
Genetic Adaptation Associated with Genome-Doubling in Autotetraploid
Genome duplication, which results in polyploidy, is disruptive to fundamental biological processes. Genome duplications occur spontaneously in a range of taxa and problems such as sterility, aneuploidy, and gene expression aberrations are common in newly formed polyploids. In mammals, genome duplication is associated with cancer and spontaneous abortion of embryos. Nevertheless, stable polyploid species occur in both plants and animals. Understanding how natural selection enabled these species to overcome early challenges can provide important insights into the mechanisms by which core cellular functions can adapt to perturbations of the genomic environment. Arabidopsis arenosa includes stable tetraploid populations and is related to well-characterized diploids A. lyrata and A. thaliana. It thus provides a rare opportunity to leverage genomic tools to investigate the genetic basis of polyploid stabilization. We sequenced the genomes of twelve A. arenosa individuals and found signatures suggestive of recent and ongoing selective sweeps throughout the genome. Many of these are at genes implicated in genome maintenance functions, including chromosome cohesion and segregation, DNA repair, homologous recombination, transcriptional regulation, and chromatin structure. Numerous encoded proteins are predicted to interact with one another. For a critical meiosis gene, ASYNAPSIS1, we identified a non-synonymous mutation that is highly differentiated by cytotype, but present as a rare variant in diploid A. arenosa, indicating selection may have acted on standing variation already present in the diploid. Several genes we identified that are implicated in sister chromatid cohesion and segregation are homologous to genes identified in a yeast mutant screen as necessary for survival of polyploid cells, and also implicated in genome instability in human diseases including cancer. This points to commonalities across kingdoms and supports the hypothesis that selection has acted on genes controlling genome integrity in A. arenosa as an adaptive response to genome doubling.
Published in the journal:
. PLoS Genet 8(12): e32767. doi:10.1371/journal.pgen.1003093
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003093
Summary
Genome duplication, which results in polyploidy, is disruptive to fundamental biological processes. Genome duplications occur spontaneously in a range of taxa and problems such as sterility, aneuploidy, and gene expression aberrations are common in newly formed polyploids. In mammals, genome duplication is associated with cancer and spontaneous abortion of embryos. Nevertheless, stable polyploid species occur in both plants and animals. Understanding how natural selection enabled these species to overcome early challenges can provide important insights into the mechanisms by which core cellular functions can adapt to perturbations of the genomic environment. Arabidopsis arenosa includes stable tetraploid populations and is related to well-characterized diploids A. lyrata and A. thaliana. It thus provides a rare opportunity to leverage genomic tools to investigate the genetic basis of polyploid stabilization. We sequenced the genomes of twelve A. arenosa individuals and found signatures suggestive of recent and ongoing selective sweeps throughout the genome. Many of these are at genes implicated in genome maintenance functions, including chromosome cohesion and segregation, DNA repair, homologous recombination, transcriptional regulation, and chromatin structure. Numerous encoded proteins are predicted to interact with one another. For a critical meiosis gene, ASYNAPSIS1, we identified a non-synonymous mutation that is highly differentiated by cytotype, but present as a rare variant in diploid A. arenosa, indicating selection may have acted on standing variation already present in the diploid. Several genes we identified that are implicated in sister chromatid cohesion and segregation are homologous to genes identified in a yeast mutant screen as necessary for survival of polyploid cells, and also implicated in genome instability in human diseases including cancer. This points to commonalities across kingdoms and supports the hypothesis that selection has acted on genes controlling genome integrity in A. arenosa as an adaptive response to genome doubling.
Introduction
The duplication of an entire set of chromosomes is a game-changing mutation. Whole-genome duplication (WGD) may create challenges for basic biological functions. For example, the regulation of gene expression, chromosome segregation, chromatin structure, and the maintenance of cellular homeostasis with altered cell size may be perturbed by duplicating an entire set of chromosomes [1]–[8]. That WGD can be challenging to organisms across kingdoms is evidenced by observations of dysfunction in very different contexts, such as reduced fertility observed in many newly formed plant autopolyploids, and mitotic instability in polyploid cancer cells [1], [5], [9]. Despite potential roadblocks, polyploid species are abundant in nature and genome doubling has been implicated in speciation and adaptive radiations [10]. Polyploids are especially well known among plants, but also occur in a diverse array of animals, including vertebrates [11].
The short-term consequences of WGD have been extensively studied in both natural and synthetic polyploids, especially in plants. These studies indicate that chromosome structural changes and rearrangements are common following WGD, as are abnormalities in mitosis and meiosis; in some cases changes in gene expression have also been observed (e.g. see [1]–[8]). These observations support the idea that polyploidy can pose challenges to aspects of gene regulation, chromosome organization and chromosome segregation. A yeast mutant screen indicates that some of these challenges are common across kingdoms. Genes encoding proteins implicated in the maintenance of genome integrity, including homologous recombination, DNA repair, sister chromatid cohesion and mitotic spindle function were identified as essential genes specifically in tetraploids [12].
The existence of stable, fertile polyploid species in different kingdoms demonstrates that the challenges that genome-doubled organisms may face at their inception are not insurmountable, and suggests that genome-doubled lineages should experience a period of compensatory genetic adaptation to their genome-doubled state. In sharp contrast to our understanding of the early transcriptional or genomic responses of organisms to WGD [1]–[8], very little is known about what molecular mechanisms might contribute to longer-term stabilization of polyploids or adaptation to a genome-doubled state. In plants, a single gene important for polyploid stabilization has been molecularly characterized: the homologous pairing suppressor (Ph1) from allohexaploid wheat. Allopolyploids like wheat have hybrid origins and carry already somewhat divergent sets of chromosomes. Ph1 enhances meiotic pairing preferences of chromosomes for more similar (homologous) chromosomes over less similar (homeologous) ones, resulting in bivalent pairing and stable meiosis [13]. This work provides an important molecular insight into the process of meiotic stabilization in allopolyploids.
However, not all polyploids stabilize meiosis by developing pairing preferences. Autopolyploids arise from within-species genome duplications and thus carry four homologs of each chromosome [1], [3]–[5], [14]. Established autopolyploids often have cytologically diploidized meiosis (forming primarily bivalent associations), but show polysomic inheritance at genetic markers, which is possible if the chromosomes lack pairing preferences and partner randomly at meiosis [4], [5], [14]. Thus there must be at least two mechanisms by which polyploids can stabilize meiosis, one that involves enhancing pairing preferences (as is common in allopolyploids like wheat) and one that ensures bivalent formation without affecting pairing preference.
