
The Essential Function of RNase III Is to Silence Foreign Toxin Genes
RNase III–related enzymes play key roles in cleaving double-stranded RNA in many biological systems. Among the best-known are RNase III itself, involved in ribosomal RNA maturation and mRNA turnover in bacteria, and Drosha and Dicer, which play critical roles in the production of micro (mi)–RNAs and small interfering (si)–RNAs in eukaryotes. Although RNase III has important cellular functions in bacteria, its gene is generally not essential, with the remarkable exception of that of Bacillus subtilis. Here we show that the essential role of RNase III in this organism is to protect it from the expression of toxin genes borne by two prophages, Skin and SPβ, through antisense RNA. Thus, while a growing number of organisms that use RNase III or its homologs as part of a viral defense mechanism, B. subtilis requires RNase III for viral accommodation to the point where the presence of the enzyme is essential for cell survival. We identify txpA and yonT as the two toxin-encoding mRNAs of Skin and SPβ that are sensitive to RNase III. We further explore the mechanism of RNase III–mediated decay of the txpA mRNA when paired to its antisense RNA RatA, both in vivo and in vitro.
Published in the journal:
. PLoS Genet 8(12): e32767. doi:10.1371/journal.pgen.1003181
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003181
Summary
RNase III–related enzymes play key roles in cleaving double-stranded RNA in many biological systems. Among the best-known are RNase III itself, involved in ribosomal RNA maturation and mRNA turnover in bacteria, and Drosha and Dicer, which play critical roles in the production of micro (mi)–RNAs and small interfering (si)–RNAs in eukaryotes. Although RNase III has important cellular functions in bacteria, its gene is generally not essential, with the remarkable exception of that of Bacillus subtilis. Here we show that the essential role of RNase III in this organism is to protect it from the expression of toxin genes borne by two prophages, Skin and SPβ, through antisense RNA. Thus, while a growing number of organisms that use RNase III or its homologs as part of a viral defense mechanism, B. subtilis requires RNase III for viral accommodation to the point where the presence of the enzyme is essential for cell survival. We identify txpA and yonT as the two toxin-encoding mRNAs of Skin and SPβ that are sensitive to RNase III. We further explore the mechanism of RNase III–mediated decay of the txpA mRNA when paired to its antisense RNA RatA, both in vivo and in vitro.
Introduction
Ribonuclease III is a key enzyme for double-stranded (ds) RNA processing reactions in both bacterial and eukaryotic systems. In bacteria, it is best known for its role in ribosomal RNA maturation [1] and more recently has been shown to be involved in the regulation by small RNAs [2]–[4]. The enzyme was first identified and characterized for its roles in phage RNA (f2, T7 and lambda) processing in E. coli (for recent review, see [5]). Recently, RNase III has shown to be involved in bacterial gene silencing by processing CRISPR RNAs, generated as part of a host defense mechanism against phage DNA in many species [6]. In eukaryotes, enzymes with RNase III domains, such as Dicer and Drosha, play fundamental roles in the processes of RNA interference and in the generation of microRNAs [7], [8].
The recent influx of high resolution RNA transcriptome data has revealed a high level of antisense RNA transcription in many species and this has further stimulated interest in how cells deal with dsRNA on a larger scale. A recent study in Staphylococcus aureus, showed a relatively high level of pervasive transcription that is removed by RNase III [9]. Antisense RNAs to a large proportion of coding RNAs have also been revealed in Helicobacter pylori, Synechocystis and other organisms [10]–[12]. RNase III is the most likely candidate for controlling the level of dsRNA in these systems.
It has long been a mystery why RNase III, encoded by the rnc gene, is essential in B. subtilis [13]. It is possible to delete rnc in E. coli and other bacteria [14]–[16], although such mutations are often accompanied by a decrease in growth rate. Within the Firmicutes, it has been shown that the rnc gene is non-essential in S. aureus [2]. Furthermore, an RNase III-encoding gene is naturally lacking from Deinococcus radiodurans [17] and throughout the archaeal kingdom, although many archaea possess an enzyme of analogous function, called bulge-helix-bulge (BHB) or splicing endonuclease [18]. In eukaryotes, it has been shown that it is possible to delete the RNase III gene rnt1 in yeast, albeit with severe growth defects [19]. The essential role of RNase III in B. subtilis is not related to its function in rRNA metabolism or in maturation of the scRNA, part of the essential 4.5S particle involved in co-translational insertion of proteins in the cellular membrane [13]. Indeed, only trace amounts of 30S rRNA precursor, much lower than in E. coli, are observed in the absence of RNase III in B. subtilis. This indicates that the enzymes that catalyze the final steps of rRNA maturation, RNase J1 [20], Mini-III [21] and RNase M5 [22], function efficiently without prior RNase III action.
We recently performed a tiling array analysis of B. subtilis strains depleted for RNase III and observed a surprisingly minor contribution of RNase III to the stability of many known antisense RNAs [23]. Furthermore, the effect of RNase III-depletion on many of the specific mRNAs tested was shown to be at the transcriptional level. The whole SigW regulon was up-regulated in this way under conditions of RNase III-deficiency, for example. One of the few RNAs tested that did show an effect on mRNA stability was the txpA mRNA. The txpA gene encodes a short (59 amino acids) hydrophobic peptide that causes cell lysis when overexpressed in B. subtilis [24]. It is part of a type I toxin/antitoxin (TA) system. TA systems were initially discovered as part of plasmid and transposon maintenance mechanisms and have more recently been found to be widespread on chromosomes, where they are thought to play important roles in adaptive responses to stress, including phenomena such as bacterial persistence and programmed cell death (for reviews, see [25], [26]). Typically, the toxin is a relatively stable molecule that targets some fundamental cellular function, such as membrane integrity, DNA replication or provokes RNA degradation, while the antitoxin is an unstable entity that needs to be constantly synthesized to counteract the toxin. In type II TA systems, both toxin and antitoxins are proteins, coded in an operon under control of a single promoter, whereas in type I TA systems, the antitoxin is an antisense (as) RNA, expressed from its own promoter on the opposite strand to the toxin gene (for recent review, see [27]). The antitoxin for the txpA mRNA is the RatA RNA and the 3′ ends of the two RNAs overlap [24]. Overexpression of RatA leads to degradation of the txpA toxin RNA, by an unknown mechanism. In this study, we show that the essential role of RNase III in B. subtilis, is the degradation of two type I toxin-encoding mRNAs, the txpA mRNA and the yonT mRNA from the Skin and SPβ prophages, respectively. In the absence of these two prophages or these two toxin-encoding mRNAs, deletion of the RNase III gene is possible and the growth rate of the resulting strains is hardly affected. We probe the role of RNase III in the mechanism of RatA mediated degradation of txpA mRNA both in vivo and in vitro.
