
Construction of a Global Pain Systems Network Highlights Phospholipid Signaling as a Regulator of Heat Nociception
The ability to perceive noxious stimuli is critical for an animal's survival in the face of environmental danger, and thus pain perception is likely to be under stringent evolutionary pressure. Using a neuronal-specific RNAi knock-down strategy in adult Drosophila, we recently completed a genome-wide functional annotation of heat nociception that allowed us to identify α2δ3 as a novel pain gene. Here we report construction of an evolutionary-conserved, system-level, global molecular pain network map. Our systems map is markedly enriched for multiple genes associated with human pain and predicts a plethora of novel candidate pain pathways. One central node of this pain network is phospholipid signaling, which has been implicated before in pain processing. To further investigate the role of phospholipid signaling in mammalian heat pain perception, we analysed the phenotype of PIP5Kα and PI3Kγ mutant mice. Intriguingly, both of these mice exhibit pronounced hypersensitivity to noxious heat and capsaicin-induced pain, which directly mapped through PI3Kγ kinase-dead knock-in mice to PI3Kγ lipid kinase activity. Using single primary sensory neuron recording, PI3Kγ function was mechanistically linked to a negative regulation of TRPV1 channel transduction. Our data provide a systems map for heat nociception and reinforces the extraordinary conservation of molecular mechanisms of nociception across different species.
Published in the journal:
. PLoS Genet 8(12): e32767. doi:10.1371/journal.pgen.1003071
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003071
Summary
The ability to perceive noxious stimuli is critical for an animal's survival in the face of environmental danger, and thus pain perception is likely to be under stringent evolutionary pressure. Using a neuronal-specific RNAi knock-down strategy in adult Drosophila, we recently completed a genome-wide functional annotation of heat nociception that allowed us to identify α2δ3 as a novel pain gene. Here we report construction of an evolutionary-conserved, system-level, global molecular pain network map. Our systems map is markedly enriched for multiple genes associated with human pain and predicts a plethora of novel candidate pain pathways. One central node of this pain network is phospholipid signaling, which has been implicated before in pain processing. To further investigate the role of phospholipid signaling in mammalian heat pain perception, we analysed the phenotype of PIP5Kα and PI3Kγ mutant mice. Intriguingly, both of these mice exhibit pronounced hypersensitivity to noxious heat and capsaicin-induced pain, which directly mapped through PI3Kγ kinase-dead knock-in mice to PI3Kγ lipid kinase activity. Using single primary sensory neuron recording, PI3Kγ function was mechanistically linked to a negative regulation of TRPV1 channel transduction. Our data provide a systems map for heat nociception and reinforces the extraordinary conservation of molecular mechanisms of nociception across different species.
Introduction
Although studies in inbred mouse strains and in human twin cohorts have indicated that pain has a strong genetic component [1]–[5], with an estimated heritability of ∼50%, little is known about the specific genes involved in regulating pain sensitivity across phyla. Further, conservation of gene function between species and across evolutionary time acts as a useful tool to develop an understanding of core genetic mechanisms relative to more specialized programs and how they influence behavior [6]. Drosophila is an excellent model organism for characterizing genetic regulators of behavior such as nociception [7]. Use of Drosophila genetics has highlighted a conserved role for multiple genes in the detection and avoidance of noxious heat [8], [9], and recent work on mechanosensation suggests the genetics of this process is likely also highly conserved across phyla [10]. We have previously reported a global in vivo RNAi screen for avoidance of noxious heat in Drosophila, and identification of hundreds of novel genes required in the adult fly, for its manifestation [8]. To interrogate this resource, we have now constructed a global systems network of heat pain. Our goal was to identify potentially conserved genes and pathways involved in pain perception, to provide a tool for focusing research on key pain molecular pathways.
