed under a 100× objective, in an upright fluorescent microscope (Olympus) in a 20 minutes range to avoid damage for long exposure to sodium azide.
We used the uIs31 strain (Pmec-17mec-17::gfp) as a control for the time of appearance of the postembryonic cells (AVM and PVM). We established the following morphological categories for somas and axons: Somas, SoW: soma wild-type, SoV: soma vacuolated, swollen several times the wild type size; SoØ: soma absent. Axons, AxW: axon wild-type; AxB: beaded axon of wild type length; AxT: axon truncated, of smaller length than wild type and sometimes detached from soma; AxØ: axon absent. When the axon was truncated and beaded it was scored as AxT.
Since we used gfp expression as the only morphological measure of neuronal integrity, there is a possibility that neuronal degeneration assessed by this mean follows a different degeneration dynamic than when studied by other techniques. The fact that TRN axons degenerate in mec-4d mutants, has been solidly established by electron microscopy analysis [13].
Touch sensitivity
Worms were touched gently with an eyebrow hair [19] with alternative anterior and posterior touches (five times each) to determine an average response. These experiments were done in triplicates for each time point. Because we scored the functionality of the AVM touch neuron, which participates in the anterior touch circuit, only anterior responses (5 in total) are reported here. When AVM is intact (as in mec-4d dauer) the maximum anterior response is of three touches. Only worms that moved when prodded with a platinum wire pick or responded to the nose touch, were assayed. To test the function of the PVC neurons, a posterior (tail) touch was performed in deg-1(u38) animals at various times. deg-1(u38) animals have normal locomotion and are sensitive to anterior touch, which permits unequivocal interpretation of a negative posterior touch response.
Quantification neuronal degeneration in dauer larvae
Mixed-stage worms were grown until all food exhausted to induce the formation of large amounts of dauer [73]. Plates were observed periodically every 24 hours for the appearance of dauer larvae. The first day dauers appeared on the plate was considered the day zero of dauer and from then a week or a month was counted. At the appropriate time, all worms were collected in M9, centrifuged and incubated in 1% SDS for 30 minutes to eliminate all but dauer larvae [21]. Quantification of AVM integrity was carried out in the same way as non-dauer worms (see above).
Touch tests: wild type (N2), uIs31;mec-4d(e1611) or deg-1(u38) SDS treated dauers were placed on a plate without food and allowed to disperse before touching them. All strains were touched 10 times in a head to tail fashion. In deg-1(e38) mutants, which are characterized by tail insensitivity due to death of the PVC neuron, we scored the posterior response [32]. mec-4d(e1611) mutants were assessed for the rescue of the anterior response, as a measure of functionality of the AVM. Responsive mec-4d(e1611) worms were separated then from non-responsive and both groups observed separately in the microscope for AVM integrity.
Dauer recovery: One week and one month dauers were treated with SDS, plated on food and scored every 24 hours for 120 hours. Each time point was done in triplicates of 30 worms each.
Temperature-sensitive experiments
Experiments to restrict the expression of daf-2 in CB1370 [daf-2(e1370ts)III] and WCH4 (daf-2(e1370ts)III; uIs31(Pmec-17gfp); mec-4d(e1611)X were done at 25°C. L3–L4 worms grown at 20°C were passed to 25°C and allowed to grow for 2 days and lay eggs. We synchronized worms by washing off the plates and leaving eggs that remained attached to the bacterial lawn or agar. Two hours later, 50 newly hatched larvae were picked with a mouth pipette to each new plate and kept at 25°C. Each time point was done in triplicates of 30 worms each.
Cell-autonomous RNAi
Bacteria expressing dsRNA from the Ahringer library [74], [75] were grown on LB plates supplemented with ampicillin at 37°C overnight. Next morning a large amount of bacteria was inoculated in LB liquid supplemented with ampicillin and grown for 6–8 h. The resulting culture was seeded onto 1-day-old NGM-IPTG-carbenicillin plates and allowed to dry for 24 hours. For combinatorial RNAi, each bacterial culture was grown separately overnight in liquid LB. Before seeding the plates, the contents of each culture were spun down, concentrated in half the volume and mixed in equal parts to reconstitute the same concentration used in single RNAi experiments. We added 30–50 newly hatched L1 with a mouth pipette onto each plate and grew them at 20°C.