Results
Some toxin-encoding mRNAs are stabilized in RNase III–depleted strains
In a recently performed analysis of B. subtilis strains depleted for the essential ribonucleases RNase Y, J1 or III, we noticed a significant increase in the steady-state levels of the type I toxin mRNA txpA in strains depleted for RNase III, while the RatA antitoxin RNA levels were elevated upon depletion of the single-strand specific RNase Y [23]. This was an intriguing result, because we had anticipated that the levels of both sense and antisense partners would be sensitive to the double-strand specific enzyme RNase III. We confirmed the results of the tiling array experiment by Northern blot analysis at times after the addition of rifampicin to inhibit new transcription initiation and showed that the increased expression in both cases was due to increased RNA stability (Figure 1). In an RNase J1 mutant, a degradation intermediate of the RatA asRNA accumulated. To determine whether this was a general phenomenon, we examined the expression patterns of two other suspected type I toxin/antitoxin cassettes, bsrH/as-bsrH, which lies adjacent to txpA/RatA on the chromosome, and bsrG/as-bsrG(SR4), which has recently been studied by the Brantl group [28]. In both cases, the asRNA was stabilized in strains depleted for the single-strand specific enzyme RNase Y (Figures S1 and S2). However, while the bsrG toxin mRNA was significantly stabilized in the RNase III mutant, as seen by Jahn et al. [28], the bsrH toxin mRNA was not. Rather, it showed increased levels in the strain depleted for RNase J1. Thus, even among related type I TA systems, turnover mechanisms differ.

The RNase III gene can be inactivated in strains lacking Skin and SPβ prophages
The fact that RNase III is essential in B. subtilis, while the rnc gene can be deleted in other species, suggests that the B. subtilis enzyme has a specific function that explains why it cannot be removed. We wondered whether this essential function might be related to its role in toxin mRNA turnover. Both the txpA/RatA and bsrH/as-bsrH cassettes belong to the prophage known as Skin, while the bsrG/as-bsrG(SR4) pair is found on the SPβ prophage. SPβ also contains a second known type I TA system named yonT/as-yonT [27] and a TA pair that could be classified as type II, consisting of the precursor for the bacteriocidal sublancin peptide SunA and its immunity protein SunI [29]. Both of these toxin mRNAs are over-expressed in RNase III depleted strains, yonT through increased mRNA stability and sunA through higher transcriptional levels (Figure 2 and Figure S3). The as-yonT transcript is highly stable regardless of whether RNase III is present or not (Figure 2C).

B. subtilis strains lacking SPβ, Skin and a third prophage, PBSX, have been constructed previously [30]. We therefore asked whether it was possible to delete the rnc gene in strains lacking one (SPβ), two (SPβ and Skin) or all three of these prophages. Using chromosomal DNA from a B. subtilis strain (CCB 302), in which most of the rnc open-reading frame (ORF) has been replaced by an ORF encoding spectinomycin resistance (rnc::spc) and which survives through the presence of a xylose-dependent promoter expressing RNase III integrated at the amyE locus (amyE::Pxyl-rnc CmR), we transformed each of the three prophage deficient strains and determined whether we could obtain spectinomycin resistant (SpcR) colonies. As a control for competence levels in each strain, we spread an aliquot of the transformation mixture on plates containing chloramphenicol (Cm) and counted the number of colonies having acquired the Cm-resistant construct at amyE. In wild-type cells or in cells lacking SPβ, very few SpcR colonies were obtained, whereas in cells lacking both SPβ and Skin, or those lacking all three prophages, hundreds of colonies grew overnight (Figure 3A). When normalized for transformation efficiency (number of SpcR colonies divided by number of CmR colonies), at least 100-fold more SpcR colonies were obtained in strains lacking SPβ and Skin prophages compared to wild-type strains or strains lacking SPβ alone (Figure 3B). These results suggest it is possible to delete the RNase III gene from strains lacking SPβ and Skin.

Natural suppressors of the rnc::spc mutation have excised Skin
We next asked what was the nature of the few SpcR transformants obtained with the wild-type and SPβ-deficient strains. Given the result above, it was conceivable that suppressor strains might be obtained by excision of the prophages under selective pressure. We therefore analyzed some of the SpcR transformants obtained in wild-type and SPβ-deficient strains from a number of different experiments to determine their genotypes. First, none of about 100 colonies tested (23 wild-type; 84 ΔSPβ) were CmR, indicating that their viability was not due to simultaneous transfer of the Pxyl-rnc construct at amyE. We then examined the genotype of about 30 SpcR transformants by multiplex colony PCR using oligonucleotide pairs specific for the rnc, sigK and ypqP genes. The Skin and SPβ prophages interrupt the sigK and ypqP genes, respectively, and thus spontaneous excision events can be observed by restoration of these genes and the generation of 567 and 880 bp PCR products. An intact rnc gene is indicated by a 347 bp PCR product. Three different classes of SpcR transformants were obtained. A number of SpcR transformants in wild-type strains retained an intact rnc gene (10 of 19 tested; see Figure 4A for examples), indicating a background level of spontaneous spectinomycin resistance in this strain. This was also evident from the growth of a significant number of SpcR colonies in control transformations with water instead of CCB302 DNA (data not shown). All of the wild-type SpcR transformants in which rnc had been successfully inactivated (9 of 19 tested) had also excised the Skin prophage (see Figure 4A for examples). It is therefore possible to obtain suppressors of the rnc::spc mutation by excising Skin. Curiously, none of the suppressors tested had excised SPβ, suggesting that of the two prophages, the presence of Skin is the most detrimental in an RNase III mutant strain. Over time, these suppressors show a pseudolysis phenotype on plates, however, suggesting all is not well in these strains. Almost all SpcR transformants of the ΔSPβ strain had also excised Skin (13 of 14 tested; see Figure 4A for examples), confirming the idea that Skin is more deleterious than SPβ in strains lacking RNase III. The only suppressor that had not excised Skin (sup5) had a frame shift-mutation in the txpA gene (Figure 4B), suggesting that the TxpA peptide is the key Skin-encoded toxic moiety in the absence of RNase III.