One pathway highlighted in this global systems network was phosphatidylinositol signaling. Phosphatidylinositol signaling is a second messenger cascade involving the sequential phosphorylation of phosphatidylinositol 4-phosphate (PIP) to generate phosphatidylinositol 4,5-bisphosphate (PIP2) via PIP5' kinases (phosphatidylinositol-5-OH kinase; PIP5K), and then phosphorylation of PIP2 via PI3 kinases (phosphatidylinositol-3-OH kinase; PI3K) to generate phosphatidylinositol 3,4,5-trisphosphate (PIP3). In mammalian systems phospholipid signaling has been implicated in regulating pain perception [11]–[13], TRPV1 function [14]–[21], and itch [22], but how phosphatidylinositol signaling is involved in mammalian nociception is controversial, with data suggesting for example that PIP2 may either increase or decrease TRPV1 function [23], [24]. Based on the suggested involvement of phospholipid signaling in our conserved functional pain network, and in context of the controversial role for phosphatidylinositol signaling in pain perception, we employed genetic approaches to evaluate the role of phosphatidylinositol signaling in mammalian nociception.
Results/Discussion
To construct a global systems network of heat pain we first identified potential mouse and human orthologs of fly candidate pain genes (Figure S1). Of the 580 candidate fly thermal nociception genes we had previously identified [8], 399 had human orthologs (Table S1), many of which are known mammalian pain genes (Table S2). Gene ontology (GO) analyses of the human and mouse orthologs of the fly thermal nociception hits showed a marked enrichment of genes involved in neurotransmission and secretion, housekeeping systems such as mitochondrial structure, ATP synthesis, metabolism, or calcium signaling (Figure S2A–S2C and Table S3). We next generated an interaction map based on first-degree binding partners for the fly thermal nociception hits (Table S4). All binding partners were identified in yeast-2-hybrid screens and reported in the biomolecular interaction network database BIND, i.e. binding partners experimentally confirmed to interact with the candidate genes. Among the first degree binding partners in this interaction network, we found fly allatostatin C receptor 1, which has homology with mammalian opioid receptors, a fly homolog of Lmx1b, which regulates central serotonergic responses to opioids [25], a fly gene related to the nuclear factor-erythroid 2-related factor 2, which has anti-nociceptive effects [26] by inducing upregulation of heme oxygenase-1, and tyrosine hydroxylase, the enzyme required for dopamine and catecholamine production [27]. Thus, our genome-wide functional screen for thermal nociception in flies has generated a human gene network that includes orthologs of several known mammalian pain genes, in addition to numerous uncharacterized genes and pathway not previously associated with pain perception.