Intermittent fasting
We designed an intermittent fasting protocol with 12 hour-shifts that consisted of 3 hours with ad libitum food followed by 9 hours without food, for 72 hours at 20°C. We began with 1 month-old dauers, selected by SDS treatment as detailed above. After the 3 hours on food, worms were washed off using M9 medium and spun down for 3 minutes at 2000 rpm. Three washes were performed to eliminate bacteria. For the 9 hours without food, worms were placed on NGM plates carrying ampicillin to avoid any growth of E. coli OP50.
C. elegans drug treatment
Drugs were administered in the food, by mixing the appropriate concentrations of the chemical with E. coli OP50. New plates were made every 24 hours to keep the chemical fresh, and worms changed by hand picking on the next morning. The scoring was always done at 72 hours post-hatching. The following drugs and concentrations were used; EGTA (Sigma, 50 mM), cyclosporin A (CsA, LC Laboratories, 50 µM), trolox (Sigma, 100 µM), ascorbic acid (Sigma, 50 mM).
Mice handling, treatment, and surgery
Mice of the wild-type (WT, C57BL/6J) strain were obtained from the University animal house. Transgenic YFP (B6.CG-T6, THY1-YFP) mice strain were obtain from Jackson laboratories (USA). Adult mice 11-week old, weighting 20–25 g were used. Experiments with mice followed protocols approved by the Institutional Animal Care and Use Committees and complied with NIH guidelines.
Ascorbic acid was diluted in the mice drinking water at a concentration of 0,5 g/L. A pretreatment of 10 days was performed prior to the nerve crush. As mice consumes between 4–5 ml of water per day (as measured daily in our experimental groups), the systemic dose of ascorbic acid corresponds to 2,5 mg/g per day. We used the body surface area (BSA) normalization method [76] to calculate the mice dose administration of ascorbic acid from the human dose, in which the Human Equivalent Dose (HED, 10 mg/kg) is multiplied by the ratio between the Km factor of the human (37) and the Km factor of the mouse (3) (Office of New Drugs, 2005), resulting in an animal dose of 123,33 mg/kg. Considering a 20 mg mouse the daily dosage of ascorbic acid given was 2,47 mg/animal, dissolved in its drinking water according to an intake per day of 5 ml/animal (data not shown). No difference in the intake of water with or without ascorbic acid was observed. For dietary restriction, mice were fasted ad libitum every other day with regular mice food for 10 days before nerve crush. Mice weights were daily measured in all experimental groups with no significant differences between them, except in the dietary restricted group, which shows a regular decrease of 3 g after the non-feeding day, but with a net weight increase comparable to the other experimental groups.
For nerve crush, mice were anaesthetized with Avertin (0.37 mg/g) and the sciatic nerve was exposed at the mid-thigh and crushed three times for 5 seconds each, using Dumont #5 forceps, the crush site was marked with graphite powder applied in the forceps and the wound was closed using surgical clips.
Immunofluorescence and electron microscopy of sciatic nerves
For immunofluorescence analysis, sciatic nerves were fixed by immersion in 4% paraformaldehyde in 0.1 M phosphate buffer saline (1× PBS, pH 7.4) for 1 hour. Followed by 3×10 minutes washes in 1× PBS, sucrose gradient (5%, 10%, 20% in 1× PBS) and then embedded in OCT (Sakura Finetek). Cryostat sections from the middle of the explants were cut transversely at 10 µm thickness and mounted on Superfrost Plus slides (Thermo Scientific). Sections were washed in 1× PBS for 10 minutes and then blocked/permeabilized in 0.1% Triton X-100, 2% fish skin gelatin (Sigma Aldrich) in 1× PBS for 1 hour at room temperature (RT). Sections were incubated in primary antibodies in blocking/permeabilizing solution overnight at 4°C, washed in 1× PBS 3×10 minutes, and incubated in secondary antibodies for 2 hours at RT. Sections were washed 3×10 minutes in 1× PBS and mounted in Vectashield (Vector laboratories). Number of axons per area of nerve tissue was assessed in images of neurofilament-immunostained explant sections using the particle analysis macro of ImageJ.
The following antibodies were used for immunofluorescence analysis: rabbit anti-neurofilament heavy chain (Sigma-Aldrich, #N4142) at 1∶1000; chicken anti-neurofilament medium chain (Chemicon International, #AB5753) at 1∶2000; donkey anti-rabbit TRITC (Jackson Immunoresearch Lab. Inc., #711-025-152) at 1∶300; and, goat anti-chicken FITC (Invitrogen, #A-21449) at 1∶300.