Expression of the txpA and yonT toxin mRNAs account for the lethality of the Skin and SPβ prophages in RNase III mutants
To determine which prophage genes were responsible for the toxic effects in the absence of RNase III, we constructed strains no longer expressing the different RNase III-sensitive toxin mRNAs. Expression of the txpA gene was abolished by making a markerless deletion of its -10 promoter region (txpA -10Δ) using the pMAD system [31] and confirmed by Northern blot (data not shown), while the bsrG, sunA and yonT genes were inactivated by antibiotic resistance cassettes (Table S2). Transformation of the txpA, sunA, bsrG or yonT single mutants, with CCB302 chromosomal DNA had little effect on the number of SpcR colonies or the SpcR/CmR ratio obtained overnight compared to wild-type (Figure 5). Based on the observation made above with the suppressor strain sup5, we assumed that TxpA was the relevant toxin in Skin and combined the txpA -10Δ mutation with that of each of the three toxin genes of SPβ. Only transformation of the txpA -10Δ yonT double mutant with CCB302 DNA yielded a similar SpcR/CmR colony ratio to strains lacking both Skin and SPβ prophages, suggesting these two toxin-encoding mRNAs are the primary determinants of cell toxicity in the absence of RNase III. To confirm this, we compared the growth rates of the ΔSkin ΔSPβ Δrnc and txpA -10Δ ΔyonT Δrnc strains with those of their parental strains lacking prophages or toxin genes. Deletion of the rnc gene in these two genetic contexts had no significant effect on doubling time in LB medium (Table 1), confirming that the major role of RNase III in B. subtilis under these growth conditions is to protect against the expression of these two foreign toxin-encoding mRNAs.


The degradation pathway of RatA
We decided to further investigate the mechanism of RatA-induced degradation of txpA by RNase III, by studying the degradation pathways of the individual RNAs and the RatA/txpA complex. We were also curious about the sensitivity of the RatA antitoxin to the single-strand specific enzyme RNase Y, as we had expected that both toxin and antitoxin RNAs would be sensitive to RNase III.
The stabilization of RatA under conditions of RNase Y depletion suggested that the initial endonucleolytic cleavage of this asRNA is performed by RNase Y. When RNase J1 was depleted, a number of 3′ proximal RatA degradation intermediates accumulated on high resolution gels, clustered around 130 nts and 75 nts in length (Figure S4A). This suggests that the major cleavage site by RNase Y and entry point for RNase J1 is around nt 90 of RatA, just upstream of the region of complementarity with txpA. The accumulation of the ∼75-nt species could either be due to the stabilization of the downstream product of a secondary endonucleolytic cleavage by an unknown enzyme or due to the incomplete depletion of RNase J1 and a stalling of the small amounts of remaining enzyme at the base of secondary structure. The 5′ ends of the ∼130-nt and ∼75-nt species were mapped by primer extension to nts 91 and to two clusters of bands around nts 97–99, and nts 145–147, respectively (Figure S5), both at the base of secondary structures (see below). In the absence of PNPase, the major 3′-5′ exoribonuclease in B. subtilis, an ∼80 nt 5′ proximal species accumulated (Figure S6), also consistent with an initial cleavage by RNase Y around nt 90 and a difficulty of the remaining 3′-5′ exoribonucleases in degrading past stem loop 2 (see below). A model for the degradation pathway of RatA is shown in Figure 6A, with cleavage by RNase Y around nt 90 giving access to both PNPase and RNase J1.

We next examined the degradation pattern of RatA in the absence of txpA, using the strain in which the -10 region of the txpA promoter was deleted. Surprisingly, the degradation pattern of RatA did not change; we observed the same stabilization of RatA in the absence of RNase Y and the accumulation of the same 3′ proximal degradation intermediates of around 130 nts and 75 nts in cells depleted for RNase J1 (Figure S4B). There were two possible explanations for this result; either the structure of RatA does not change significantly upon hybridization to txpA, or RatA is produced in large excess over txpA, in which case the degradation pathway observed in wild-type cells is primarily that of RatA alone, rather than the RatA/txpA hybrid. Quantitative Northern blotting using known quantities of in vitro transcribed RatA and txpA, showed that RatA is present in about 15-fold excess over txpA in wild-type cells (Figure S7), in favor of the latter hypothesis.
Degradation of txpA is dependent on both RatA and RNase III
In wild-type cells, turnover of txpA is primarily dependent on RNase III. The fact that RatA is present in large excess over txpA in this strain suggests that, in the case of txpA, we primarily measured an effect of enzyme depletion on the RatA/txpA hybrid. We failed in attempts to make a deletion of the -10 region of the RatA promoter to study the degradation of wild-type txpA in the absence of RatA in vivo, presumably because of TxpA toxicity. To circumvent this problem, we made an untranslated derivative of the txpA mRNA by changing its start codon to AAG. This mutation falls outside of the region of RatA/txpA complementarity and is not anticipated to affect the secondary structure of txpA (see below) or the ability of these two RNAs to form a duplex. Like the wild-type txpA mRNA, the AUG→AAG mRNA was stabilized in strains depleted for RNase III (Figure 7), showing that the degradation of this non-translated derivative is still RNase III dependent. The difference in half-life compared to that measured with the wild-type txpA in Figure 1 (11.5 mins vs. >20 mins) is most likely explained by different RNase III depletion efficiencies in the two experiments.