The molecular mechanisms that underlie long-term polyploid stabilization and evolution remain largely mysterious. To help fill this gap, we undertook a population genomic analysis of an established autotetraploid plant, Arabidopsis arenosa. This species is closely related to two sequenced Arabidopsis diploids: its sister taxa A. lyrata and the model system A. thaliana [15]–[17]. Like A. lyrata, A. arenosa is obligately outcrossing, and abundant throughout Europe [16], [17]. Tetraploid A. arenosa is cytologically diploidized, with primarily bivalent chromosome associations at meiosis [18]. We sequenced the genomes of twelve tetraploid A. arenosa individuals from four populations in Germany and Austria and tested for allele frequency patterns suggestive of selective sweeps. We identified 192 genes in the A. arenosa genome with patterns of polymorphism indicative of recent or ongoing selective sweeps. Several functional classes represented among these genes are consistent with adaptation to WGD. We provide candidate genes that will help boost our mechanistic understanding of these processes, while also suggesting new hypotheses. Similarities of the functional classes we identified with those identified in a yeast mutant study [12] indicate that at least some challenges are shared across kingdoms, and suggests that the functions targeted by selection in A. arenosa are especially critical in tetraploids.
Results
Genome analysis of A. arenosa
We selected 12 A. arenosa individuals grown from seeds collected at four sites in Austria and Germany (Figure 1) for genome sequencing. Cytological and flow cytometric analyses demonstrated that A. arenosa populations throughout these regions are tetraploid [19], [20]. We confirmed ploidy for at least one individual from each population by flow cytometric analysis of nuclear DNA content (Figure S1), and performed testcrosses for the remainder. We aligned DNA sequence data to the publicly available reference genome of A. lyrata [15]. After filtering for sequence and mapping quality, overall genome coverage per sequenced individual averaged 25× across the eight A. lyrata chromosome scaffolds (Figure S2). We focused subsequent analyses on coding regions. We used a maximum likelihood method to infer tetraploid genotypes for each single nucleotide polymorphism (SNP) in each individual.

We generated three-species alignments with consensus sequences from all sites in the A. arenosa sample that had at least 4× coverage per individual, with homologs from both A. thaliana and A. lyrata. In total 26,655,179 bp were aligned, representing 20,889 homologous genes. The final dataset contains 3,148,695 segregating sites (Table 1). The average divergence of A. arenosa from A. lyrata per site was 8.7×10−4, 2.7×10−4, and 9.6×10−4 for synonymous, non-synonymous, and intronic positions, respectively. In addition, there were 13,634 fixed differences in A. arenosa consensus sequences relative to both A. thaliana and A. lyrata, distributed among 5,855 protein-coding genes, 2,147 of which contained at least one non-synonymous fixed difference relative to the A. lyrata reference.

Other studies have previously found that polymorphism in A. arenosa is higher than in A. lyrata [21], [22]. Consistent with this, we found high levels of segregating variation genome-wide in A. arenosa (Table 1), and synonymous site diversity approximately double that estimated for diploid A. lyrata [21]. This is consistent with the prediction that equilibrium genetic variation in an outcrossing autotetraploid population with tetrasomic inheritance should be approximately double that of a diploid population of similar size [23]. The site frequency spectrum (SFS) of non-synonymous SNPs showed a significant skew toward low-frequency mutations compared to the synonymous SFS (Mann-Whitney U Test p<7×10−8), consistent with widespread purifying selection (Figure 2). Importantly, the sequencing error rate we estimated from the data (0.1–0.2%; see Methods) was an order of magnitude below our estimates of Theta for all classes of sites, and the likelihood function in our genotyping algorithm explicitly accounted for errors. Thus, sequencing errors were unlikely to have contributed significantly to our estimates of diversity.

Estimation of mode of inheritance
Inheritance can vary in tetraploids from disomic to tetrasomic. Disomic inheritance results when chromosomes have pairing partner preferences (genes thus behave as duplicates segregating two alleles each). Tetrasomic inheritance occurs in species that lack pairing preferences among the four homologous copies of each chromosome, in which case each locus segregates four alleles. Whether populations have tetrasomic or disomic inheritance has significant implications for population genetic analyses of tetraploids [3]. Therefore we investigated historic and ongoing modes of inheritance in A. arenosa by comparing our sequence data to simulated datasets.
We used coalescent simulations to generate expected neutral SFS and genotype frequencies under different historical scenarios and inheritance models. Our observed data did not differ significantly from simulated SFS for the tetrasomic model, but did differ from both disomic models (p<0.01 Mann-Whitney U test; Table S1). Similar results were obtained for inferred genotypic classes (Figure S3; Table S1). Importantly, we do not observe an excess of duplex (AAaa) genotypes, or a high number of SNPs with frequency ∼50% in the data, both of which are expected if the A. arenosa sample had been evolving under disomic inheritance for a significant amount of time (Figure S3). These results strongly support the hypothesis that A. arenosa has tetrasomic inheritance. Together with prior findings that this species has bivalent chromosome associations at meiosis [18], this places A. arenosa on a growing list of established tetraploids with cytologically, but not genetically diploidized meiosis [14]. Importantly, tetraploid A. arenosa will display patterns of polymorphism typical of a population of diploids with twice the effective size [23], [24]. Therefore, signatures of adaptive evolution are detectable using methods developed for diploids.
Signatures of selection in A. arenosa
We used diploid A. lyrata and A. thaliana reference genomes [15], [25] to identify 20,265 genes that had >80% sequence identity among all three species. These genes comprise the dataset used in all analyses described below. The sampled individuals originate from four populations with distinct habitats (Figure 1). We tested for population structure or habitat-associated differentiation by pairwise FST comparisons across the genome [26]. Overall there was low differentiation among populations. Genome wide pairwise FST at synonymous sites ranged from 0.047 to 0.063 (Table S2), which is an order of magnitude lower than average pairwise FST measured between populations of A. lyrata [22]. This suggests that A. arenosa lacks strong local population differentiation in this geographic region.
During the formation and early establishment of an autotetraploid, alleles that contribute to tetraploid formation or are important for the success of the tetraploid lineage should experience strong selection. To perform genome-wide tests for selection in tetraploid A. arenosa we identifed genes for which SFS were skewed toward high frequency derived haplotypes [27] and genes in which polymorphism was low. The two measures were uncorrelated genome-wide (R2 = 0.014) and together provide evidence of past selective sweeps. There were 192 genes that were both within the 5% most skewed SFS and the 5% lowest polymorphism (Table S3).
In most cases, candidate selected genes were unlinked. There were only eight instances where genes separated by less than 10 kb both showed signatures of selection. As a result, almost all potential selective sweep signatures in A. arenosa are sufficiently narrow to identify single candidate genes based on homology to A. thaliana (www.arabidopsis.org). Several gene ontology categories are over-represented among these genes (Fisher's Exact Test p<0.005 for each category) compared to their representation within the entire genome. These include functions related to the regulation of basal transcription, epigenetic regulation, sister chromatid cohesion, homologous recombination, DNA repair, cell cycle, cell morphogenesis and cell growth. The genes representing the most enriched categories are summarized in Table S4. We focus below on two general categories in more detail: transcriptional regulation and meiosis.
