
, a Gene Involved in Axonal Pathfinding, Is Mutated in Patients with Kallmann Syndrome
Kallmann syndrome (KS) associates congenital hypogonadism due to gonadotropin-releasing hormone (GnRH) deficiency and anosmia. The genetics of KS involves various modes of transmission, including oligogenic inheritance. Here, we report that Nrp1sema/sema mutant mice that lack a functional semaphorin-binding domain in neuropilin-1, an obligatory coreceptor of semaphorin-3A, have a KS–like phenotype. Pathohistological analysis of these mice indeed showed abnormal development of the peripheral olfactory system and defective embryonic migration of the neuroendocrine GnRH cells to the basal forebrain, which results in increased mortality of newborn mice and reduced fertility in adults. We thus screened 386 KS patients for the presence of mutations in SEMA3A (by Sanger sequencing of all 17 coding exons and flanking splice sites) and identified nonsynonymous mutations in 24 patients, specifically, a frameshifting small deletion (D538fsX31) and seven different missense mutations (R66W, N153S, I400V, V435I, T688A, R730Q, R733H). All the mutations were found in heterozygous state. Seven mutations resulted in impaired secretion of semaphorin-3A by transfected COS-7 cells (D538fsX31, R66W, V435I) or reduced signaling activity of the secreted protein in the GN11 cell line derived from embryonic GnRH cells (N153S, I400V, T688A, R733H), which strongly suggests that these mutations have a pathogenic effect. Notably, mutations in other KS genes had already been identified, in heterozygous state, in five of these patients. Our findings indicate that semaphorin-3A signaling insufficiency contributes to the pathogenesis of KS and further substantiate the oligogenic pattern of inheritance in this developmental disorder.
Published in the journal:
. PLoS Genet 8(8): e32767. doi:10.1371/journal.pgen.1002896
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002896
Summary
Kallmann syndrome (KS) associates congenital hypogonadism due to gonadotropin-releasing hormone (GnRH) deficiency and anosmia. The genetics of KS involves various modes of transmission, including oligogenic inheritance. Here, we report that Nrp1sema/sema mutant mice that lack a functional semaphorin-binding domain in neuropilin-1, an obligatory coreceptor of semaphorin-3A, have a KS–like phenotype. Pathohistological analysis of these mice indeed showed abnormal development of the peripheral olfactory system and defective embryonic migration of the neuroendocrine GnRH cells to the basal forebrain, which results in increased mortality of newborn mice and reduced fertility in adults. We thus screened 386 KS patients for the presence of mutations in SEMA3A (by Sanger sequencing of all 17 coding exons and flanking splice sites) and identified nonsynonymous mutations in 24 patients, specifically, a frameshifting small deletion (D538fsX31) and seven different missense mutations (R66W, N153S, I400V, V435I, T688A, R730Q, R733H). All the mutations were found in heterozygous state. Seven mutations resulted in impaired secretion of semaphorin-3A by transfected COS-7 cells (D538fsX31, R66W, V435I) or reduced signaling activity of the secreted protein in the GN11 cell line derived from embryonic GnRH cells (N153S, I400V, T688A, R733H), which strongly suggests that these mutations have a pathogenic effect. Notably, mutations in other KS genes had already been identified, in heterozygous state, in five of these patients. Our findings indicate that semaphorin-3A signaling insufficiency contributes to the pathogenesis of KS and further substantiate the oligogenic pattern of inheritance in this developmental disorder.