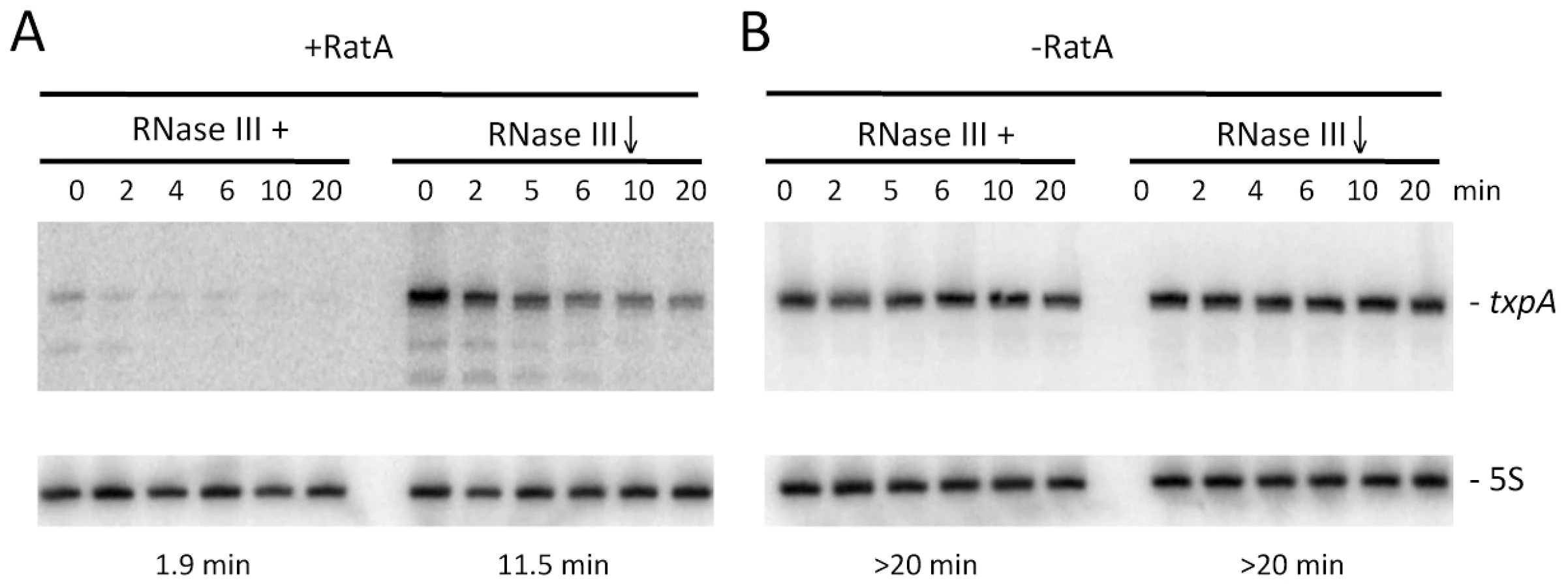
We were able to successfully replace the ratA promoter region with an antibiotic resistance cassette in the context of the txpA (AUG→AAG) mutation, showing that the peptide rather than the mRNA is toxic in B. subtilis, and allowing us to study the txpA mRNA independently of RatA. In the absence of RatA, the txpA (AUG→AAG) mRNA was highly stabilized, regardless of whether or not RNase III was present. These experiments show that the rapid turnover of the txpA mRNA is dependent on both RatA and RNase III; in the absence of either one, the txpA mRNA is extremely stable.
txpA and RatA form an extended hybrid that is a substrate for RNase III cleavage
To study the degradation mechanisms of RatA and txpA further, we turned to in vitro experiments. We made 5′ labeled RatA and txpA RNAs in vitro using T7 RNA polymerase and first probed the secondary structures of the individual RNAs and the RatA/txpA hybrid using the single-stranded endonuclease activity of RNase J1. We have previously shown that this property of RNase J1 can be exploited to determine both known and unknown RNA folding patterns [32]. The structure probing data are shown in Figures S8, S9, and S10 and the predicted secondary structures of the individual RatA and txpA RNAs and the RatA/txpA hybrid are shown in Figure 6. The RatA asRNA forms four stem-loop structures (labeled SL1–4) in addition to the transcription terminator (ter). The txpA RNA forms up to six helical structures (SL1–6) in addition to the terminator in vitro. The significantly reduced intensity of RNase J1 cleavages between nts 137 and 169 of RatA in the RatA/txpA hybrid (Figure S8) is consistent with an extended duplex comprising most of the last 120 nts of each RNA. The duplex does not appear to extend all the way to the 3′ end however, as an RNase J1 hypersensitive region appears in nts corresponding to the downstream strand of the RatA terminator sequence (marked with an asterisk in Figure S8B) when RatA is complexed with txpA.
Incubation of the txpA mRNA with purified RNase III revealed a major cleavage site at nts 96–98 and a corresponding cleavage site at nts 218–220, consistent with cleavage on both sides of the helical structure SL4 by RNase III (Figure 8A). When txpA was hybridized to RatA, these cleavages disappeared completely and different set of RNase III cleavage sites was seen between nts 170–179 of txpA. Thus, binding of RatA changes the structure of txpA, creating new RNase III-sensitive sites. Corresponding cleavages were seen between nts 198–206 of the RatA RNA (Figure 8B), suggesting that both sense and antisense partners are simultaneously cleaved by RNase III. A second pair of RNase III cleavage sites was observed at nts 217/218 of txpA and nt 159 of RatA.