To construct a mammalian systems map of thermal nociception, we performed an enrichment analysis (using KEGG pathways and Broad Institute C2 gene sets) on the mouse and human orthologs of the fly pain genes and their first degree binding partners (Figure S1; Table S5 and Table S6). We found significant enrichment of genes (hypergeometric enrichment >90%) involved in mitochondria, metabolism, calcium signaling, inflammation, cell adhesion, RNA processing, and neurotransmission. Finally, to generate a comprehensive conserved network map of thermal nociception, the KEGG pathways from Drosophila, mouse and human were combined with relevant gene sets from the C2 annotations to create a global putative “nociception network” (Figure 1; Table S7). From this combined systems network, we identified gene sets or pathways known to play key roles in many major neural systems.
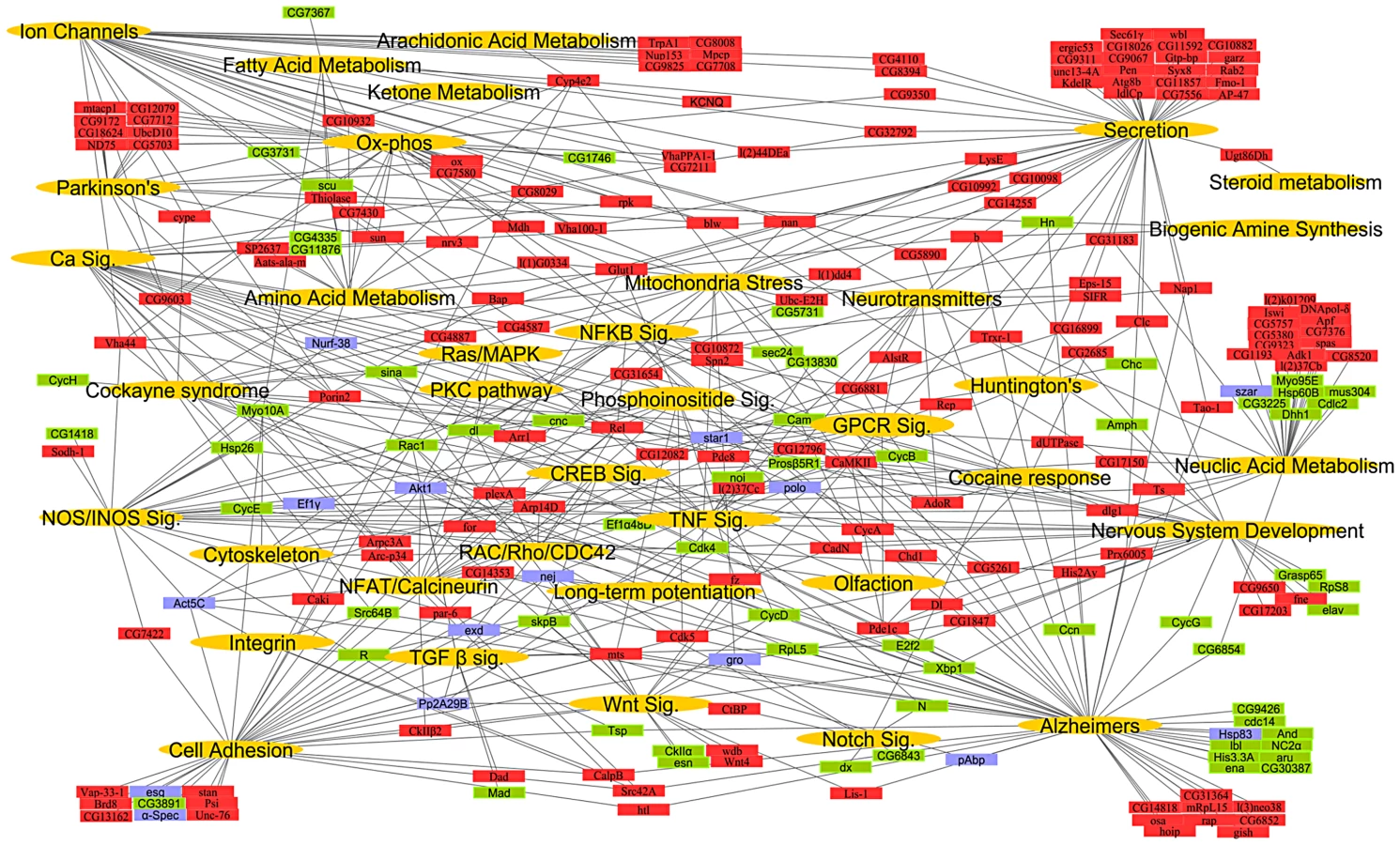
The connectivity of the entire systems map remains intact even after omitting all binding partners (Figure S3), i.e., expansion of the “pain” network map by including binding partners does not introduce a bias. Moreover, all except one pathway in the network map has >50% representation from the Drosophila heat pain hits alone. To test whether this approach can predict mammalian pain genes, we measured the overlap of the direct candidate pain hits and their binding partners with previous rat microarray expression profiling data generated from pain studies [28] and all “pain” annotated genes from OMIM (Online Mendelian Inheritance in Man, NCBI). Intriguingly, we found a 38.55% overlap of our direct pain hits and a 42.33% overlap of their binding partners with the microarray and OMIM data for pain. In contrast, 100 random gene lists gave a maximum of 6% overlapping genes and a minimum of 0% (Table 1; Table S8). Thus, our hypothesis-free systems map is markedly enriched for genes known to be associated with rodent and human pain. Considering the complexity of the neuronal network involved in generating nociception in the periphery and CNS, these pathways may operate across several neurons; however, our genome-wide functional fly pain screen and in silico data mining provide a road map for conserved molecular components and pathways putatively involved in heat nociception globally across phyla.

We next wanted to validate whether this “nociception network” has the power to identify conserved pathways involved in nociception, and if this pathway information can then help to pinpoint key mammalian pain genes. To this end we focused our first efforts on phosphatidylinositol signaling, one of the major nodes predicted from the pain systems map, and a heat pain precedented pathway. Phosphatidylinositol signaling has been implicated in heat nociception and regulation of TRPV1 by multiple groups [11], [13], [16], [18], however its precise role has been controversial [16], [18], [23], and the specific participation of different phospholipid kinases has never been evaluated genetically, which we now decided to do.