Introduction
Kallmann syndrome (KS, MIM 147950, 244200, 308700, 610628, 612370, 612702) is an inherited neurodevelopmental disorder defined as the association of hypogonadotropic hypogonadism, due to gonadotropin-releasing hormone (GnRH) deficiency, and the inability to smell (anosmia or hyposmia), related to abnormal development of the peripheral olfactory system (olfactory nerves and olfactory bulbs). The genetics of KS involves various modes of transmission, specifically, autosomal recessive, autosomal dominant with incomplete penetrance, X-chromosome linked, and also oligogenic inheritance [1], [2]. Pathohistological studies of fetuses with olfactory bulb agenesis have shown that the reproductive phenotype of KS results from a pathological sequence in embryonic life, whereby premature interruption of the olfactory, vomeronasal and terminal nerve fibers in the frontonasal region disrupts the migration of neuroendocrine GnRH cells, which normally migrate from the nose to the brain along these nerve fibers [3], [4]. What causes the primary failure of these fibers to establish proper contact with the forebrain is, however, still unknown. Since KS is genetically heterogeneous, identification of the various genes involved and the study of appropriate animal models are expected to provide valuable clues. Barely 30% of the KS patients have mutations in any of the eight genes known so far, specifically, KAL1 (ID 3730) [5]–[7], FGFR1 (ID 2260) [8], FGF8 (ID 2253) [9], PROKR2 (ID 128674), PROK2 (ID 60675) [10], WDR11 (ID 55717) [11], HS6ST1 (ID 9394) [12], CHD7 (ID 55636) [13], [14], and current efforts thus concentrate on the identification of other genes that contribute to this disorder. One strategy is based on close pathohistological examination of targeted mutant mice that may reproduce the human KS phenotype. Here, we show that Nrp1sema/sema mutant mice, which are defective for the semaphorin-binding domain of the membrane coreceptor neuropilin-1, have a KS-like phenotype, and we provide genetic evidence that insufficient semaphorin-3A signaling can contribute to the KS phenotype in man.
Results/Discussion
Neuropilin-1 expression delineates the migratory route of embryonic GnRH cells in mice and humans
In the mouse, GnRH cells begin to leave the epithelium of the medial olfactory pit around embryonic day 11.5 (E11.5). They migrate in the frontonasal region in close association with growing fibers of the vomeronasal and terminal nerves, then penetrate into the rostral forebrain together with the central processes of these nerves, and continue their migration towards the hypothalamic region along a branch of the vomeronasal nerve that projects to the basal forebrain or along fibers of the terminal nerve itself [15]–[17] (Figure 1A). Proper navigation of growing axons depends on guidance cues, which include semaphorins, a large and diverse family of secreted and membrane-associated proteins [18]. Among these, there is semaphorin-3A (Sema3A), a secreted protein with repulsive effects on primary olfactory axons expressing the coreceptor neuropilin-1 (Nrp1) [19]–[21]. The role of semaphorins in the navigation of vomeronasal/terminal axons and embryonic GnRH cells is still unclear, but previous studies in rodents have shown that migrating GnRH cells are morphologically associated with Nrp1-immunoreactive axons and are themselves immunoreactive [22], [23]. Indeed, we were able to confirm these findings in E14.5 mouse embryos, and extend them to a 9-week old human fetus (Figure 1B–1D), using specific antibodies to Nrp1 (Figure S1) in immunohistofluorescence experiments. Notably, the caudal branch of the vomeronasal nerve that accompanies GnRH cells in their intracerebral path was also Nrp1-immunoreactive in the mouse embryos (Figure 1C). These observations suggested that semaphorin signaling through Nrp1 imparts guidance information to axons of the vomeronasal neurons and migrating GnRH cells.