Regulation of transcription
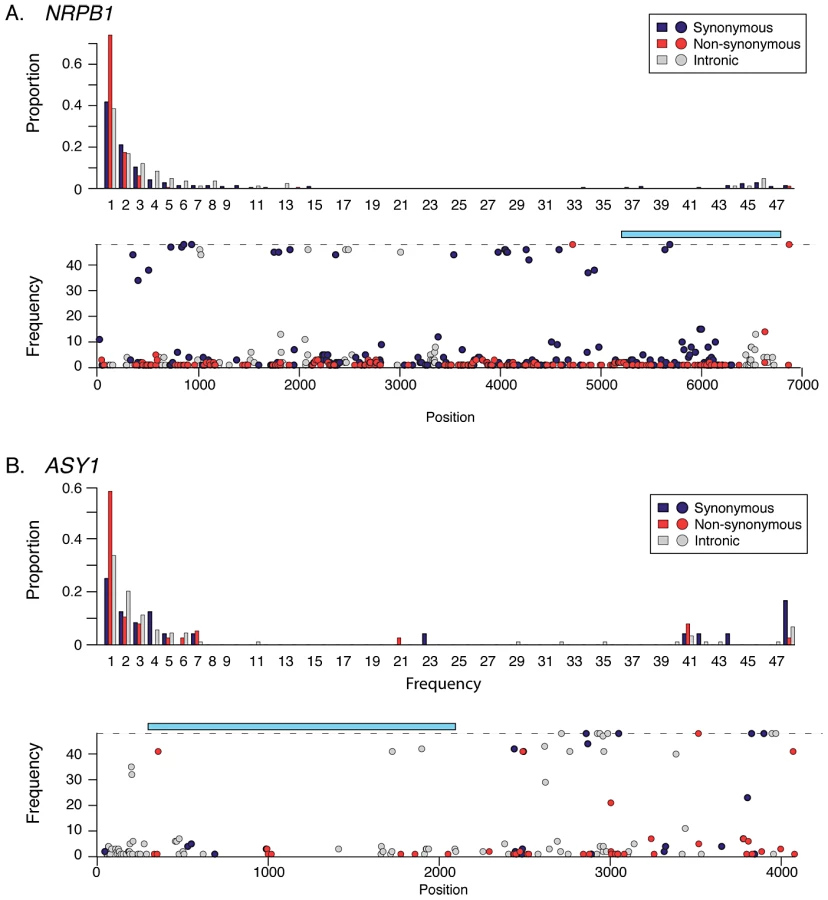
A “retuning” of basal transcription in response to increased cell size may be important in polyploids for maintaining a balance between expression from additional chromosome copies and altered cell size and/or nuclear membrane surface to volume ratio [1], [3]. In this light, it is intriguing that numerous genes showing indications of selection in A. arenosa encode proteins implicated in basal transcription, including the large subunits of two of the core DNA-dependent RNA Polymerases (Pol) II and III (Tables S3, S4). The gene encoding the large subunit of Pol II (NRPB1) has numerous high frequency SNP differences in A. arenosa relative to A. lyrata and A. thaliana. These include two fixed amino acid differences flanking either side of the highly conserved long C-terminal tail (CTD; Figure 3A). The CTD consists of a series of heptad repeats whose phosphorylation state regulates the activity of the Pol II complex [28]. In yeast, phosphorylation of the CTD is orchestrated by three cyclin dependent kinases, CDK 7, 8 and 9 [29]. A homolog of CDK8, HUA ENHANCER 3 (HEN3) [29], also shows evidence of having undergone a selective sweep in A. arenosa. Two other CTD-interactors, PRE-MRNA PROCESSING PROTEIN 40A (PRP40A) and GENERAL TRANSCRIPTION FACTOR B1 (GTB1) also show evidence of selective sweeps (Table S4).
In addition to the CTD-interactors, other genes encoding regulators of Pol II activity or recruitment also show signatures of selection in A. arenosa (Table S4). These include genes encoding core transciption factors such as two TRANSCRIPTION FACTOR IIS (TFIIS) family genes and TBP-ASSOCIATED FACTOR 5 (TAF5), which encodes a subunit of TFIID. TFIID and TFIIS are general transcription factors that associate with Pol II and promote its movement during transcription [28]. We also find evidence of selection on STRUWWELPETER and CENTER CITY, which encode subunits of RNA Pol II-recruiting mediator complexes [30], [31]. Together, the signatures in these genes, as well as epigenetic regulators including genes implicated in RNA-mediated silencing, histone modification and chromatin remodeling (Table S4), suggest that a global re-tuning of transcription may have been very important in the history of A. arenosa.
Meiosis
Autopolyploids also face an important handicap in meiosis: They are equipped with meiotic machinery inherited from diploid ancestors optimized over evolutionary time to segregate pairs of homologous chromosomes. That an increase to four homologs presents an obstacle is evident in newly formed tetraploids, which often show high rates of sterility due to failures of chromosome segregation in meiosis [1]–[5]. In A. arenosa, eight loci homologous to genes essential for meiosis fit our selective sweep criteria. These have predicted roles in chromosome synapsis, cohesion and homologous recombination (Tables S3, S4). These genes include SISTER CHROMATID COHESION2 (SCC2), which encodes an adherin that loads cohesins during meiosis [32], and one of its substrates, the cohesin subunit STRUCTURAL MAINTENANCE OF CHROMOSOMES 3 (SMC3) [33], [34]. SMC5 and SMC6a are also among the eight meiosis-related genes that show signatures of selective sweeps. These encode proteins that function together in sister chromatid alignment, cohesion, DNA repair and homologous recombination during mitosis [35]. Recently the SMC5/6 complex was also shown to play an essential role in meiosis [36]. While sister chromatid cohesion has not previously been specifically discussed as a possible challenge for tetraploid plants, genes involved in sister chromatid cohesion were also shown to be crucial for survival of tetraploid yeast [12].
We compiled a list of 59 genes annotated in TAIR10 (www.arabidopsis.org) as playing a role in meiosis that also had clear homologs in A. lyrata as well as in our A. arenosa sample (Table S5). This set of genes showed enrichment for the signatures of positive selection. Among the 59 genes, 17 (29%) showed a significantly skewed SFS and nine showed low polymorphism in A. arenosa (Table S5). Eight of these genes (13.5%) were among the 192 that were in both the upper 5% tail of the CLR distribution as well as the lower 5% tail of the π/site distribution (Table S3), which is a 10-fold enrichment (Fisher's exact test p≪0.001). Six meiosis-related genes with skewed SFS in A. arenosa (top 5% genome-wide) are homologous to genes that were also identified as critical for survival in tetraploid yeast [12]. These are RAD54, MEIOTIC RECOMBINATION 11 (MRE11), RECQ4A, TOPOISOMERASE3 (TOP3), SMC1 and SEPARASE (ESP) (Fisher's Exact Test p<0.001). This indicates again that fundamental aspects of chromosome biology present challenges upon genome doubling in very different species and that sister chromatid cohesion, homologous recombination and DNA repair are key shared processes.
In A. arenosa, the chromosome synapsis gene ASYNAPSIS1 (ASY1) [37] has a strongly skewed SFS, low polymorphism and an abundance of high frequency derived SNPs relative to A. lyrata and A. thaliana (Figure 3B). A high-frequency derived SNP in the tetraploid A. arenosa population sample of ASY1 causes an amino acid change in the conserved HORMA domain. This alters an ancestral positively charged lysine (K) to a negatively charged glutamic acid (E) in the derived allele. We examined other ASY1 sequences reported to date in Genbank and found that this amino acid position is conserved in a wide range of vascular plants (Figure 4). Only two other plant species have amino acid changes at this residue. Both replaced the lysine with a polar uncharged asparagine (N).