Phosphatidylinositol signaling involves the generation of PIP2 via PIP5K, and then phosphorylation of PIP2 via PI3K to generate PIP3. PIP5Kα is highly expressed in the nervous system but no neuronal function for this kinase has been established [29]. PIP5Kα mutant mice are viable and exhibit an exaggerated anaphylactic immune reaction in response to Fc-receptor engagement [30]. We find that PIP5Kα mutant mice exhibit a significant hyper-responsiveness to radiant heat (Figure 2A) and contact heat, when compared to littermate controls (Figure 2B). In mammals, TRPV1 is the prototypical noxious heat thermo-receptor, and is also the receptor for capsaicin, the active ingredient in chili peppers [31]. We therefore tested whether PIP5Kα mutant mice exhibit exaggerated TRPV1 agonist responses. Indeed, following capsaicin injection, PIP5Kα mutant mice display heightened reactivity compared to littermate controls (Figure 2C) but exhibited no difference in mechanical pain threshold using the von Frey test (Figure 2D).
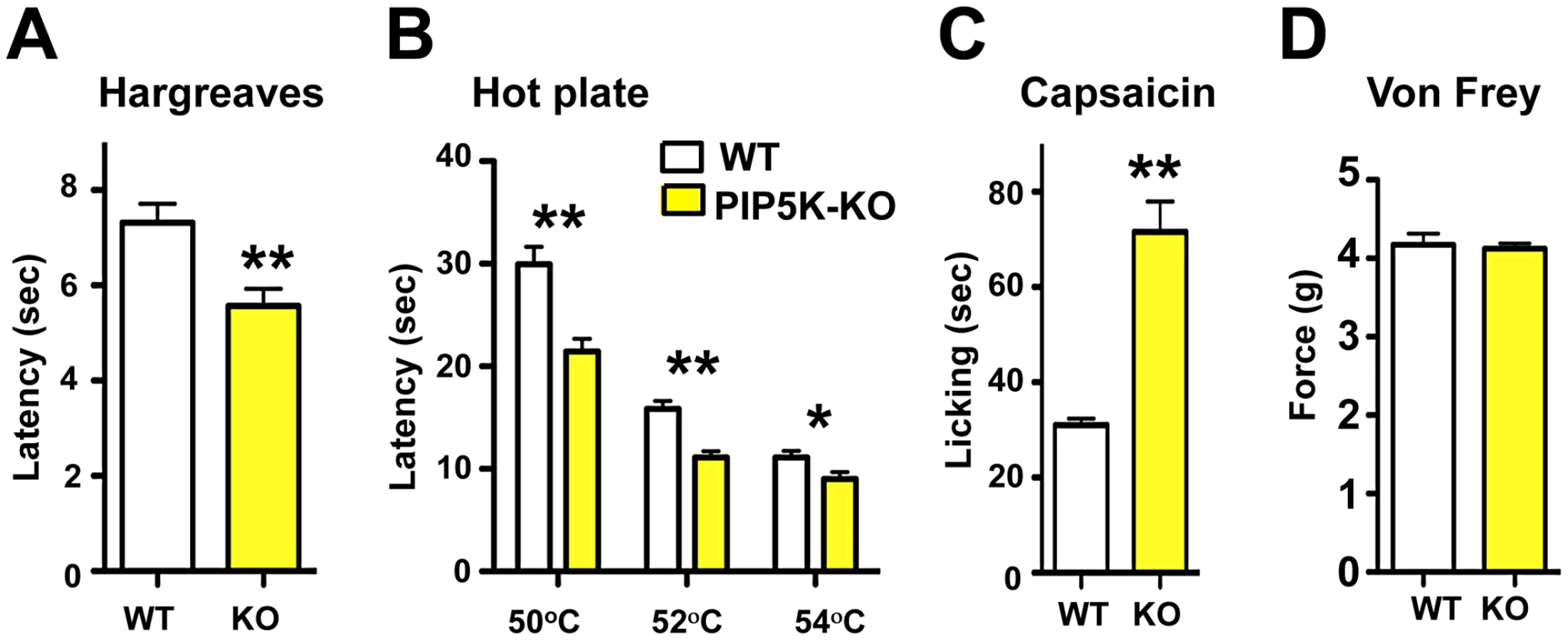
PI3Kγ, the only G-protein coupled PI3K, is expressed in TRPV1-positive peripheral sensory neurons in both rats and mice [32], [33] and has been implicated in morphine-induced peripheral analgesia [32] and morphine tolerance [33]. Use of non-specific inhibitors, like wortmannin, has suggested a role for PI3' kinases in producing NGF-mediated TRPV1 sensitization [21],[34]. We therefore tested thermal nociception in PI3Kγ (p110γ) mutant mice [35] and found that these mice, like the PIP5Kα null mice, also exhibit an exaggerated behavioral response to radiant heat plantar stimulation using the Hargreaves test (Figure 3A). This enhanced pain sensitivity was confirmed using the hot plate assay (Figure 3B). PI3Kγ mutant mice also exhibit an enhanced pain response to a capsaicin challenge (Figure 3C). Similar to PIP5Kα null mice, the mechanical pain threshold using the von Frey test (Figure 3D), and the behavioral responses to acetone application (a cooling sensation) (Figure 3E) were comparable between control and PI3Kγ−/− littermates. Of note we found a similar thermal hyperalgesia phenotype in a second independent PI3Kγ−/− mutant mouse strain [36] (not shown). Thus, genetic loss of PI3Kγ and PIP5Kα results in enhanced pain responses to heat and capsaicin, providing evidence that phosphatidylinositol signaling acts as a negative regulator of heat pain perception and TRPV1 reactivity in vivo.