For EM analyses, nerves were fixed overnight by immersion in 2.5% glutaraldehyde, 0.01% picric acid, 0.1 M cacodylate buffer (pH 7.4). Nerves were rinsed in the same buffer, immersed in 1% OsO4 for 1 hour followed by in block incubation with 2% uranyl acetate for 2 hours. Nerves were dehydrated with a graded series of ethanol, propylene oxide and infiltrated with Epon (Ted Pella Inc.). Ultrathin sections from the middle of the explants were contrasted with 1% uranyl acetate and lead citrate. Grids were examined with a Philips Tecnai 12 electron microscope operated at 80 kV. Negative films were developed and scanned.
Ex vivo axonal degeneration
Sciatic nerve explants were performed as previously described [7]. Briefly, sciatic nerve segment were dissected from adult mice and cultured in Neurobasal medium supplemented with 2% B27 (Invitrogen), 0.3% L-glutamine, and 1% streptomycin/penicillin. C57BL/6J nerves were incubated in culture medium alone (Vehicle) or with trolox (Sigma-Aldrich, 1 mM). The explant culture medium was daily replaced.
Lipoperoxides content measurement
Sciatic nerves were frozen in liquid nitrogen and homogenized in 10% trichloroacetic acid (TCA). The homogenate was subjected to centrifugation at 10.000 rpm for 10 min at room temperature, the resulting supernatant was used as the lipoperoxide fraction. Oxidized lipids were detected by mixing the supernatant with a reaction buffer (0,5% thiobarbituric acid and 20% TCA). The reaction solution was then incubated at 100°C for 30 min and thereafter the absorbance was measured at 512 nm. Lipoperoxide content was determined using the extinction coefficient of 155 mM−1×cm−1 and normalized to the starting nerve dry weight (modified from reference [77]).
ROS detection in C. elegans and mice
Worm embryos were collected after hypochlorite treatment and embedded in OCT (Sakura Finetek) without a fixation step. Cryostat sections were cut at 10 µm thickness and mounted on Superfrost Plus slides (Thermo Scientific). Sections were washed in 1× PBS for 10 minutes and then incubated at room temperature in the fluorogenic probe CellROX Deep Red Reagent (Invitrogen, C10422) at a final concentration of 20 µM for 30 min. After washing in 1× PBS, sections were fixed in 4% paraformaldehyde in 1× PBS for 10 minutes followed by 3×10 minutes washes in 1× PBS. Sections were mounted in DAPI-containing Vectashield (Vector laboratories).
Sciatic nerves from wild type and YFP expressing mice were dissected at different times post injury and the perineurium was removed under a dissecting microscope. Nerve fiber bundles were attached to 3-aminopropyltriethoxysilane (Sigma-Aldrich) coated slides and incubated with the fluorogenic probe CellROX Deep Red Reagent (Invitrogen, C10422) at final concentration of 20 µM for 1 h at 37°C. After washing in 1×PBS, nerve fibers were mounted in Fluoromount-G (Southern Biotech) and visualized by confocal microscopy.
Zdroje
1. ColemanMP, PerryVH (2002) Axon pathology in neurological disease: a neglected therapeutic target. Trends Neurosci 25 : 532–537.
2. SaxenaS, CaroniP (2007) Mechanisms of axon degeneration: from development to disease. Prog Neurobiol 83 : 174–191.
3. ColemanM (2005) Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci 6 : 889–898.
4. WangJT, MedressZA, BarresBA (2012) Axon degeneration: molecular mechanisms of a self-destruction pathway. J Cell Biol 196 : 7–18.
5. ColemanMP, FreemanMR (2010) Wallerian degeneration, wld(s), and nmnat. Annu Rev Neurosci 33 : 245–267.
6. TaoJ, RollsMM (2011) Dendrites have a rapid program of injury-induced degeneration that is molecularly distinct from developmental pruning. J Neurosci 31 : 5398–5405.
7. BarrientosSA, MartinezNW, YooS, JaraJS, ZamoranoS, et al. (2011) Axonal degeneration is mediated by the mitochondrial permeability transition pore. J Neurosci 31 : 966–978.
8. SchlaepferWW, BungeRP (1973) Effects of calcium ion concentration on the degeneration of amputated axons in tissue culture. J Cell Biol 59 : 456–470.