We tested whether this polymorphism is differentiated between diploid and tetraploid cytotypes within A. arenosa using a PCR marker. We genotyped 38 plants from two diploid populations collected from the Carpathian Mountains in Slovakia (SN and CA in Figure 1B). We found that the derived allele is present, but rare in the diploids (at a frequency of ∼4%). In sharp contrast, in tetraploid A. arenosa, the derived allele represents 41 of the 48 assayed sequences in our genome resequencing data (85%) and in a wider sample of 75 tetraploids from five additional populations, the derived allele has a frequency ∼90%.
Gene interactions
We next asked whether any of the selected genes in A. arenosa are predicted to interact using the AtPIN database [38]. Forty-six (∼24%) of the 192 candidate selected proteins are known or predicted to interact with at least one other on the list (). Twelve genes encode products indicated in pairwise interactions. A set of four forms a small network associated with TARGET OF RAPAMYCIN (TOR) and RAPTOR, which regulate a variety of processes associated with cell proliferation [39]–[41]. A set of three is associated with a ubiquitin protein ligase, UPL4 [42] (Table S6).
All of the remaining 27 genes are linked in a single network of predicted interactions, many with multiple connections per node (Figure 5). The two most connected are NRPB1 (9 connections) and HEN3 (6 connections). Many of the additional genes linked to these encode regulators of basal transcription, chromatin structure and cell cycle. This includes several additional interactors of the CTD tail of NRPB1, core transcription factor components such as TAF5 [28], [43], and histone modifiers implicated in the regulation of transcription, including HISTONE ACETYLTRANSFERASE 5 and TAF1 [28], [43], [44] (Figure 5). Shared links through nuclear-cytoplasmic trafficking via EXPORTIN1B connect the network surrounding NRPB1 and HEN3 to a small group of genes involved in regulation of chromatin structure and cohesion in meiosis, including SMC3 and SCC2. None of these 27 genes are closely linked in the genome, suggesting that multiple components of this interaction network have been under selection.

Discussion
Here we report results from a population genomic analysis in autotetraploid A. arenosa. We show that A. arenosa has high genetic diversity, little population structure, and allele and genotype frequencies consistent with a history of tetrasomic inheritance, in which four alleles segregate at each genomic locus. We identified 192 genes that exhibit two signatures of selective sweeps: reduced diversity and a SFS skewed toward high frequency derived alleles. It is important to note that our analysis could not identify loci contributing to polyploid stabilization strictly via adaptive changes in gene expression pattern, unless accompanied by a signature of selection that extended into coding regions. Identification of such loci would require comparative analysis of gene expression patterns among diploids and tetraploids, and/or analysis of sequence evolution in intergenic regions. Nevertheless, our focus on adaptive evolution within protein-coding regions allowed identification of putatively selected genes that have clear orthologs in A. thaliana, and for which functional information is therefore available.
This work suggests candidate genes and processes that may have been important for compensatory adaptation of A. arenosa to its genome-doubled state. The functional annotations of the A. thaliana homologs of these genes point to the modulation of fundamental biological processes, including the regulation of core transcription, epigenetic regulation, DNA repair, cell division and morphogenesis, chromosome synapsis and cohesion, homologous recombination, and chromosome segregation. Several of these categories represent functions that have been previously demonstrated or hypothesized to be problematic for neo-polyploids, but for which the mechanisms of longer-term stabilization have not been studied [1]–[5].
Several functional classes represented among candidate selected genes in A. arenosa, particularly chromosome cohesion, segregation and repair, show considerable overlap with genes necessary for survival specifically in polyploid yeast [12]. Moreover, six genes with SFS indicative of selection are the closest (or only) Arabidopsis homologs of the genes identified in the yeast screen. These are RAD54, MRE11, SMC1, TOP3, RECQ4A, and ESP. That these genes are truly fundamental in genome maintenance is also underlined by the fact that all of them have been implicated in numerous human diseases associated with genome instability, including cancer, Ataxia-Telangiectasia-like disorders, Bloom Syndrome and others, e.g. [45]–[50]. This indicates that at least some of the fundamental challenges to the maintenance of genome integrity that organisms face after genome perturbations, including whole genome duplication, are broadly shared across kingdoms. It also provides corroborative evidence that at least some of the signatures of selection in A. arenosa are indeed attributable to adaptation to a doubled genome.
There have been numerous studies of gene expression in response to whole genome duplication (see e.g. [2], [6]–[8]). Though most have focused on allopolyploids, several have directly compared gene expression in diploids and their autotetraploid derivates (e.g. [51]–[57]). In most cases, there is little or no overlap with the functional classes or specific genes identified in expression studies and those we identified in our study. This suggests that the genes and functional classes involved in short-term responses to genome duplication are largely distinct from those that may be under selection during longer-term polyploid evolution. There are some exceptions: In Paspalum notatum, gene expression changes in new polyploids occur in some of the same gene classes as those we identified here, including transcription, DNA repair and chromatin structure regulation [51]. Thus in some cases early gene expression responses do occur in genes or functional classes that may be under selection in longer-term polyploid evolution, suggesting that some of the selection acting on polyploid genomes may be a compensatory response to early shifts in gene expression. One of the genes we identified as putatively under selection in A. arenosa, RAD54, which is involved in DNA repair as well as homologous recombination [58], [59], has also been reported to be upregulated in response to genome duplication in autotetraploid A. thaliana [39] (though see [54]).
Another feature of the putatively selected genes in A. arenosa is that many are known or predicted to interact. This is especially true of genes implicated in the regulation of basal transcription. That multiple functionally connected, but unlinked genes may have experienced selective sweeps suggests that these loci either contribute incrementally to fitness through modifications of a common process or have been selected together as a functional module. Entire networks can experience selection effectively as units if epistatic interactions are synergistic and alter the selective environment for mutations at functionally related loci, allowing a larger coordinated response to selection [60], [61]. Indeed, findings in other species support the idea that genetic modules encoding networks of interacting proteins can in some circumstances respond to selection as units [60]–[65]. Whether interaction effects have driven selection on a functional module surrounding basal transcription in A. arenosa, or whether the polymorphisms contribute additively to a selected phenotype merits further exploration. Interestingly, in yeast it has also been noted that genes important in tetraploid survival are predicted to interact extensively [12], suggesting that this, too, may be a shared feature of polyploids across kingdoms.
Processes such as core transcription are interlinked with other cellular functions. For some genes we have identified it will be possible to clearly hypothesize what the selected function is. However, for other genes, it is less clear what function selection has acted to modulate, or if there are pleiotropic effects. For example, GTB1, which shows evidence of selection in A. arenosa, binds the C-terminal extension of Pol II and participates in regulation of Pol II processivity [28]. Thus it is reasonable to suppose it might have been under selection for its contribution to the regulation of basal transcription. However, GTB1 has also been predicted to interact with ARGONAUTE (AGO) proteins which function in the processing of small RNAs [66]. AGO1 also shows evidence of a selective sweep in A. arenosa (Tables S3, S4), and AGO4 also shows evidence of adaptive protein evolution (not shown). This however, may not be due to polyploidy per se, since AGO genes show evidence of selective sweeps in diploid species as well. For example, successive selective sweeps in an Argonaute gene in Drosophila species have been suggested to be associated with host-pathogen co-evolution [67].