Migration of GnRH cells to the basal forebrain is defective in Nrp1sema/sema mutant mice
We thus analyzed Nrp1sema/sema mutant mice that harbor inactivating aminoacid substitutions in the semaphorin-binding domain of Nrp1. Unlike Nrp1−/− knockout mice, which die around E12.5 [24], these mice survive until birth [25]. In Nrp1sema/sema newborn mice (n = 4), many axons of olfactory receptor neurons were stuck at the dorsal aspect of the cribriform plate and did not project to the olfactory bulb glomeruli (Figure 2A). Olfactory cues are thought to play an important role in suckling behavior [26]. Analysis of six litters at postnatal day 1 (P1) indeed showed that 7 out of 8 Nrp1sema/sema pups had little or no milk in their stomachs, whereas most Nrp1+/+ and Nrp1sema/+ littermates (18 out of 21) had full stomachs. These findings account for the decreased survival rate of homozygous, but not heterozygous, mutant pups [25], and strongly suggest that the sense of smell is affected in Nrp1sema/sema mice. Most importantly, DiI axonal labeling at E14.5 showed abnormal projection of the vomeronasal nerve to the ventral forebrain in the homozygous mutant embryos (n = 4) (Figure 2B). Since this projection forms the axonal scaffold for the intracerebral migration of GnRH cells [17], [27], we analyzed the distribution of these cells in E14.5 and newborn mice. At E14.5, a significant accumulation of GnRH cells in the nasal compartment and concomitant decreased cell number within the brain already indicated abnormal cell migration in the mutants (n = 4) (Figure 2E). In addition, while GnRH cells normally turn ventrally towards the basal forebrain, in Nrp1sema/sema embryos, many GnRH cells were found to migrate dorsally and medially towards the cortex and the thalamus, respectively, along aberrantly projecting axonal fibers (Figure 2C, Figure S2). Incidentally, conditional mutant mice that lack Nrp1 only in GnRH cells (GnRH::cre; Nrp1loxP/loxP mice) displayed a normal distribution of these cells between the nose and the brain at E14.5 as well as a normal number of these cells in the adult brain (Figure S3 and data not shown), thus confirming that the defective migration we found in Nrp1sema/sema embryos is not a cell-autonomous trait. The migration defect was still conspicuous at birth (Figure 2D), a time when neuroendocrine GnRH cells have completed their migration in normal mice [3]. The ventral forebrain region of Nrp1sema/sema newborn mice (n = 4) indeed contained 38% fewer GnRH cells, which were dispersed, while there was a 36% increase in the number of GnRH cells detected in the rostral forebrain compared with Nrp1+/+ littermates (n = 5, p<0.01 for both comparisons) (Figure 2E). This GnRH-cell migration defect in Nrp1sema/sema animals resulted in decreased GnRH immunoreactivity in the median eminence of the hypothalamus (Figure 2D), which is the projection field of neuroendocrine GnRH cells.

Of the Nrp1sema/sema newborn mice, only four males and two females survived into adulthood. Both females had delayed pubertal activation, specifically, the first ovulation occurred more than 10 days later than in Nrp1sema/+ heterozygous littermates, and monitoring of the ovarian cycle from P60 showed that one female stayed in the diestrous stage (a stage with low gonadotropin outputs) throughout the 3-week study period, while the other female had disrupted ovarian cyclicity (data not shown). Male reproductive capacity was assessed by breeding the young adult (P90) Nrp1sema/sema males with confirmed wild-type dams, and monitoring the occurrence of litters over 10–13 months. While Nrp1sema/+ males (n = 4) produced about one litter per month, as did Nrp1+/+ males, the fertility index (number of litters per month) was markedly reduced in the Nrp1sema/sema males, which only gave birth to 2 to 4 litters (fertility index: 0.29±0.04 vs. 1.08±0.12 in Nrp1sema/+; Student's t-test, p<0.001). Moreover, neuroanatomical analysis of Nrp1sema/sema adult brains showed significantly reduced GnRH cell populations in the preoptic and hypothalamic regions (384±67 GnRH cells, n = 4) compared to Nrp1sema/+ littermates (767±49 GnRH cells, n = 4; Student's t-test, p<0.001), whereas Nrp1sema/+ mice did not differ from Nrp1+/+ mice (701±11 GnRH cells, n = 4; Student's t-test, p>0.05). Therefore, the GnRH cell migration defect found in Nrp1sema/sema mouse embryos was not corrected during later development, and caused subfertility in adult homozygous mutants.