9. MackTG, ReinerM, BeirowskiB, MiW, EmanuelliM, et al. (2001) Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci 4 : 1199–1206.
10. FerriA, SanesJR, ColemanMP, CunninghamJM, KatoAC (2003) Inhibiting axon degeneration and synapse loss attenuates apoptosis and disease progression in a mouse model of motoneuron disease. Curr Biol 13 : 669–673.
11. KanekoS, WangJ, KanekoM, YiuG, HurrellJM, et al. (2006) Protecting axonal degeneration by increasing nicotinamide adenine dinucleotide levels in experimental autoimmune encephalomyelitis models. J Neurosci 26 : 9794–9804.
12. SajadiA, SchneiderBL, AebischerP (2004) Wlds-mediated protection of dopaminergic fibers in an animal model of Parkinson disease. Curr Biol 14 : 326–330.
13. HallDH, GuG, García-AñoverosJ, GongL, ChalfieM, et al. (1997) Neuropathology of degenerative cell death in Caenorhabditis elegans. J Neurosci 17 : 1033–1045.
14. XuK, TavernarakisN, DriscollM (2001) Necrotic cell death in C. elegans requires the function of calreticulin and regulators of Ca(2+) release from the endoplasmic reticulum. Neuron 31 : 957–971.
15. SyntichakiP, XuK, DriscollM, TavernarakisN (2002) Specific aspartyl and calpain proteases are required for neurodegeneration in C. elegans. Nature 419 : 939–944.
16. TothML, SimonP, KovacsAL, VellaiT (2007) Influence of autophagy genes on ion-channel-dependent neuronal degeneration in Caenorhabditis elegans. J Cell Sci 120 : 1134–1141.
17. VandenabeeleP, GalluzziL, Vanden BergheT, KroemerG (2010) Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11 : 700–714.
18. FinchCE, RuvkunG (2001) The genetics of aging. Annu Rev Genomics Hum Genet 2 : 435–462.
19. ChalfieM, SulstonJ (1981) Developmental genetics of the mechanosensory neurons of Caenorhabditis elegans. Dev Biol 82 : 358–370.
20. ChalfieM, SulstonJE, WhiteJG, SouthgateE, ThomsonJN, et al. (1985) The neural circuit for touch sensitivity in Caenorhabditis elegans. J Neurosci 5 : 956–964.
21. CassadaRC, RussellRL (1975) The dauerlarva, a post-embryonic developmental variant of the nematode Caenorhabditis elegans. Dev Biol 46 : 326–342.
22. BurnellAM, HouthoofdK, O'HanlonK, VanfleterenJR (2005) Alternate metabolism during the dauer stage of the nematode Caenorhabditis elegans. Exp Gerontol 40 : 850–856.
23. HouthoofdK, BraeckmanBP, LenaertsI, BrysK, De VreeseA, et al. (2002) Ageing is reversed, and metabolism is reset to young levels in recovering dauer larvae of C. elegans. Exp Gerontol 37 : 1015–1021.
24. LarsenPL (1993) Aging and resistance to oxidative damage in Caenorhabditis elegans. Proc Natl Acad Sci USA 90 : 8905–8909.
25. VanfleterenJR, De VreeseA (1995) The gerontogenes age-1 and daf-2 determine metabolic rate potential in aging Caenorhabditis elegans. FASEB J 9 : 1355–1361.
26. GottliebS, RuvkunG (1994) daf-2, daf-16 and daf-23: genetically interacting genes controlling Dauer formation in Caenorhabditis elegans. Genetics 137 : 107–120.
27. TulletJM, HertweckM, AnJH, BakerJ, HwangJY, et al. (2008) Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell 132 : 1025–1038.
28. HondaY, HondaS (1999) The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J 13 : 1385–1393.
29. McMahonM, ItohK, YamamotoM, ChanasSA, HendersonCJ, et al. (2001) The Cap'n'Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res 61 : 3299–3307.
30. AnJH, BlackwellTK (2003) SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes Dev 17 : 1882–1893.
31. ChelurDS, ErnstromGG, GoodmanMB, YaoCA, ChenL, et al. (2002) The mechanosensory protein MEC-6 is a subunit of the C. elegans touch-cell degenerin channel. Nature 420 : 669–673.