The picture may be even more complex, since small RNAs have also been implicated in DNA double-strand break repair [68], [69], meiotic chromosome pairing [70], and mitotic and meiotic chromosome structure and segregation [71]–[73]. Indeed, AGOs have themselves been directly implicated in maintaining chromatin silencing during meiosis [71], [73]. These are fundamental genome maintenance processes strongly implicated in polyploid stabilization. Thus the true causes of selection on genes like GTB1 or AGO1 that are implicated in multiple distinct but interlocked processes provide extensive opportunities for follow-up studies to unravel the complexities of selection acting on interconnected pleiotropic genes, more than one of which may be under selection for different reasons.
For the chromosome synapsis gene ASY1, we confirmed differentiation among A. arenosa cytotypes of an amino acid substitution at a conserved position. ASY1 is related to the Hop1 gene in yeast, which plays important roles in the assembly of the synaptonemal complex and the regulation of homologous recombination [74]. In plants, these functions are conserved, e.g. [37], [75]. Synapsis is a process that has been hypothesized to play a role in meiotic stabilization of tetraploids [1], [4], and ASY1 itself has been functionally implicated in polyploid meiosis. Expression of wheat TaASY1 is affected by Ph1, and transgenic downregulation of TaASY1 results in reduced synapsis but strengthened associations of homeologs at metaphase I [76]. If the derived ASY1 allele in A. arenosa was important in polyploid evolution, as the signature of selection suggests, this implies that this gene may play a role in promoting meiotic stability in both allo - and autopolyploids. The presence of the derived ASY1 allele at low frequency in the diploid gene pool suggests that standing variation for ASY1, rather than de novo mutation, may have been important for a rapid response to selection during tetraploid stabilization. This is consistent with findings in other species that genetic variation in diploids can affect meiotic stability after artificial genome doubling, e.g. [77].
Overall our data indicate that selection has acted on numerous genes in the tetraploid A. arenosa genome, providing specific candidate genes and mutations for mechanistic follow-up work. Some of this selection may have been on standing genetic variation in diploid A. arenosa that contributes to polyploid formation, for example by promoting unreduced (diploid) gamete formation. However, many of these selected alleles are likely to have been involved in the stabilization of fundamental biological processes after whole genome duplication. Our analysis implicates several fundamental processes and functions in adaptation to polyploidy, both supporting previous hypotheses about polyploid stabilization, such as modulation of meiosis, and suggesting new ones, such as involvement of a network associated with the regulation of core transcription. Finally, our analysis reveals an overlap of putatively selected genes and functions in A. arenosa with genes identified as essential in tetraploid yeast [12] and implicated in disease-associated failures of genome maintenance in humans. This suggests that key challenges faced by polyploids are shared across kingdoms and understanding how natural selection can circumvent these problems in a variety of species will provide important insights.
Materials and Methods
Plant material
Plants were grown directly from seeds collected from wild populations in the summers of 2009 and 2010. Seeds were collected in late June 2009 from the railway station in Triberg (TBG) in the Black Forest of southwestern Germany, and from a limestone outcrop near the Upfinger Steige (US), between Upfingen and Bad Urach in the Swabian Alb region of southwestern Germany. Seeds were collected in June 2010 from Kasparstein castle, in southern Austria (KA) and Berchtesgaden railway station (BGS) in southeastern Germany. Seeds were surface sterilized with 70% ethanol/0.05% Triton X-100, and then stratified at 4°C in the dark for six to eight days on 1/2×MS plates with 8% agar. Seeds were germinated in a tissue culture incubator at 16°C with 16 hour long days, and then transferred to soil (50% Sunshine Mix #4/50% fine vermiculite) and grown in a growth chamber with 16-hour long-day light cycles. Ploidy was verified by flow cytometry on at least one individual per population, and by testcrosses to known diploid and tetraploid individuals (the Streçno castle site in Slovakia, from which we also collected in 2010, was previously identified and confirmed as diploid by Luca Comai, UC Davis). Flow cytometry was also used to confirm that plants in our Streçno and Carpathians collections are diploid.
Sequencing
Genomic DNA was extracted from one gram of leaf and inflorescence material from 6 to 10-week old plants using a DNeasy Maxi-Prep kit (Qiagen). We chose three individuals each from the TBG, US, BGS, and KA populations for sequencing. For all individuals, cluster generation and sequencing were performed using standard protocols provided with the kits used. Three of the genomic sequencing libraries were prepared using the Illumina Genomic Sample Preparation Kit for sequencing on the Illumina Genome Analyzer II (GAII). Each of the three individuals was sequenced on a single GAII lane for 85 sequencing cycles. The remaining nine libraries were prepared following the Illumina TruSeq Genomic Sample Preparation protocol for sequencing on the Illumina HiSeq 2000. For sequencing on the HiSeq, each sample was bar-coded and all nine samples were run across seven lanes for 100 sequencing cycles. Sequencing results in the form of FASTQ files were used as input for read mapping and analysis.
Read mapping and error rate calculation
Short read mapping and processing was performed using SHORE version 5.0 [78]. Reads were mapped to the published Arabidopsis lyrata genome sequence using GenomeMapper, called by the SHORE subprogram mapflowcell (a list of all SHORE commands used during data processing are given in Text S2). Prior to mapping, we imposed a Sanger quality score cutoff of 30 for base calling. In addition, because errors can arise from both sequencing and read mapping, we assessed the full error rate by calculating the observed divergence between A. arenosa reads, the A. lyrata sequence mapped to, and the orthologous A. thaliana sequences following [79]. We selected a sample of 500,000 uniquely mapping reads from each individual, and produced local alignments of each read and the corresponding A. lyrata sequence [15] to the published A. thaliana TAIR 9 genome sequence using BLAST. For sequences that had a unique match to the A. thaliana reference (E-value cutoff = 1e−5), we then counted the number of changes between the A. thaliana sequence and the A. lyrata sequence or A. arenosa read, respectively. Because the number of observed changes reflects both evolutionary divergence and sequencing error, the excess number of changes on the A. arenosa reads relative to the A. lyrata sequence gives an estimate of error stemming from both sequencing and mapping (the contribution of sequencing error in the A. lyrata reference genome was assumed to be negligible). The estimated error rates were low, ranging from 0.1–0.2%.
Genotyping
Consensus sequence outputs for each individual were produced using the SHORE consensus sub-program specifying the -v to write all intermediate data to a file. This allowed selection of only uniquely mapping reads exceeding the quality cutoff (see above), upon which all subsequent analysis was based. All downstream parsing of files was performed using custom PERL scripts (see Text S2).