Since PIP5Kα and PI3Kγ cooperate to sequentially generate PIP3, and both mutant mice exhibit a similar hypersensitivity phenotype, we focused further on the role of PI3Kγ and PIP3 generation in setting the threshold for heat pain perception. PI3Kγ is highly expressed in haematopoietic cells and functions as key mediator of inflammatory cell migration to the site of injury [35], [37]. We therefore tested PI3Kγ mutant mice for potential defects in inflammation-induced pain sensitization, i.e. thermal hyperalgesia. PI3Kγ−/− and control mice developed comparable levels of thermal hyperalgesia (Figure S4A and S4B) following plantar CFA injection. CFA-induced inflammation, as determined by paw swelling, was also comparable between mutant and control mice (Figure S4C). To further exclude a potential role of haematopoietic cells, we transplanted wild type bone marrow into PI3Kγ mutant mice (WT→KO) and PI3Kγ mutant bone marrow into wild type mice (KO→WT). The presence of a wild type haematopoietic system did not rescue the enhanced sensitivity to thermal pain in the PI3Kγ mutant background (Figure 3F), i.e. the requirement for PI3Kγ in thermal sensing maps to non-haematopoeitic cells. PI3Kγ has been shown to act both in a kinase-dependent fashion, through conversion of PIP2 to PIP3, and in a kinase-independent manner [38]. We therefore tested the behavioral response of PI3Kγ kinase-dead (KD) knock-in mice. PI3Kγ KD mice exhibited a heightened reaction to noxious heat with a reduced thermal nociception latency (Figure 3G), comparable to the enhanced heat pain responses observed in complete PI3Kγ mutant mice. Thus, the kinase activity generating PIP3 modulates heat pain. We also assessed the general neurological phenotypes of PI3Kγ−/− mice, all of which appeared normal (Figure 3H; Figure S5A–S5G). Furthermore, the overall morphology and histology of the central nervous system appeared normal in PI3Kγ−/− mice. These data demonstrate that generation of PIP3 through PI3Kγ negatively regulates pain sensitivity in vivo.
To test if the phosphatidylinositol signaling pathway acts in primary sensory nociceptors, we employed electrophysiology on isolated wild type and PI3Kγ−/− dorsal root ganglion (DRG) neurons. PI3Kγ−/− DRG neurons responded to a thermal ramp (Figure 4A) with a significantly increased steepness in the inward current response to increasing temperature when compared to control neurons. This translated into a substantial increase in the Q10 value (Figure 4B), a measure of temperature-dependent rate change in channel conductivity, indicating that PI3Kγ−/− DRG cells exhibit massive hyper-activation in response to noxious heat, although initiation of this response occurs at a slightly elevated temperature (44.77°C for PI3Kγ−/− vs 42.22°C for wild type DRG neurons, Figure 4A). Since we also observed an enhanced response to capsaicin in PI3Kγ−/− mice in vivo, we directly tested TRPV1 reactivity to capsaicin in sensory neurons in vitro. In accordance with our behavioral data, isolated PI3Kγ−/− DRG neurons exhibited augmented sensitivity to capsaicin (Figure 4C and 4D). Thus, PI3Kγ functions as a negative regulator of TRPV1 responses in nociceptive neurons.