Since the RatA asRNA is a substrate for RNase Y in vivo, we also subjected both RNAs to RNase Y cleavage in vitro. Incubation of the RatA mRNA with purified RNase Y, revealed a number of minor sites of endonucleolytic cleavage, indicative of a relatively relaxed specificity for this enzyme in vitro (Figure 8B). The site most relevant to the pathway seen in vivo is likely to be that at nt 93, close to the major site of RNase J1 access to the RatA RNA in vivo (nt 91). Upon hybridization to txpA, the in vitro cleavage by RNase Y at nt 93 was considerably weaker while that at nt 103, immediately upstream of the duplex, was significantly enhanced. Furthermore, the clusters of cleavages from nt 137 to 159 were lost, consistent with the creation of an extended RatA/txpA duplex in this region. Although a relatively prominent RNase Y cleavage site was observed at nt 201 of the txpA mRNA alone (Figure 8A), this site is in the region of complementarity to RatA and is not likely to exist very much in vivo under conditions of a large excess of RatA. Indeed, cleavage at this site no longer occurs when txpA is hybridized to RatA in vitro.
Discussion
In this paper we have determined that the essential function for RNase III in B. subtilis is to protect it from the expression of toxin genes borne by the Skin and SPβ prophages, via antisense RNA. The cleavage of double-stranded RNA by RNase III-related enzymes as part of host defense mechanisms against virus infection is well-documented. The role played by Dicer in the generation of siRNAs in the process of RNA interference, for example, is a fundamental component of this innate viral defense system (for recent review, see [33]). A recent study has also shown that the CRISPR system, initially characterized as a self-contained defense mechanism against prokaryotic plasmids and phages, uses the host enzyme RNase III to help generate the short protective crRNAs in some bacteria [6]. Although B. subtilis does not contain a CRISPR cassette, our data shows that it nonetheless relies on RNase III to protect it from prophage gene expression, through antisense RNA.
The mode of action of the TxpA and YonT toxins is not known. Both encode short peptides of 59 and 58 amino acids, respectively, with little sequence homology between them. Both have hydrophobic N-termini, predicted to form a transmembrane domain [27], and hydrophilic C-termini; the last 20 amino acids of YonT are very highly positively charged (75% arginine or lysine residues). TxpA has been shown to cause cell-lysis in B. subtilis [24] and YonT to cause growth arrest upon induction in E. coli [27]. We had previously noticed that TxpA (previously known as YqdB) and YonT have the two strongest Shine-Dalgarno (SD) sequences in B. subtilis [34], with 11–12 possible base-pairs with the anti-SD 3′ end of 16S rRNA. It is not known what effect this has on toxin translation levels; while a very strong SD should help ribosome recruitment, these ribosomes may have difficulty escaping the SD. The observation that suppressor colonies, obtained upon transformation of either wild-type or ΔSPβ cells with the rnc::spc construct, have excised the Skin prophage or mutated TxpA, suggests that TxpA is the more toxic of the two peptides (it also has the stronger match to the anti-SD in 16S rRNA).
Paradoxically, we were able to obtain suppressor strains lacking only the Skin prophage, but for optimal transformation efficiency with the rnc::spc construction, it was necessary to remove both Skin and SPβ. Similarly, one of the suppressor strains had a frame-shift mutation in txpA, yet the transformation efficiency of the txpA -10Δ mutant was much lower than that of the txpA yonT double mutant. We cannot rule out the possibility that the suppressors have additional mutations that affect the levels or expression of SPβ genes (see below). However, it is also important to consider that the pressure on the cell to produce colonies is not the same in both cases. In the transformation efficiency assay, cells are additionally being asked to go through the process of becoming competent, a complex developmental program put in place by starving cells. Although the specific effect of competence on toxin gene expression is not known, expression of yonT has recently been shown to reach a peak about 30 mins after glucose starvation [35] and this could have a significant effect on the number of colonies recovered in the transformation assay. The fact that the Δskin SPβ+ suppressors of the Δrnc::spc mutation show a pseudolysis phenomenon on plates suggests that they are not completely healthy, consistent with the idea that the lack of both prophages and their encoded toxins is the optimal configuration for growth and survival in the absence of RNase III.
The Bechhofer laboratory has previously isolated two suppressor strains (BG322 and BG323) in which the rnc gene was successfully inactivated [13]. We also examined both of these strains for the presence of the Skin and SPβ prophages. Both BG322 and BG323 have excised Skin, but appear to have different propensities to excise SPβ during growth (Figure S11). BG323 consistently has a greater proportion of cells lacking SPβ than BG322, but only a minor fraction of cells in freshly plated colonies of either strain have lost SPβ (data not shown), suggesting that this prophage is not stably cured and that these strains have additional mutations that influence their ability to excise SPβ.
We performed in vivo and in vitro experiments to probe the mechanism of RatA-mediated destabilization of txpA mRNA catalyzed by RNase III. We showed that degradation of a non-translated derivative of txpA (AUG→AAG) is dependent on both RatA and RNase III in vivo and that hybridization of the txpA and RatA RNAs generates a highly sensitive substrate for RNase III in vitro. We also determined the secondary structures of the two individual RNAs and the RatA/txpA hybrid. The apical loops of SL3 and the transcription terminator of RatA are complementary to those of the terminator of txpA and SL6, respectively (Figure 6). The formation of sense/antisense RNA complexes is often initiated through loop-loop or ‘kissing’ interactions [36], [37] and RatA and txpA can potentially use the same mechanism to initiate hybrid formation. The extent to which the duplex extends in either direction from the initial interaction site depends on the RNAs in question. For the copA/copT sense/antisense pair, involved in copy number control of the plasmid R1, the duplex does not extend very far from the initial interaction site before getting trapped in a four-way junction with side-by-side helices [37]. The RatA/txpA duplex, on the other hand, seems to extend over most of the 120 nucleotides of complementarity between the two RNAs.