To estimate the tetraploid genotype from each individual, we used a modification of the genotyping algorithm described in [80] modified to account for the three heterozygous states possible in a tetraploid (AAAa, AAaa, Aaaa), and also designed to call homozygotes (AAAA and aaaa) in the presence of sequencing errors. Given the uncertainty regarding the mode of inheritance and demographic history of A. arenosa populations, both of which can affect expected genotype frequencies, we estimated individual genotypes directly from the pileup of bases for each individual (see Text S1). Following [79], we defined the probability of the data D (the pileup of bases) given the genotype G for a given reference position



Genomic analysis
All downstream data analysis made use of custom PERL and R scripts (see Text S2), in tandem with other software listed below. Summary statistics were generated using the libsequence evolutionary genetics software package [81]. Alignments of A. arenosa consensus sequences with A. thaliana and A. lyrata protein coding regions were generated using CLUSTALW 2.0. Alignments with <80% sequence identity among the three species were not included in further analysis.
All statistical tests were done using R version 2.11.0, and custom R scripts were written to perform genome wide analysis and tests for selection (see Text S2). Nucleotide diversity, π, is equivalent here to expected gametic heterozygosity estimated from inferred genotypes, where gametic heterozygosity equals the number of differences between any two sequences in a population sample [23]. Gametic heterozygosity was used for pairwise FST and diversity summaries.
To test for selective sweeps, we first implemented a non-parametric test for atypical site frequency spectra (SFS) [27]. This test is particularly well suited to identifying regions with SFS skewed toward high frequency derived SNPs. We used both the A. thaliana and A. lyrata reference genomes to obtain the unfolded SFS for all SNPs in the A. arenosa data, assuming that sites identical in both outgroup sequences represent the ancestral state. To implement the test, we divided each of the genes in the dataset into 100 snp windows, and calculated the composite likelihood ratio (CLR) score for each window separately, and then identified outliers relative to the genome-wide distribution (Figure S4). To test for a local reduction in genetic diversity, we measured π/basepair and again identified outliers from the genome-wide pattern. We then made a list of the strongest candidates for selective sweeps by selecting the set of genes that fell both in the lowest 5% of the genome-wide distribution of π/basepair and also had one or more 100 bp windows that scored in the top 5% for CLR score. π and CLR were uncorrelated in our data (R2 = 0.014).
Gene interaction predictions were examined using the atPIN database [38] (http://bioinfo.esalq.usp.br/atpin/atpin.pl). We confirmed predicted interactions with literature searches and removed all that were based purely on phylogenetic relatedness of genes, but included all predicted as well as experimentally verified interactions with experimental support in A. thaliana or other species.
Simulation analysis of mode of inheritance
We used coalescent simulations to generate neutral datasets using the software ms [82]. We used Watterson's estimator of 4Neu from the A. arenosa data to set realistic values of the population mutation rate. For each simulation, theta was set (using the -t switch) equal to the silent theta value from a randomly selected gene from the A. arenosa data (sampling with replacement). For all simulations, the sample size was set to 48 chromosomes (our A. arenosa sample size).
Disomic inheritance occurs when genetic diploidization effectively isolates homeologous chromosomes via consistent meiotic pairing preferences; this may happen immediately in allotetraploids, but in autotetraploids, pairing preferences may evolve much later. To model disomic inheritance, we simulated two sub-populations isolated for different lengths of time, and then drew two chromosomes from each per individual (representing the two homeologous chromosome pairs). We simulated data with the time since evolution of disomic inheritance (td; in units of 4N generations) set over a range of values from td = 1 to td = 0.2, stepping by 0.2. For simulation of the evolution of disomic inheritance, the -I switch was used to simulate two sub-populations of sample size 24, and the -eN switch was used to specify the time in the past when the sub-populations split from the ancestral population – forward in time, this represented the isolation of homeologs (i.e. the evolution of disomic inheritance). Two chromosomes from each sub-population were then assigned to each simulated individual, representing the two pairs of homeologous chromosomes. We also simulated a fully tetrasomic set of chromosomes with no sub-division of chromosome pools. Simulated tetraploid genotypes were generated by randomly assigning four chromosomes to each of twelve “individuals”. The distribution of genotype frequencies for 12 “individuals” from fully tetrasomic simulations accurately recapitulated theoretical expectations for tetrasomic inheritance with bivalent pairing (Figure S3). For all models, we performed 500,000 simulation runs and pooled SNPs across all runs.
PCR analysis of ASY1
We designed PCR primers to amplify a conserved region in the HORMA domain of ASY: 5′TTTGGTTTTCGTTTTGCTGA3′ and 5′GAGATTCAGCGTCCATAGGC3′. The high frequency SNP in this region causes a restriction site polymorphism for XmnI. Fragments were amplified from DNA from progeny of wild plants from five populations (Spisska, Slovakia; Carpathian Mountains, Tatras range, Slovakia; Gulsen, Austria; Koßelbach, Austria Berchtesgaden, Germany) using Taq polymerase (New England Biolabs) with an annealing temperature of 56°C. 10 µl of each product was digested with XmnI (New England Biolabs) and visualized on 1.5% agarose gels.
Supporting Information
Zdroje
1. ComaiL (2005) The advantages and disadvantages of being polyploid. Nat Rev Genet 6 : 836–846.
2. OsbornTC, PiresJC, BirchlerJA, AugerDL, ChenZJ, et al. (2003) Understanding mechanisms of novel gene expression in polyploids. Trends Genet 19 : 141–147.
3. OttoSP (2007) The evolutionary consequences of polyploidy. Cell 131 : 452–462.
4. ParisodC, HoldereggerR, BrochmannC (2010) Evolutionary consequences of autopolyploidy. New Phytol 186 : 5–17.
5. RamseyJ, SchemskeDW (2002) Neopolyploidy in flowering plants. Ann Rev Ecol Systemat 33 : 589–639.
6. AdamsKL, WendelJF (2005) Novel patterns of gene expression in polyploid plants. Trends Genet 21 : 539–543.
7. ChenZJ, NiZ (2006) Mechanisms of genomic rearrangements and gene expression changes in plant polyploids. Bioessays 28 : 240–252.
8. ChenZJ (2007) Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu Rev Plant Biol 58 : 377–406.
9. StorchovaZ, PellmanD (2004) From polyploidy to aneuploidy, genome instability and cancer. Nat Rev Mol Cell Biol 5 : 45–54.
10. WoodTE, TakebayashiN, BarkerMS, MayroseI, GreenspoonPB, et al. (2009) The frequency of polyploid speciation in vascular plants. Proc Natl Acad Sci U S A 106 : 13875–13879.
11. GregoryTR, MableBK (2005) Polyploidy in animals. The Evolution of the Genome 171 : 427–517.
12. StorchováS, BrenemanA, CandeJ, DunnJ, BurbankK, et al. (2006) Genome-wide genetic analysis of polyploidy in yeast. Nature 443 : 541–547.
13. GriffithsS, SharpR, FooteTN, BertinI, WanousM, et al. (2006) Molecular characterization of Ph1 as a major chromosome pairing locus in polyploid wheat. Nature 439 : 749–752.