32. ChalfieM, WolinskyE (1990) The identification and suppression of inherited neurodegeneration in Caenorhabditis elegans. Nature 345 : 410–416.
33. BrockiePJ, MadsenDM, ZhengY, MellemJ, MaricqAV (2001) Differential expression of glutamate receptor subunits in the nervous system of Caenorhabditis elegans and their regulation by the homeodomain protein UNC-42. J Neurosci 21 : 1510–1522.
34. LinK, HsinH, LibinaN, KenyonC (2001) Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat Genet 28 : 139–145.
35. Arantes-OliveiraN, ApfeldJ, DillinA, KenyonC (2002) Regulation of life-span by germ-line stem cells in Caenorhabditis elegans. Science 295 : 502–505.
36. DurieuxJ, WolffS, DillinA (2011) The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144 : 79–91.
37. HsinH, KenyonC (1999) Signals from the reproductive system regulate the lifespan of C. elegans. Nature 399 : 362–366.
38. LibinaN, BermanJR, KenyonC (2003) Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell 115 : 489–502.
39. LehnerB, TischlerJ, FraserAG (2006) RNAi screens in Caenorhabditis elegans in a 96-well liquid format and their application to the systematic identification of genetic interactions. Nat Protoc 1 : 1617–1620.
40. MurphyCT, McCarrollSA, BargmannCI, FraserA, KamathRS, et al. (2003) Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424 : 277–283.
41. SohalRS (2002) Role of oxidative stress and protein oxidation in the aging process. Free Radic Biol Med 33 : 37–44.
42. AdalbertR, GillingwaterTH, HaleyJE, BridgeK, BeirowskiB, et al. (2005) A rat model of slow Wallerian degeneration (WldS) with improved preservation of neuromuscular synapses. Eur J Neurosci 21 : 271–277.
43. HoopferED, MclaughlinT, WattsRJ, SchuldinerO, O'LearyDDM, et al. (2006) Wlds protection distinguishes axon degeneration following injury from naturally occurring developmental pruning. Neuron 50 : 883–895.
44. MacDonaldJM, BeachMG, PorpigliaE, SheehanAE, WattsRJ, et al. (2006) The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron 50 : 869–881.
45. ZhaiRG, CaoY, HiesingerPR, ZhouY, MehtaSQ, et al. (2006) Drosophila NMNAT maintains neural integrity independent of its NAD synthesis activity. PLoS Biol 4: e416 doi:10.1371/journal.pbio.0040416.
46. FengY, YanT, ZhengJ, GeX, MuY, et al. (2010) Overexpression of Wlds or Nmnat2 in mauthner cells by single-cell electroporation delays axon degeneration in live zebrafish. J Neurosci Res 88 : 3319–3327.
47. ReedTT (2011) Lipid peroxidation and neurodegenerative disease. Free Radic Biol Med 51 : 1302–1319.
48. O'HaganR, ChalfieM, GoodmanMB (2005) The MEC-4 DEG/ENaC channel of Caenorhabditis elegans touch receptor neurons transduces mechanical signals. Nat Neurosci 8 : 43–50.
49. HendersonST, JohnsonTE (2001) daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr Biol 11 : 1975–1980.
50. TatarM, BartkeA, AntebiA (2003) The endocrine regulation of aging by insulin-like signals. Science 299 : 1346–1351.
51. MattsonMP, MagnusT (2006) Ageing and neuronal vulnerability. Nat Rev Neurosci 7 : 278–294.
52. MartinB, MattsonMP, MaudsleyS (2006) Caloric restriction and intermittent fasting: two potential diets for successful brain aging. Ageing Res Rev 5 : 332–353.
53. McCayCM, CrowellMF, MaynardLA (1989) The effect of retarded growth upon the length of life span and upon the ultimate body size. 1935 155–171; discussion 172 p.
54. YuZF, MattsonMP (1999) Dietary restriction and 2-deoxyglucose administration reduce focal ischemic brain damage and improve behavioral outcome: evidence for a preconditioning mechanism. J Neurosci Res 57 : 830–839.
55. DuanW, RangnekarVM, MattsonMP (1999) Prostate apoptosis response-4 production in synaptic compartments following apoptotic and excitotoxic insults: evidence for a pivotal role in mitochondrial dysfunction and neuronal degeneration. J Neurochem 72 : 2312–2322.