SEMA3A loss-of-function mutations in Kallmann syndrome patients
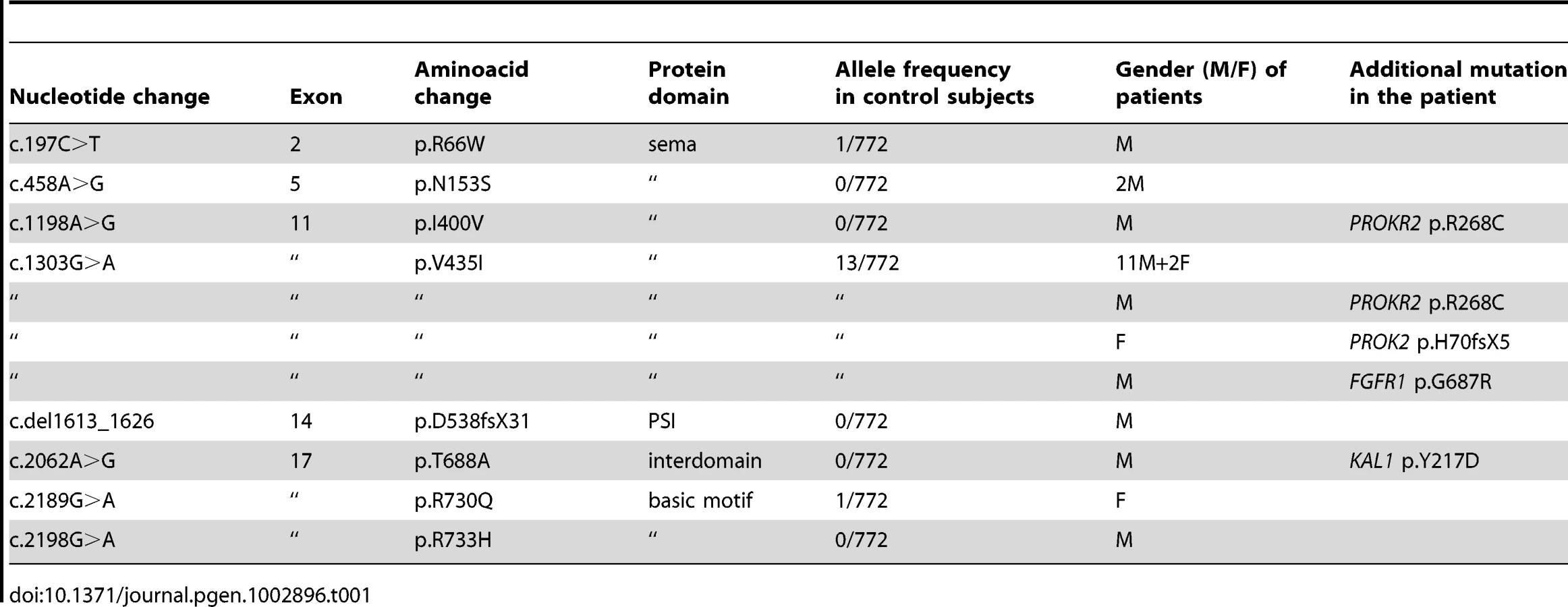
The KS-like phenotype of Nrp1sema/sema mice, and that, even more pronounced, of Sema3a−/− mice [22], prompted us to ask whether insufficient Sema3A signaling through Nrp1 might also be involved in the human disorder. We sought mutations, by Sanger sequencing, in the 17 coding exons of SEMA3A (ID 10371) and flanking splice sites, in 386 unrelated KS patients (297 males and 89 females). All of them had confirmed hypogonadotropic hypogonadism and anosmia or hyposmia, and some already harbored a mutation in one of the five KS genes we had previously analyzed, specifically, in KAL1 (13 patients), FGFR1 (30 patients), FGF8 (3 patients), PROKR2 (30 patients), or PROK2 (12 patients). Nonsynonymous mutations in SEMA3A were found in 24 patients (20 males and 4 females), all in heterozygous state (Table 1). They consist of a frameshifting deletion of 14 nucleotides (c.del1613_1626; p.D538fsX31), and seven different missense mutations (p.R66W, p.N153S, p.I400V, p.V435I, p.T688A, p.R730Q, p.R733H) that affect evolutionarily conserved aminoacid residues located in different domains of the protein (Figure 3). In addition, the p.R730Q and p.R733H mutations, which both remove basic residues in the C-terminal basic motif of Sema3A, are predicted to affect in vivo proteolytic processing by furin-like endoproteases at residue R734 [28]. Notably, all the missense mutations, but not the frameshifting mutation, have been reported in the Exome Variant Server database, with allele frequencies in the European American population below 0.03% except for p.N153S (0.4%) and p.V435I (1.3%). Three of these mutations (p.R66W, p.V435I, p.R730Q) were also detected in our sample of 386 unrelated Caucasian controls (see Table 1). We thus studied the effects of the eight mutations on the signaling activity of Sema3A using the GN11 cell line, derived from murine embryonic GnRH cells [29]–[31], and conditioned media from transfected COS-7 cells producing Sema3A either from the wild-type SEMA3A cDNA or from cDNAs harboring the mutations. We found that the conditioned medium from COS-7 cells transfected with the wild-type SEMA3A cDNA was as potent at inducing phosphorylation of FAK (focal adhesion kinase) and ERK1/2 (extracellular signal-regulated kinases 1 and 2) in GN11 cells as the purified recombinant human Sema3A (100 µg/L). By contrast, Sema3As harboring the N153S, I400V, T688A, or R733H missense mutations were ineffective, despite normal production and secretion of the proteins by COS-7 cells, shown by western blot analysis of the conditioned media. The R66W and V435I mutant proteins were not detected in the conditioned medium, which indicates defective