14. SoltisDE, SoltisPS, SchemskeDW, HancockJF, ThompsonJN, et al. (2007) Autopolyploidy in angiosperms: Have we grossly underestimated the number of species? Taxon 56 : 13–30.
15. HuTT, PattynP, BakkerEG, CaoJ, ChengJ-F, et al. (2011) The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nature Genet 43 : 476–481.
16. Al-ShehbazIA, O'KaneSL (2002) Taxonomy and phylogeny of Arabidopsis (Brassicaceae). Arabidopsis Book 1: e0001.
17. KochMA, MatschingerM (2007) Evolution and genetic differentiation among relatives of Arabidopsis thaliana. Proc Natl Acad Sci U S A 104 : 6272–6277.
18. CarvalhoA, DelgadoM, BarãoA, FrescatadaM, RibeiroE, et al. (2010) Chromosome and DNA methylation dynamics during meiosis in the autotetraploid Arabidopsis arenosa. Sex Plant Reprod 23 : 29–37.
19. JørgensenMH, EhrichD, SchmicklR, KochMA, BrystingAK (2011) Interspecific and interploidal gene flow in Central European Arabidopsis (Brassicaceae). BMC Evol Biol 11 : 346.
20. SchmicklR, KochMA (2011) Arabidopsis hybrid speciation processes. Proc Natl Acad Sci U S A 108 : 14192–14197.
21. Ross-IbarraJ, WrightSI, FoxeJP, KawabeA, DeRose-WilsonL, et al. (2008) Patterns of polymorphism and demographic history in natural populations of Arabidopsis lyrata. PLoS ONE 3: e2411 doi:10.1371/journal.pone.0002411..
22. WrightSI, LaugaB, CharlesworthD (2003) Subdivision and haplotype structure in natural populations of Arabidopsis lyrata.. Mol Ecol 12 : 1247–1263.
23. MoodyME, MuellerLD, SoltisDE (1993) Genetic variation and random drift in autotetraploid populations. Genetics 134 : 649–657.
24. ArnoldB, BombliesK, WakeleyJ (2012) Extending coalescent theory to autotetraploids. Genetics 192 : 195–204.
25. Arabidopsis genome initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408 : 796–815.
26. Weir BS (1996) Genetic Data Analysis II. Sunderland, MA: Sinauer Associates.
27. NielsenR, WilliamsonS, KimY, HubiszMJ, ClarkAG, et al. (2005) Genomic scans for selective sweeps using SNP data. Genome Res 15 : 1566–1575.
28. HahnS (2004) Structure and mechanism of the RNA polymerase II transcription machinery. Nat Struct Mol Biol 18 : 2437–2468.
29. WangW, ChenX (2004) HUA ENHANCER3 reveals a role for a cyclin-dependent protein kinase in the specification of floral organ identity in Arabidopsis.. Development 131 : 3147–3156.
30. AutranD, JonakC, BelcramK, BeemsterGTS, KronenbergerJ, et al. (2002) Cell numbers and leaf development in Arabidopsis: a functional analysis of the STRUWWELPETER gene. EMBO J 21 : 6036–6049.
31. GillmorCS, ParkMY, SmithMR, PepitoneR, KerstetterRA, et al. (2010) The MED12-MED13 module of mediator regulates the timing of embryo patterning in Arabidopsis.. Development 137 : 113–122.
32. SebastianJ, RaviM, AndreuzzaS, PanoliAP, MarimuthuMPA, et al. (2009) The plant adherin AtSCC2 is required for embryogenesis and sister-chromatid cohesion during meiosis in Arabidopsis. Plant J 59 : 1–13.
33. LamWS, YangX, MakaroffCA (2005) Characterization of Arabidopsis thaliana SMC1 and SMC3: evidence that AtSMC3 may function beyond chromosome cohesion. J Cell Sci 118 : 3037–3048 (2005).
34. SchubertV, WeißlederA, AliH, FuchsJ, LermontovaI, et al. (2009) Cohesin gene defects may impair sister chromatid alignment and genome stability in Arabidopsis thaliana. Chromosoma 118 : 591–605.
35. WatanabeK, PacherM, DukowicS, SchubertV, PuchtaH, et al. (2009) The STRUCTURAL MAINTENANCE OF CHROMOSOMES 5/6 Complex Promotes Sister Chromatid Alignment and Homologous Recombination after DNA Damage in Arabidopsis thaliana.. Plant Cell 21 : 2688–2699.
36. BickelJS, ChenL, HaywardJ, YeapSL, AlkersAE, et al. (2010) Structural Maintenance of Chromosomes (SMC) proteins promote homolog-independent recombination repair in meiosis crucial for germ cell genomic stability. PLoS Genet 6: e1001028 doi:10.1371/journal.pgen.1001028..
37. CarylAP, ArmstrongSJ, JonesGH, FranklinFCH (2000) A homologue of the yeast HOP1 gene is inactivated in the Arabidopsis meiotic mutant asy1. Chromosoma 109 : 62–71.
38. BrandãoMM, DantasLL, Siva-FilhoMC (2009) AtPIN: Arabidopsis thaliana protein interaction network. BMC Bioinformatics 10 : 454.
39. HaraK, MarukiY, LongX, YoshinoK-I, OshiroN, et al. (2002) Raptor, a binding partner of Target of Rapamycin (TOR), mediates TOR action. Cell 110 : 177–189.
40. MenandB, DesnosT, NussameL, BergerF, BouchezD, et al. (2002) Expression and disruption of the Arabidopsis TOR (target of rapamycin) gene. Proc Natl Acad Sci U S A 99 : 6422–6427.
41. AndersonGH, VeitB, HansonMR (2005) The Arabidopsis AtRaptor genes are esssential for post-embryonic plant growth. BMC Biology 3 : 12.
42. ParkHJ, ParkHC, LeeSY, BohnertHJ, YunD-J (2011) Ubiquitin and ubiquitin-like modifiers in plants. J Plant Biol 54 : 275–285.
43. LagoC, ClericiE, MizziL, ColomboL, KaterMM (2004) TBP-associated factors in Arabidopsis. Gene 342 : 231–241.
44. EarleyKW, ShookMS, Brower-TolandB, HicksL, PikaardCS (2007) In vitro specificities of Arabidopsis co-activator histone acetyltransferases: implications for histone hyperacetylation in gene activation. Plant J 52 : 615–626.
45. KhannaKK, JacksonSP (2001) DNA double-strand breaks: signaling, repair and the cancer connection. Nature Genet 27 : 247–254.
46. MatsudaM, MiyagawaK, TakahashiM, FukudaT, KataokaT, et al. (1999) Mutations in the RAD54 recombination gene in primary cancers. Oncogene 18 : 3427–3430.
47. LiuJ, KrantzID (2008) Cohesin and human disease. Ann Rev Genom Human Genet 9 : 303–320.
48. ManniniL, MengaS, MusioA (2010) The expanding universe of cohesin functions: a new stability caretaker involved in human disease and cancer. Human Mut 31 : 623–630.