Our data provide a conserved functional systems network map for pain behaviour. This network map revealed many pathways and gene sets previously reported to be involved in mammalian nociception, including multiple genes annotated as candidate pain genes in the human OMIM database. Thus, our systems approach, starting from a functional whole genome fly screen and bioinformatic construction of a conserved pain network map, has the power to identify regulators of mammalian nociception. Our network pointed to a key role for phosphatidylinositol signaling in noxious heat nociception. Positive as well as negative regulatory functions of phosphatidylinositol signaling on the thermal nociceptive sensor TRPV1 have been reported [15], [16], [19], [20], [23], [24]. Our results provide genetic data that the phosphatidylinositol signaling pathway is relevant to heat pain sensitivity in vivo. In particular, we find that the lipid kinases PIP5α and PI3Kγ are involved in regulating heat nociception responses by acting as negative modulators of thermal pain perception and TRPV1 activity.
Our data reinforce the extraordinary evolutionary conservation of the neurobiological mechanisms of nociception, from its manifestation as an acute damage avoidance response in simple organisms like flies to the complex sensation of pain in mammals. When used in conjunction with additional complimentary approaches (e.g. published literature, gene expression profiling, or genetic association studies), this systems network map should be a valuable tool to further pinpoint and prioritize novel candidate nociception genes in mammals.
Materials and Methods
Bioinformatics analysis
Identification of fly orthologs in mouse and human was done using pre-computed orthology predictions [39]. Gene Ontology (GO) analysis was performed using GOstat. Binding partner identification was done using GeneSpring GX. Hypergeometric tests were used to identify over-represented gene lists (BROAD Institute) and pathways (KEGG) amongst the pain hits and to generate a conserved systems map. Pain genes and binding partners in the system map that have been annotated as pain genes in the Online Mendelian Inheritance in Man database or by our previous Microarray experiments [28] were also identified.
Mouse behavioral tests
PI3Kγ (p110γ) knock out [35], [36], kinase dead PI3Kγ knock-in [38], and PIP5Kα mutant mice [30] and have been previously described. Thermal and mechanical sensitivities were assessed using the Hargreaves, hot plate, and von Frey tests. Capsaicin behavior was assessed over 5 minutes following intraplantar injection of capsaicin.
Single DRG neuron recordings
Lumbar dorsal root ganglia (DRG) were harvested as previously reported [40], [41]. Patch-clamp recordings were performed using the whole-cell voltage-clamp configuration of the patch-clamp technique as previously described [40], [41].
Ethics statement
All mice were bred and maintained according to an ethical animal license protocol complying with the current Austrian law.
Detailed Materials and Methods are available in Text S1.
Supporting Information
Zdroje
1. LivshitsG, PophamM, MalkinI, SambrookPN, MacgregorAJ, et al. (2011) Lumbar disc degeneration and genetic factors are the main risk factors for low back pain in women: the UK Twin Spine Study. Ann Rheum Dis 70 : 1740–1745.
2. LariviereWR, WilsonSG, LaughlinTM, KokayeffA, WestEE, et al. (2002) Heritability of nociception. III. Genetic relationships among commonly used assays of nociception and hypersensitivity. Pain 97 : 75–86.
3. MogilJS, WilsonSG, BonK, LeeSE, ChungK, et al. (1999) Heritability of nociception I: responses of 11 inbred mouse strains on 12 measures of nociception. Pain 80 : 67–82.
4. NielsenCS, StubhaugA, PriceDD, VassendO, CzajkowskiN, et al. (2008) Individual differences in pain sensitivity: genetic and environmental contributions. Pain 136 : 21–29.
5. HartvigsenJ, NielsenJ, KyvikKO, FejerR, VachW, et al. (2009) Heritability of spinal pain and consequences of spinal pain: a comprehensive genetic epidemiologic analysis using a population-based sample of 15,328 twins ages 20–71 years. Arthritis Rheum 61 : 1343–1351.
6. ReaumeCJ, SokolowskiMB (2011) Conservation of gene function in behaviour. Philos Trans R Soc Lond B Biol Sci 366 : 2100–2110.