secretion. Likewise, the c.del1613_1626 (p.D538fsX31) frameshifting mutation resulted in the absence of protein secretion, as expected (Figure 4). From these results, we were able to conclude that all the mutations, except p.R730Q, are loss-of-function mutations that affect the secretion or signaling activity of Sema3A, which strongly argues in favor of their pathogenic effect in the KS patients. In addition, the p.R730Q mutation may still have a pathogenic effect not detected in our experimental system, especially since this mutation is expected to impair proteolytic processing of Sema3A in vivo, as mentioned previously. Notably, the patients carrying the p.T688A and p.I400V mutations, and three patients carrying the p.V435I mutation also carry, in heterozygous state, p.Y217D, p.R268C (two patients), p.H70fsX5, and p.G687N pathogenic mutations in KAL1, PROKR2, PROK2, and FGFR1, respectively (Table 1), which further substantiates the digenic/oligogenic mode of inheritance of KS [1], [2]. Based on the seemingly normal reproductive phenotype of Sema3a+/− heterozygous mice [21], [22], we suggest that the monoallelic mutations in SEMA3A are not sufficient to induce the abnormal phenotype in the patients, but contribute to the pathogenesis of KS through synergistic effects with mutant alleles of other disease-associated genes. Accordingly, the other KS patients who carry monoallelic mutations in SEMA3A are also expected to carry at least one pathogenic mutation in another gene (see footnote). Although NRP1 (ID 8829) might be viewed as one of the best candidates, we did not find a mutation within its 17 coding exons and flanking splice sites in any of these patients, nor did we in a group of 100 KS patients without SEMA3A mutations, which indicates that mutations in NRP1, if any, are infrequent. It is also possible that some of the additional mutations affect other proteins involved in Sema3A-signaling, such as members of the plexin family of transmembrane receptors or neuropilin-2 [18], [22]. A whole-exome sequencing strategy should prove useful to explore the spectrum of genes which, when mutated, can lead to a KS phenotype in conjunction with SEMA3A mutations.


Note
While this article was under review, Young et al. reported the coexistence of KS and a large deletion in SEMA3A, in heterozygous state, in two siblings and their clinically affected father (Hum. Reprod., 2012; 27 : 1460–1465). Our findings do not support mere autosomal dominant Mendelian inheritance in this family, and suggest that another, as yet unidentified genetic hit combines with SEMA3A haploinsufficiency to produce the disease phenotype.
Materials and Methods
Ethics statement
This study was approved by the national research ethics committee (agence de biomédecine, Paris, France).