56. KregelKC, ZhangHJ (2007) An integrated view of oxidative stress in aging: basic mechanisms, functional effects, and pathological considerations. Am J Physiol Regul Integr Comp Physiol 292: R18–36.
57. FontanaL, MeyerTE, KleinS, HolloszyJO (2004) Long-term calorie restriction is highly effective in reducing the risk for atherosclerosis in humans. Proc Natl Acad Sci U S A 101 : 6659–6663.
58. MeyerTE, KovacsSJ, EhsaniAA, KleinS, HolloszyJO, et al. (2006) Long-term caloric restriction ameliorates the decline in diastolic function in humans. J Am Coll Cardiol 47 : 398–402.
59. CivitareseAE, CarlingS, HeilbronnLK, HulverMH, UkropcovaB, et al. (2007) Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med 4: e76 doi:10.1371/journal.pmed.0040076.
60. SohalRS, WeindruchR (1996) Oxidative stress, caloric restriction, and aging. Science 273 : 59–63.
61. AlyKB, PipkinJL, HinsonWG, FeuersRJ, DuffyPH, et al. (1994) Chronic caloric restriction induces stress proteins in the hypothalamus of rats. Mech Ageing Dev 76 : 11–23.
62. EhrenfriedJA, EversBM, ChuKU, TownsendCMJr, ThompsonJC (1996) Caloric restriction increases the expression of heat shock protein in the gut. Ann Surg 223 : 592–597; discussion 597–599.
63. HeydariAR, YouS, TakahashiR, GutsmannA, SargeKD, et al. (1996) Effect of caloric restriction on the expression of heat shock protein 70 and the activation of heat shock transcription factor 1. Dev Genet 18 : 114–124.
64. LeeCK, KloppRG, WeindruchR, ProllaTA (1999) Gene expression profile of aging and its retardation by caloric restriction. Science 285 : 1390–1393.
65. SchulzTJ, ZarseK, VoigtA, UrbanN, BirringerM, et al. (2007) Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab 6 : 280–293.
66. VlachosM, TavernarakisN (2010) Non-apoptotic cell death in Caenorhabditis elegans. Dev Dyn 239 : 1337–1351.
67. ChungS, GumiennyTL, HengartnerMO, DriscollM (2000) A common set of engulfment genes mediates removal of both apoptotic and necrotic cell corpses in C. elegans. Nat Cell Biol 2 : 931–937.
68. GilleyJ, ColemanMP (2010) Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol 8: e1000300 doi:10.1371/journal.pbio.1000300.
69. BrennerS (1974) The genetics of Caenorhabditis elegans. Genetics 77 : 71–94.
70. ChelurDS, ChalfieM (2007) Targeted cell killing by reconstituted caspases. Proc Natl Acad Sci U S A 104 : 2283–2288.
71. MelloCC, KramerJM, StinchcombD, AmbrosV (1991) Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. Embo J 10 : 3959–3970.
72. OkkemaPG, FireA (1994) The Caenorhabditis elegans NK-2 class homeoprotein CEH-22 is involved in combinatorial activation of gene expression in pharyngeal muscle. Development 120 : 2175–2186.
73. GoldenJW, RiddleDL (1984) The Caenorhabditis elegans dauer larva: developmental effects of pheromone, food, and temperature. Dev Biol 102 : 368–378.
74. FraserAG, KamathRS, ZipperlenP, Martinez-CamposM, SohrmannM, et al. (2000) Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature 408 : 325–330.
75. KamathRS, FraserAG, DongY, PoulinG, DurbinR, et al. (2003) Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421 : 231–237.
76. Reagan-ShawS, NihalM, AhmadN (2008) Dose translation from animal to human studies revisited. FASEB J 22 : 659–661.
77. ContrerasL, MellaD, MoenneA, CorreaJA (2009) Differential responses to copper-induced oxidative stress in the marine macroalgae Lessonia nigrescens and Scytosiphon lomentaria (Phaeophyceae). Aquat Toxicol 94 : 94–102.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 12
Nejčtenější v tomto čísle
- Population Genomics of Sub-Saharan : African Diversity and Non-African Admixture
- Dnmt3a Protects Active Chromosome Domains against Cancer-Associated Hypomethylation
- Excessive Astrocyte-Derived Neurotrophin-3 Contributes to the Abnormal Neuronal Dendritic Development in a Mouse Model of Fragile X Syndrome
- Pre-Disposition and Epigenetics Govern Variation in Bacterial Survival upon Stress