49. JohnsonFB, LombardDB, NeffNF, MastrangeloM-A, DewolfW, et al. (2000) Association of the Bloom Syndrome protein with Topoisomerase IIIa in somatic and meiotic cells. Cancer Res 60 : 1162–1167.
50. MeyerR, FofanovV, PanigrahiAK, MerchantF, ZhangN, et al. (2009) Overexpression and mislocalization of the chromosomal segregation protein separase in multiple human cancers. Clin Cancer Res 15 : 2703–2710.
51. MartelottoLG, OrtizJPA, SteinJ, EspinozaF, QuarinCL, et al. (2005) A comprehensive analysis of gene expression alterations in a newly synthesized Paspalum notatum autotetraploid. Plant Science 169 : 211–220.
52. WangJ, TianL, LeeHS, WeiNE, JiangH, et al. (2006) Genome-wide nonadditive gene regulation in Arabidopsis allotetraploids. Genetics 172 : 507–517.
53. StuparRM, BhaskarPB, YandellBS, RensinkWA, HartAL, et al. (2007) Phenotypic and transcriptomic changes associated with potato autopolyploidization. Genetics 176 : 2055–2067..
54. PignattaD, DilkesBP, YooS-Y, HenryIM, MadlungA, et al. (2010) Differential sensitivity of the Arabidopsis thaliana transcriptome and enhancers to the effects of genome doubling. New Phytologist 186 : 194–206..
55. YuZ, HabererG, MatthesM, RatteiT, MayerKFX, et al. (2010) Impact of natural genetic variation on the transcriptome of autotetraploid Arabidopsis thaliana.. Proc Natl Acad U S A 107 : 17809–17814.
56. NgDW-K, ZhangC, MillerM, ShenZ, BriggsSP, et al. (2012) Proteomic divergence in Arabidopsis autopolyploids and allopolyploids and their progenitors. Heredity 108 : 419–430.
57. WangJ, TianL, MadlungA, LeeH-S, ChenM, et al. (2004) Stochastic and epigenetic changes of gene expression in Arabidopsis polyploids. Genetics 167 : 1961–1973.
58. ShakedH, Avivi-RagolskyN, LevyAA (2006) Involvement of the Arabidopsis SWI2/SNF2 chromatin remodeling gene family in DNA damage response and recombination. Genetics 173 : 985–994.
59. OsakabeK, AbeK, YoshiokaT, OsakabeY, TodorikiS, et al. (2006) Isolation and characterization of the RAD54 gene from Arabidopsis thaliana. Plant J 48 : 827–842.
60. ØstmanB, HintzeA, AdamiC (2012) Impact of epistasis and pleiotropy on evolutionary adaptation. Proc R Soc B 279 : 247–256.
61. TakahasiKR (2009) Coalescent under the evolution of coadaptation. Mol Ecol 18 : 5018–5029.
62. HittingerCT, GonçalvesP, SampaioJP, DoverJ, JohnstonM, et al. (2010) Remarkably ancient balanced polymorphisms in a multi-locus gene network. Nature 464 : 54–58.
63. BremRB, StoreyJD, WhittleJ, KruglyakL (2005) Genetic interactions between polymorphisms that affect gene expression in yeast. Nature 436 : 701–703.
64. SteinerCC, WeberJN, HoekstraHE (2007) Adaptive variation in beach mice produced by two interacting pigmentation genes. PLoS Biol 5: e219 doi:10.1371/journal.pbio.0050219..
65. GerkeJ, LorenzK, CohenB (2009) Genetic interactions between transcription factors cause natural variation in yeast. Science 323 : 498–501.
66. KarlowskiWM, ZielezinskiA, CarrèreJ, PontierD, LagrangeT, et al. (2010) Genome-wide computational identification of WG/GW Argonaute-binding proteins in Arabidopsis. Nucl Acids Res 38 : 4231–4245.
67. ObbardDJ, JigginsFM, BradshawNJ, LittleTJ (2011) Recent and recurrent selective sweeps of the antiviral RNAi gene Argonaute-2 in three species of Drosophila. Mol Biol Evol 28 : 1043–1056.
68. LeeH-C, ChangS-S, ChoudharyS, AaltoAP, MaitiM, et al. (2009) qiRNA is a new type of small interfering RNA induced by DNA damage. Nature 459 : 274–278.
69. WeiW, BaZ, GaoM, WuY, MaY, et al. (2012) A role for small RNAs in DNA double-strand break repair. Cell 149 : 101–112.
70. DingD-Q, OkamasaK, YamaneM, TsutsumiC, HaraguchiT, et al. (2012) Meiosis-specific noncoding RNA mediates robust pairing of homologous chromosomes in meiosis. Science 336 : 732–736.
71. Durand-DubiefM, BastinP (2003) TbAGO1, an Argonaute protein required for RNA interference, is involved in mitosis and chromosome segregation in Trypanosoma brucei. BMC Biol 1 : 2.
72. HallIM, NomaK-I, GrewalSI (2003) RNA interference machinery regulates chromosome dynamics during mitosis and meiosis in fission yeast. Proc Natl Acad Sci U S A 100 : 193–198.
73. LeeDW, PrattRJ, McLaughlinM, AramayoR (2003) An Argonaute-like protein is required for meiotic silencing. Genetics 164 : 821–828.
74. HollingsworthNM, ByersB (1989) HOP1: a yeast meiotic pairing gene. Genetics 121 : 445–462.
75. NonomuraK, NakanoM, EiguchiM, SuzukiT, KurataN (2006) PAIR2 is essential for homologous chromosome synapsis in rice meiosis. J Cell Sci 119 : 217–225.
76. BodenSA, LangridgeP, SpangenbergG, AbleJA (2009) TaASY1 promotes homologous chromosome interactions and is affected by deletion of Ph1. Plant J 57 : 487–497.
77. AviviL (1976) The effect of genes controlling different degrees of homoeologous pairing on quadrivalent frequency in induced autotetraploid lines of Triticum longissimum. Can J Genet Cytol 18 : 357–364.
78. OssowskiS, SchneebergerK, ClarkRM, LanzC, WarthmannN, et al. (2008) Sequencing of natural strains of Arabidopsis thaliana with short reads. Genome Res 18 : 2024–2033.
79. GreenRE, KrauseJ, BriggsAW, MaricicT, StenzelU, et al. (2010) A draft sequence of the Neandertal genome. Science 328 : 710–722.
80. McKennaA, HannaM, BanksE, SivachenkoA, CibulskisK, et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20 : 1297–1303.
81. ThorntonK (2003) libsequence: a C++ class library for evolutionary genetic analysis. Bioinformatics 19 : 2325–2327.
82. HudsonRR (2002) Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18 : 337–338.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 12
Nejčtenější v tomto čísle
- Population Genomics of Sub-Saharan : African Diversity and Non-African Admixture
- Dnmt3a Protects Active Chromosome Domains against Cancer-Associated Hypomethylation
- Excessive Astrocyte-Derived Neurotrophin-3 Contributes to the Abnormal Neuronal Dendritic Development in a Mouse Model of Fragile X Syndrome
- Pre-Disposition and Epigenetics Govern Variation in Bacterial Survival upon Stress