Animals and human fetus
All experiments on mice were carried out in accordance with Directive 86/609/EEC of the Council of the European Communities regarding the mammalian research and French bylaw. Nrp1sema/+ mice (B6.129(C)-Nrp1tm1Ddg/J) [25] were purchased from the Jackson laboratory (Maine, USA), maintained on a controlled 12 h∶12 h light cycle, provided with food and water ad libitum, and genotyped as described previously [25]. E14.5 (plug day, E0.5), P0, and adult Nrp1+/+, Nrp1sema/+ and Nrp1sema/sema mice were obtained and processed for immunohistofluorescence analyses as previously described [30]. In addition, homozygous Nrp1loxP/loxP mice (B6.129(SJL)-Nrp1tm2Ddg/J) [25] from the Jackson laboratory were crossed with a transgenic mouse line expressing the cre recombinase under the control of the GnRH gene promoter (GnRH::cre mice) [32], a gift from C. Dulac (Harvard university, Cambridge, USA), to obtain GnRH::cre; Nrp1loxP/loxP mice that lack Nrp1 in GnRH cells only. Nrp1loxP/loxP and GnRH::cre; Nrp1loxP/loxP mice were used for immunohistofluorescence analyses at E14.5 and adult stages.
The human fetus was obtained from a voluntary terminated pregnancy, with parent's written informed consent. Gestational age was established by crown-rump length measurement. The fetus was fixed in 4% paraformaldehyde in 0.1 M phosphate buffered saline (PBS), pH 7.4, for three weeks at 4°C, and then immersed in 0.1 M PBS containing 30% sucrose for two days at 4°C. The head was embedded in OCT embedding medium (Tissue-Tek), frozen, and sagittal cryosections (20 µm thick) were cut and processed for immunohistofluorescence.
Immunohistofluorescence
Immunohistofluorescence experiments were carried out as described previously [30]. Primary antibodies were: rabbit anti-GnRH (dilution 1∶3000), a gift from G. Tramu (University of Bordeaux, France); rabbit anti-peripherin (dilution 1∶1000), AB1530 (Millipore); goat anti-neuropilin1 (dilution 1∶400), AF566 (R & D systems); goat anti-olfactory marker protein (dilution 1∶6000), a gift from F. L. Margolis (University of Maryland, Baltimore, USA).
DiI labeling of nerve fibers
Vomeronasal nerve fibers were traced anterogradely with the lipophilic fluorescent dye DiI (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate, Molecular Probes) as previously described [17]. After diffusion of the tracer, serial sagittal sections (100 µm thick) were cut through the forebrain, and analyzed using a LSM 710 confocal microscope (Zeiss) and the ImageJ analysis software (NIH, Bethesda, USA).
Cell cultures
COS-7 cells and GN11 cells were grown in monolayers in 5% CO2 at 37°C, in Dulbecco's modified Eagle's medium (Life Technologies, Inc.) containing 1 mM sodium pyruvate, 2 mM glutamine, 50 mM glucose, and supplemented with 10% fetal bovine serum (Invitrogen), 100 µg/ml streptomycin and 100 U/ml penicillin.
Signaling activity of wild-type and mutant Sema3A in GN11 cells
A cDNA containing the entire coding region of the human SEMA3A (GenBank NM_006080) was inserted into a pRK5 plasmid expression vector. Recombinant plasmids containing SEMA3A cDNAs harboring each of the eight mutations identified in the KS patients were then engineered using the QuickChange mutagenesis protocol (Stratagene). COS-7 cells were transiently transfected using a fast-forward protocol (Lipofectamine 2000, Invitrogen) [30]. Conditioned medium was collected 48 h after transfection, tested for the presence of Sema3A by western blot analysis using an anti-Sema3A antibody (Santa Cruz, sc-10720, dilution 1∶100), and then processed for signaling activity experiments in the GN11 cell line. Briefly, subconfluent GN11 cells were grown overnight in serum-free medium, and then stimulated for 20 min with human recombinant Sema3A (R&D systems) at 100 µg/L, or with the concentrated conditioned media from transfected COS-7 cells. Western blot experiments [30] were carried out on cell lysates using antibodies to P-ERK (#9101L) and ERK (#9102L) from Cell Signaling (dilution 1∶1000), or P-FAK (sc56901) and FAK (sc81493) from Santa Cruz (dilution 1∶500).