7. TraceyWDJr, WilsonRI, LaurentG, BenzerS (2003) painless, a Drosophila gene essential for nociception. Cell 113 : 261–273.
8. NeelyGG, HessA, CostiganM, KeeneAC, GoulasS, et al. (2010) A genome-wide Drosophila screen for heat nociception identifies alpha2delta3 as an evolutionarily conserved pain gene. Cell 143 : 628–638.
9. NeelyGG, KeeneAC, DuchekP, ChangEC, WangQP, et al. (2011) TrpA1 regulates thermal nociception in Drosophila. PLoS ONE 6: e24343 doi:10.1371/journal.pone.0024343.
10. KimSE, CosteB, ChadhaA, CookB, PatapoutianA (2012) The role of Drosophila Piezo in mechanical nociception. Nature 483 : 209–212.
11. PereiraPJ, LazarottoLF, LealPC, LopesTG, MorroneFB, et al. (2011) Inhibition of phosphatidylinositol-3 kinase gamma reduces pruriceptive, inflammatory, and nociceptive responses induced by trypsin in mice. Pain 152 : 2861–2869.
12. XuQ, FitzsimmonsB, SteinauerJ, O'NeillA, NewtonAC, et al. (2011) Spinal phosphinositide 3-kinase-Akt-mammalian target of rapamycin signaling cascades in inflammation-induced hyperalgesia. J Neurosci 31 : 2113–2124.
13. PezetS, MarchandF, D'MelloR, GristJ, ClarkAK, et al. (2008) Phosphatidylinositol 3-kinase is a key mediator of central sensitization in painful inflammatory conditions. J Neurosci 28 : 4261–4270.
14. YaoJ, QinF (2009) Interaction with phosphoinositides confers adaptation onto the TRPV1 pain receptor. PLoS Biol 7: e46 doi:10.1371/journal.pbio.1000046.
15. LiuB, ZhangC, QinF (2005) Functional recovery from desensitization of vanilloid receptor TRPV1 requires resynthesis of phosphatidylinositol 4,5-bisphosphate. J Neurosci 25 : 4835–4843.
16. PrescottED, JuliusD (2003) A modular PIP2 binding site as a determinant of capsaicin receptor sensitivity. Science 300 : 1284–1288.
17. Ufret-VincentyCA, KleinRM, HuaL, AngueyraJ, GordonSE (2011) Localization of the PIP2 sensor of TRPV1 ion channels. J Biol Chem 286 : 9688–9698.
18. KimAY, TangZ, LiuQ, PatelKN, MaagD, et al. (2008) Pirt, a phosphoinositide-binding protein, functions as a regulatory subunit of TRPV1. Cell 133 : 475–485.
19. KleinRM, Ufret-VincentyCA, HuaL, GordonSE (2008) Determinants of molecular specificity in phosphoinositide regulation. Phosphatidylinositol (4,5)-bisphosphate (PI(4,5)P2) is the endogenous lipid regulating TRPV1. J Biol Chem 283 : 26208–26216.
20. SteinAT, Ufret-VincentyCA, HuaL, SantanaLF, GordonSE (2006) Phosphoinositide 3-kinase binds to TRPV1 and mediates NGF-stimulated TRPV1 trafficking to the plasma membrane. J Gen Physiol 128 : 509–522.
21. ZhuangZY, XuH, ClaphamDE, JiRR (2004) Phosphatidylinositol 3-kinase activates ERK in primary sensory neurons and mediates inflammatory heat hyperalgesia through TRPV1 sensitization. J Neurosci 24 : 8300–8309.
22. LeeB, DescalziG, BaekJ, KimJI, LeeHR, et al. (2011) Genetic enhancement of behavioral itch responses in mice lacking phosphoinositide 3-kinase-gamma (PI3Kgamma). Mol Pain 7 : 96.
23. LukacsV, ThyagarajanB, VarnaiP, BallaA, BallaT, et al. (2007) Dual regulation of TRPV1 by phosphoinositides. J Neurosci 27 : 7070–7080.
24. ChuangHH, PrescottED, KongH, ShieldsS, JordtSE, et al. (2001) Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature 411 : 957–962.
25. ZhaoZQ, GaoYJ, SunYG, ZhaoCS, GereauRWt, et al. (2007) Central serotonergic neurons are differentially required for opioid analgesia but not for morphine tolerance or morphine reward. Proc Natl Acad Sci U S A 104 : 14519–14524.