The formation of an extended duplex between RatA and txpA generates a substrate for RNase III and this is the primary mechanism of txpA control in vivo. Indeed, we can calculate that RatA remains in about a 3.5 fold excess over txpA in strains depleted for RNase III; despite this continued excess of RatA, the increased levels of the txpA mRNA are toxic to B. subtilis, presumably because the txpA mRNA can still be translated when paired to RatA. The control of bsrG by as-bsrG(SR4) also occurs at the level of RNA turnover, mediated by RNase III [28], while the control of the type I TA system of Enterococcus faecalis, Fst, occurs primarily at the translational level [38]. The products of txpA cleavage by RNase III appear to be degraded primarily by PNPase, judging from the accumulation of many 5′ proximal txpA fragments in a pnp mutant strain in vivo (Figure S6B). The txpA mRNA is also a good substrate for RNase III in vitro even in the absence of RatA, through the formation of the double-stranded helix SL4 (Figure 6). SL4 can no longer form when txpA is hybridized to RatA. In vivo, where there is a large excess of RatA, it is therefore unlikely that this structure forms very often. Even in the absence of RatA, the txpA (AUG→AAG) mRNA is stable in cells containing RNase III (Figure 7), suggesting that this potential RNase III cleavage site in txpA is also unlikely to be accessible when such conditions arise in vivo.
Depletion of RNase III also leads to an accumulation of the yonT mRNA and a longer species that encodes both YonT and YoyJ. A ∼100-nt asRNA to yonT was detected previously by Northern blot [27] and we have seen this species is stable even in the presence of RNase III (Figure 2C). A recent tiling array analysis suggests this asRNA also overlaps the beginning of the YoyJ reading frame (Figure 2A) and may therefore control the expression of both genes. YoyJ encodes an 83 amino acid protein of unknown function.
It is not clear what the role TxpA or YonT play in the biology of the prophage. They may simply be part of a prophage maintenance system, by killing B. subtilis cells that do not constantly synthesize their asRNAs. However, they could also play a role in linking prophage biology to the physiology of the cell. It has recently been shown, for example, that levels of the bsrG toxin mRNA of SPβ are decreased about 10-fold at 48°C [28]. Interestingly, both txpA and yonT show high expression levels under conditions of glucose exhaustion [35]. TxpA expression is additionally sensitive to both high and low phosphate concentrations, while yonT is induced in the presence of mitomycin C, which induces the SOS response. It will be interesting to determine whether these conditions affect either the behavior of the prophage or the host-cell in a toxin-dependent manner. In a recent study, depletion of the Skin transcriptional repressor (SknR) was shown to cause cell death through overexpression of two proteins YqaM and YqaH, which bind to DnaA and DnaC, respectively [39]. The Skin prophage appears thus to have a variety of options that allow it to slow or halt the growth of its host cell.
Materials and Methods
Construction of bacterial strains
The B. subtilis strains used in this study were derivatives of W168 or 168 trp−. Strains lacking SPβ, Skin and PBSX prophages (168 trp− background) have been described previously [30] and were a kind gift from J.M. van Dijl. A Skin-less derivative of W168 was kindly constructed by P. Stragier and named CCB297.
Strain CCB302 (rnc::spc amyE::pX-rnc Cm) was constructed as follows. The rnc gene was amplified from B. subtilis chromosomal DNA using oligos CC816/817 (Table S1), digested with SpeI and BamHI and cloned in pX [40] cleaved with the same enzymes. The resulting plasmid, pX-rnc, was integrated in the amyE locus of Skin-less strain CCB297 to create CCB298. The rnc gene of CCB298 was then interrupted by a spectinomycin resistance cassette using chromosomal DNA from strain BG324 [13] to create strain CCB302. Growth of this strain is xylose-dependent.
Strains CCB034 (rnjA:pMUTIN-rnjA), CCB288 (rnc::spc amyE::Pspac-rnc Cm) and CCB294 (rny::spc amyE::Pspac-rny Cm) have been described previously [20], [23]. RNase depletions were performed as in [20], [23].
Strain CCB363 (SPβ::PIID-sspB kan) was constructed by transforming W168 with chromosomal DNA from MO4738 SPβ::PIID-sspB kan spoIIIE::Tc, a kind gift from P. Stragier. This chromosomal DNA was also used to transform CCB297 to create CCB364 ΔSkin SPβ::PIID-sspB kan.
Strain CCB325 (txpA -10Δ) was made by markerless mutation of the -10 promoter region of txpA on the W168 chromosome using plasmid pMAD-I according to [31]. Plasmid pMAD-I was constructed as follows. Overlapping upstream and downstream fragments containing the txpA -10 promoter deletion were amplified using oligo pairs CC907/908 and CC909/910, respectively. The overlapping fragments were then assembled in a new PCR reaction with CC907 and CC910, digested with BamHI and cloned in pMAD [31], a kind gift from M. Débarbouillé.
Strain CCB361 bsrG::kan was constructed by building a PCR fragment containing upstream and downstream regions of the bsrG gene flanking a kanamycin resistance cassette by over lapping PCR, using oligos CC1015–1020 (Table S1), and transforming in W168. Chromosomal DNA from this strain was used to transform strain CCB325 to create CCB368 txpA -10Δ bsrG::kan.
Strain CCB377 sunA::kan was made by transforming W168 by chromosomal DNA from a sunA::kan strain, kindly provided by J.M. van Dijl. This chromosomal DNA was also transformed into CCB325 to create CCB402 txpA -10Δ sunA::kan.
Strain CCB413 yonT::ery was constructed by building a PCR fragment containing upstream and downstream regions of the yonT gene, flanking an erythromycin resistance cassette using oligos CC1071–CC1077 (Table S1) and transforming in W168. Chromosomal DNA from this strain was used to transform strain CCB325 to create CCB414 txpA -10Δ yonT::ery.