DNA sequencing
Informed consent was obtained from all individuals analyzed. Genomic DNAs were prepared from white blood cells using a standard procedure. Each of the SEMA3A and NRP1 coding exons and flanking splice sites was PCR-amplified from genomic DNA using a specific primer pair (see Tables S1 and S2 for primer sequences), and sequenced using either PCR oligonucleotide as sequencing primer. The mutations were confirmed by sequencing two independent PCR products on both DNA strands. Exons 2, 5, 11, 14, and 17 of SEMA3A, which harbor the mutations identified in some patients, were analyzed by denaturing high performance liquid chromatography (DHPLC) scanning on an automated HPLC instrument (Wave technology) in 386 unrelated Caucasian controls, followed by Sanger sequencing of the exon in case of abnormal DHPLC profile.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. DodéC, HardelinJ-P (2009) Kallmann syndrome. Eur J Hum Genet 17 : 139–146.
2. SykiotisGP, PlummerL, HughesVA, AuM, DurraniS, et al. (2010) Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci U S A 107 : 15140–15144.
3. Schwanzel-FukudaM, PfaffDW (1989) Origin of luteinizing hormone-releasing hormone neurons. Nature 338 : 161–164.
4. TeixeiraL, GuimiotF, DodéC, Fallet-BiancoC, MillarRP, et al. (2010) Defective migration of neuroendocrine GnRH cells in human arrhinencephalic conditions. J Clin Invest 120 : 3668–3672.
5. FrancoB, GuioliS, PragliolaA, IncertiB, BardoniB, et al. (1991) A gene deleted in Kallmann's syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature 353 : 529–536.
6. HardelinJ-P, LevilliersJ, BlanchardS, CarelJ-C, LeuteneggerM, et al. (1993) Heterogeneity in the mutations responsible for X chromosome-linked Kallmann syndrome. Hum Mol Genet 2 : 373–377.
7. LegouisR, HardelinJ-P, LevilliersJ, ClaverieJ-M, CompainS, et al. (1991) The candidate gene for the X-linked Kallmann syndrome encodes a protein related to adhesion molecules. Cell 67 : 423–435.
8. DodéC, LevilliersJ, DupontJ-M, De PaepeA, Le DuN, et al. (2003) Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet 33 : 463–465.
9. FalardeauJ, ChungWC, BeenkenA, RaivioT, PlummerL, et al. (2008) Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J Clin Invest 118 : 2822–2831.
10. DodéC, TeixeiraL, LevilliersJ, FouveautC, BouchardP, et al. (2006) Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet 2: e175 doi:10.1371/journal.pgen.0020175.
11. KimHG, AhnJW, KurthI, UllmannR, KimHT, et al. (2010) WDR11, a WD protein that interacts with transcription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet 87 : 465–479.
12. TornbergJ, SykiotisGP, KeefeK, PlummerL, HoangX, et al. (2011) Heparan sulfate 6-O-sulfotransferase 1, a gene involved in extracellular sugar modifications, is mutated in patients with idiopathic hypogonadotrophic hypogonadism. Proc Natl Acad Sci U S A 108 : 11524–11529.
13. JongmansMC, van Ravenswaaij-ArtsCM, PitteloudN, OgataT, SatoN, et al. (2009) CHD7 mutations in patients initially diagnosed with Kallmann syndrome: the clinical overlap with CHARGE syndrome. Clin Genet 75 : 65–71.
14. KimHG, KurthI, LanF, MelicianiI, WenzelW, et al. (2008) Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet 83 : 511–519.
15. WiermanME, Kiseljak-VassiliadesK, TobetS (2011) Gonadotropin-releasing hormone (GnRH) neuron migration: Initiation, maintenance and cessation as critical steps to ensure normal reproductive function. Front Neuroendocrinol 32 : 43–52.
16. WrayS (2010) From nose to brain: development of gonadotropin-releasing hormone-1 neurones. J Neuroendocrinol 22 : 743–753.
17. YoshidaK, TobetSA, CrandallJE, JimenezTP, SchwartingGA (1995) The migration of luteinizing hormone-releasing hormone neurons in the developing rat is associated with a transient, caudal projection of the vomeronasal nerve. J Neurosci 15 : 7769–7777.