26. RosaAO, EgeaJ, LorrioS, RojoAI, CuadradoA, et al. (2008) Nrf2-mediated haeme oxygenase-1 up-regulation induced by cobalt protoporphyrin has antinociceptive effects against inflammatory pain in the formalin test in mice. Pain 137 : 332–339.
27. HnaskoTS, SotakBN, PalmiterRD (2005) Morphine reward in dopamine-deficient mice. Nature 438 : 854–857.
28. CostiganM, BefortK, KarchewskiL, GriffinRS, D'UrsoD, et al. (2002) Replicate high-density rat genome oligonucleotide microarrays reveal hundreds of regulated genes in the dorsal root ganglion after peripheral nerve injury. BMC Neurosci 3 : 16.
29. Volpicelli-DaleyLA, LucastL, GongLW, LiuL, SasakiJ, et al. (2010) Phosphatidylinositol-4-phosphate 5-kinases and phosphatidylinositol 4,5-bisphosphate synthesis in the brain. J Biol Chem 285 : 28708–28714.
30. SasakiJ, SasakiT, YamazakiM, MatsuokaK, TayaC, et al. (2005) Regulation of anaphylactic responses by phosphatidylinositol phosphate kinase type I {alpha}. J Exp Med 201 : 859–870.
31. CaterinaMJ, SchumacherMA, TominagaM, RosenTA, LevineJD, et al. (1997) The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389 : 816–824.
32. CunhaTM, Roman-CamposD, LotufoCM, DuarteHL, SouzaGR, et al. (2010) Morphine peripheral analgesia depends on activation of the PI3Kgamma/AKT/nNOS/NO/KATP signaling pathway. Proc Natl Acad Sci U S A 107 : 4442–4447.
33. KonigC, Gavrilova-RuchO, von BanchetGS, BauerR, GrunM, et al. (2010) Modulation of mu opioid receptor desensitization in peripheral sensory neurons by phosphoinositide 3-kinase gamma. Neuroscience 169 : 449–454.
34. BonningtonJK, McNaughtonPA (2003) Signalling pathways involved in the sensitisation of mouse nociceptive neurones by nerve growth factor. J Physiol 551 : 433–446.
35. SasakiT, Irie-SasakiJ, JonesRG, Oliveira-dos-SantosAJ, StanfordWL, et al. (2000) Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science 287 : 1040–1046.
36. HirschE, KatanaevVL, GarlandaC, AzzolinoO, PirolaL, et al. (2000) Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science 287 : 1049–1053.
37. HawkinsPT, StephensLR (2007) PI3Kgamma is a key regulator of inflammatory responses and cardiovascular homeostasis. Science 318 : 64–66.
38. PatruccoE, NotteA, BarberisL, SelvetellaG, MaffeiA, et al. (2004) PI3Kgamma modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell 118 : 375–387.
39. KuzniarA, van HamRC, PongorS, LeunissenJA (2008) The quest for orthologs: finding the corresponding gene across genomes. Trends Genet 24 : 539–551.
40. ObrejaO, BiasioW, AndratschM, LipsKS, RatheePK, et al. (2005) Fast modulation of heat-activated ionic current by proinflammatory interleukin 6 in rat sensory neurons. Brain 128 : 1634–1641.
41. ObrejaO, RatheePK, LipsKS, DistlerC, KressM (2002) IL-1 beta potentiates heat-activated currents in rat sensory neurons: involvement of IL-1RI, tyrosine kinase, and protein kinase C. Faseb J 16 : 1497–1503.
42. Lacroix-FralishML, LedouxJB, MogilJS (2007) The Pain Genes Database: An interactive web browser of pain-related transgenic knockout studies. Pain 131 : 3 e1–4.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 12
Nejčtenější v tomto čísle
- Population Genomics of Sub-Saharan : African Diversity and Non-African Admixture
- Dnmt3a Protects Active Chromosome Domains against Cancer-Associated Hypomethylation
- Excessive Astrocyte-Derived Neurotrophin-3 Contributes to the Abnormal Neuronal Dendritic Development in a Mouse Model of Fragile X Syndrome
- Pre-Disposition and Epigenetics Govern Variation in Bacterial Survival upon Stress