Strain CCE192, used to overexpress N-terminal His tagged B. subtilis RNase Y, was constructed as follows. The RNase Y gene (ymdA/rny) lacking sequences corresponding to the N-terminal transmembrane domain was amplified by PCR using oligos CC657 and CC658 (Table S1), cleaved by NdeI and BamHI and cloned in pET28a (Novagen) cut with the same enzymes. The resulting plasmid pET28-ΔTM-YmdA was transformed into E. coli strain BL21 CodonPlus cells to yield strain CCE192.
Strain CCB456 was made by markerless mutation of the txpA start codon AUG to AAG using plasmid pMAD-III according to [31]. Plasmid pMAD-III was constructed as follows. Overlapping upstream and downstream fragments containing the AAG mutation were amplified using oligo pairs CC907/1129 and CC1130/910, respectively. The overlapping fragments were then assembled in a new PCR reaction with CC907/910, digested with BamHI and cloned in pMAD.
Strain CCB461 was made by deleting the ratA promoter region (68 nts) with an erythromycin resistance cassette in strain CCB456. Upstream and downstream homology regions were assembled on either side of the antibiotic cassette by overlapping PCR using the following oligo pairs CC1151/1148, CC1147/1149 and CC1150/1152. The 3 overlapping fragments were then assembled in a new PCR reaction amplified by oligos CC1151 and CC1152 and used to transform CCB456.
Strains CCB467 and CCB468 were made by successively transferring the amyE::Pspac-rnc Cm, rnc::spc constructs and plasmid pMAP65 to strain CCB456 and CCB461, respectively.
Northern blots and primer extension assays
Northern blots and primer extension assays were performed as described previously [20], [23].
Multiplex PCR reactions
Multiplex PCR reactions were performed in the presence of the 6 oligonucleotides CC435/1011 (rnc), CC986/987 (sigK), 990/991 (ypqP) for 25 cycles (94°C for 30 sec, 53° for 30 sec, 72°C for 1 min).
Structure probing experiments
Structure probing experiments using C-terminal His tagged RNase J1 (0.6 µg per reaction) have been described in [32]. The txpA and RatA transcripts were transcribed in vitro by T7 RNA polymerase (Ambion) using PCR templates with integrated T7 promoters (oligo pairs CC998/999 and CC1000/1001, respectively). RNAs were synthesized with a 5′OH group (using an 6-fold excess of guanosine over GTP) to facilitate 5′ labeling. RNAs were 5′ labeled using T4 polynucleotide kinase (Biolabs).
In vitro RNase cleavage assays
The purification and assay of RNase III has been described previously [21]. N-terminal His-tagged RNase Y was purified from strain CCE192 on a Ni-NTA column as described previously [41] and dialyzed against elution buffer without imidazole. The final enzyme concentration was 4 mg/ml. RNase Y was assayed in vitro in 20 mM Tris pH 8.0, 8 mM MgCl2, 100 mM NH4Cl, 0.1 mM DTT.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. NikolaevN, SilengoL, SchlessingerD (1973) A role for ribonuclease 3 in processing of ribosomal ribonucleic acid and messenger ribonucleic acid precursors in Escherichia coli. J Biol Chem 248 : 7967–7969.
2. HuntzingerE, BoissetS, SaveanuC, BenitoY, GeissmannT, et al. (2005) Staphylococcus aureus RNAIII and the endoribonuclease III coordinately regulate spa gene expression. EMBO J 24 : 824–835.
3. ViegasSC, SilvaIJ, SaramagoM, DominguesS, ArraianoCM (2011) Regulation of the small regulatory RNA MicA by ribonuclease III: a target-dependent pathway. Nucleic Acids Res 39 : 2918–2930.
4. OpdykeJA, FozoEM, HemmMR, StorzG (2011) RNase III participates in GadY-dependent cleavage of the gadX-gadW mRNA. J Mol Biol 406 : 29–43.
5. Nicholson AW, editor (2011) Ribonuclease III and the role of double-stranded RNA processing in bacterial systems. Berlin: Springer-Verlag. 269–297 p.
6. DeltchevaE, ChylinskiK, SharmaCM, GonzalesK, ChaoY, et al. (2011) CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471 : 602–607.
7. BernsteinE, CaudyAA, HammondSM, HannonGJ (2001) Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409 : 363–366.
8. LeeY, AhnC, HanJ, ChoiH, KimJ, et al. (2003) The nuclear RNase III Drosha initiates microRNA processing. Nature 425 : 415–419.
9. LasaI, Toledo-AranaA, DobinA, VillanuevaM, de los MozosIR, et al. (2011) Genome-wide antisense transcription drives mRNA processing in bacteria. Proc Natl Acad Sci U S A 108 : 20172–20177.
10. SharmaCM, HoffmannS, DarfeuilleF, ReignierJ, FindeissS, et al. (2010) The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464 : 250–255.
11. MitschkeJ, GeorgJ, ScholzI, SharmaCM, DienstD, et al. (2011) An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp. PCC6803. Proc Natl Acad Sci U S A 108 : 2124–2129.
12. GeorgJ, HessWR (2011) cis-antisense RNA, another level of gene regulation in bacteria. Microbiol Mol Biol Rev 75 : 286–300.
13. HerskovitzMA, BechhoferDH (2000) Endoribonuclease RNase III is essential in Bacillus subtilis. Mol Microbiol 38 : 1027–1033.
14. KindlerP, KeilTU, HofschneiderPH (1973) Isolation and characterization of a ribonuclease 3 deficient mutant of Escherichia coli. Mol Gen Genet 126 : 53–59.
15. PriceB, AdamidisT, KongR, ChampnessW (1999) A Streptomyces coelicolor antibiotic regulatory gene, absB, encodes an RNase III homolog. J Bacteriol 181 : 6142–6151.