18. YazdaniU, TermanJR (2006) The semaphorins. Genome Biol 7 : 211.
19. ImaiT, YamazakiT, KobayakawaR, KobayakawaK, AbeT, et al. (2009) Pre-target axon sorting establishes the neural map topography. Science 325 : 585–590.
20. PasterkampRJ, De WinterF, HoltmaatAJ, VerhaagenJ (1998) Evidence for a role of the chemorepellent semaphorin III and its receptor neuropilin-1 in the regeneration of primary olfactory axons. J Neurosci 18 : 9962–9976.
21. SchwartingGA, KostekC, AhmadN, DibbleC, PaysL, et al. (2000) Semaphorin 3A is required for guidance of olfactory axons in mice. J Neurosci 20 : 7691–7697.
22. CariboniA, DavidsonK, RakicS, MaggiR, ParnavelasJG, et al. (2011) Defective gonadotropin-releasing hormone neuron migration in mice lacking SEMA3A signalling through NRP1 and NRP2: implications for the aetiology of hypogonadotropic hypogonadism. Hum Mol Genet 20 : 336–344.
23. CariboniA, DavidsonK, DozioE, MemiF, SchwarzQ, et al. (2011) VEGF signalling controls GnRH neuron survival via NRP1 independently of KDR and blood vessels. Development 138 : 3723–3733.
24. KitsukawaT, ShimizuM, SanboM, HirataT, TaniguchiM, et al. (1997) Neuropilin-semaphorin III/D-mediated chemorepulsive signals play a crucial role in peripheral nerve projection in mice. Neuron 19 : 995–1005.
25. GuC, RodriguezER, ReimertDV, ShuT, FritzschB, et al. (2003) Neuropilin-1 conveys semaphorin and VEGF signaling during neural and cardiovascular development. Dev Cell 5 : 45–57.
26. RisserJM, SlotnickBM (1987) Nipple attachment and survival in neonatal olfactory bulbectomized rats. Physiol Behav 40 : 545–549.
27. SchwartingGA, KostekC, BlessEP, AhmadN, TobetSA (2001) Deleted in colorectal cancer (DCC) regulates the migration of luteinizing hormone-releasing hormone neurons to the basal forebrain. J Neurosci 21 : 911–919.
28. AdamsRH, LohrumM, KlostermannA, BetzH, PuschelAW (1997) The chemorepulsive activity of secreted semaphorins is regulated by furin-dependent proteolytic processing. EMBO J 16 : 6077–6086.
29. CariboniA, HickokJ, RakicS, AndrewsW, MaggiR, et al. (2007) Neuropilins and their ligands are important in the migration of gonadotropin-releasing hormone neurons. J Neurosci 27 : 2387–2395.
30. GiacobiniP, MessinaA, MorelloF, FerrarisN, CorsoS, et al. (2008) Semaphorin 4D regulates gonadotropin hormone-releasing hormone-1 neuronal migration through plexinB1-Met complex. J Cell Biol 183 : 555–566.
31. ZhenS, DunnIC, WrayS, LiuY, ChappellPE, et al. (1997) An alternative gonadotropin-releasing hormone (GnRH) RNA splicing product found in cultured GnRH neurons and mouse hypothalamus. J Biol Chem 272 : 12620–12625.
32. YoonH, EnquistLW, DulacC (2005) Olfactory inputs to hypothalamic neurons controlling reproduction and fertility. Cell 123 : 669–682.
33. GigerRJ, WolferDP, De WitGM, VerhaagenJ (1996) Anatomy of rat semaphorin III/collapsin-1 mRNA expression and relationship to developing nerve tracts during neuroembryogenesis. J Comp Neurol 375 : 378–392.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 8
Nejčtenější v tomto čísle
- Dissecting the Gene Network of Dietary Restriction to Identify Evolutionarily Conserved Pathways and New Functional Genes
- It's All in the Timing: Too Much E2F Is a Bad Thing
- A Quantitative Comparison of the Similarity between Genes and Geography in Worldwide Human Populations
- Variation of Contributes to Dog Breed Skull Diversity