16. ViegasSC, PfeifferV, SittkaA, SilvaIJ, VogelJ, et al. (2007) Characterization of the role of ribonucleases in Salmonella small RNA decay. Nucleic Acids Res 35 : 7651–7664.
17. CondonC, PutzerH (2002) The phylogenetic distribution of bacterial ribonucleases. Nucleic Acids Res 30 : 5339–5346.
18. XueS, CalvinK, LiH (2006) RNA recognition and cleavage by a splicing endonuclease. Science 312 : 906–910.
19. ChanfreauG, RotondoG, LegrainP, JacquierA (1998) Processing of a dicistronic small nucleolar RNA precursor by the RNA endonuclease Rnt1. EMBO J 17 : 3726–3737.
20. BrittonRA, WenT, SchaeferL, PellegriniO, UickerWC, et al. (2007) Maturation of the 5′ end of Bacillus subtilis 16S rRNA by the essential ribonuclease YkqC/RNase J1. Mol Microbiol 63 : 127–138.
21. RedkoY, BechhoferDH, CondonC (2008) Mini-III, an unusual member of the RNase III family of enzymes, catalyses 23S ribosomal RNA maturation in B. subtilis. Mol Microbiol 68 : 1096–1106.
22. CondonC, Brechemier-BaeyD, BeltchevB, Grunberg-ManagoM, PutzerH (2001) Identification of the gene encoding the 5S ribosomal RNA maturase in Bacillus subtilis: Mature 5S rRNA is dispensable for ribosome function. RNA 7 : 242–253.
23. DurandS, GiletL, NicolasP, BessièresP, CondonC (2012) Three essential ribonucleases, RNase Y, J1 and III, control the abundance a majority of B. subtilis mRNAs. PLoS Genet doi:10.1371/journal.pgen.1002520..
24. SilvaggiJM, PerkinsJB, LosickR (2005) Small untranslated RNA antitoxin in Bacillus subtilis. J Bacteriol 187 : 6641–6650.
25. Van MelderenL (2010) Toxin-antitoxin systems: why so many, what for? Curr Opin Microbiol 13 : 781–785.
26. YamaguchiY, InouyeM (2011) Regulation of growth and death in Escherichia coli by toxin-antitoxin systems. Nat Rev Microbiol 9 : 779–790.
27. FozoEM, MakarovaKS, ShabalinaSA, YutinN, KooninEV, et al. (2010) Abundance of type I toxin-antitoxin systems in bacteria: searches for new candidates and discovery of novel families. Nucleic Acids Res 38 : 3743–3759.
28. JahnN, PreisH, WiedemannC, BrantlS (2012) BsrG/SR4 from Bacillus subtilis - the first temperature-dependent type I toxin-antitoxin system. Mol Microbiol 83 : 579–598.
29. DuboisJY, KouwenTR, SchurichAK, ReisCR, EnsingHT, et al. (2009) Immunity to the bacteriocin sublancin 168 Is determined by the SunI (YolF) protein of Bacillus subtilis. Antimicrob Agents Chemother 53 : 651–661.
30. WestersH, DorenbosR, van DijlJM, KabelJ, FlanaganT, et al. (2003) Genome engineering reveals large dispensable regions in Bacillus subtilis. Mol Biol Evol 20 : 2076–2090.
31. ArnaudM, ChastanetA, DebarbouilleM (2004) New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl Environ Microbiol 70 : 6887–6891.
32. Daou-ChaboR, CondonC (2009) RNase J1 endonuclease activity as a probe of RNA secondary structure. RNA 15 : 1417–1425.
33. UmbachJL, CullenBR (2009) The role of RNAi and microRNAs in animal virus replication and antiviral immunity. Genes Dev 23 : 1151–1164.
34. Daou-ChaboR, MathyN, BenardL, CondonC (2009) Ribosomes initiating translation of the hbs mRNA protect it from 5′-to-3′ exoribonucleolytic degradation by RNase J1. Mol Microbiol 71 : 1538–1550.
35. NicolasP, MaderU, DervynE, RochatT, LeducA, et al. (2012) Condition-dependent transcriptome reveals high-level regulatory architecture in Bacillus subtilis. Science 335 : 1103–1106.
36. PerssonC, WagnerEG, NordstromK (1990) Control of replication of plasmid R1: formation of an initial transient complex is rate-limiting for antisense RNA–target RNA pairing. EMBO J 9 : 3777–3785.
37. KolbFA, EngdahlHM, Slagter-JagerJG, EhresmannB, EhresmannC, et al. (2000) Progression of a loop-loop complex to a four-way junction is crucial for the activity of a regulatory antisense RNA. EMBO J 19 : 5905–5915.
38. ShokeenS, PatelS, GreenfieldTJ, BrinkmanC, WeaverKE (2008) Translational regulation by an intramolecular stem-loop is required for intermolecular RNA regulation of the par addiction module. J Bacteriol 190 : 6076–6083.
39. KimuraT, AmayaY, KobayashiK, OgasawaraN, SatoT (2010) Repression of sigK intervening (skin) element gene expression by the CI-like protein SknR and effect of SknR depletion on growth of Bacillus subtilis cells. J Bacteriol 192 : 6209–6216.
40. KimL, MogkA, SchumannW (1996) A xylose-inducible Bacillus subtilis integration vector and its application. Gene 181 : 71–76.
41. CondonC, PellegriniO, MathyN, BenardL, RedkoY, et al. (2008) Assay of Bacillus subtilis ribonucleases in vitro. Methods Enzymol 447 : 277–308.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 12
Nejčtenější v tomto čísle
- Population Genomics of Sub-Saharan : African Diversity and Non-African Admixture
- Dnmt3a Protects Active Chromosome Domains against Cancer-Associated Hypomethylation
- Excessive Astrocyte-Derived Neurotrophin-3 Contributes to the Abnormal Neuronal Dendritic Development in a Mouse Model of Fragile X Syndrome
- Pre-Disposition and Epigenetics Govern Variation in Bacterial Survival upon Stress