
The Wilms Tumor Gene, , Is Critical for Mouse Spermatogenesis via Regulation of Sertoli Cell Polarity and Is Associated with Non-Obstructive Azoospermia in Humans
Azoospermia is one of the major reproductive disorders which cause male infertility in humans; however, the etiology of this disease is largely unknown. In the present study, six missense mutations of WT1 gene were detected in 529 human patients with non-obstructive azoospermia (NOA), indicating a strong association between WT1 mutation and NOA. The Wilms tumor gene, Wt1, is specifically expressed in Sertoli cells (SCs) which support spermatogenesis. To examine the functions of this gene in spermatogenesis, Wt1 was deleted in adult testis using Wt1flox and Cre-ERTM mice strains. We found that inactivation of Wt1 resulted in massive germ cell death and only SCs were present in most of the seminiferous tubules which was very similar to NOA in humans. In investigating the potential mechanism for this, histological studies revealed that the blood–testis barrier (BTB) was disrupted in Wt1 deficient testes. In vitro studies demonstrated that Wt1 was essential for cell polarity maintenance in SCs. Further studies found that the expression of cell polarity associated genes (Par6b and E-cadherin) and Wnt signaling genes (Wnt4, Wnt11) were downregulated in Wt1 deficient SCs, and that the expression of Par6b and E-cadherin was regulated by Wnt4. Our findings suggest that Wt1 is important in spermatogenesis by regulating the polarity of SCs via Wnt signaling pathway and that WT1 mutation is one of the genetic causes of NOA in humans.
Published in the journal:
. PLoS Genet 9(8): e32767. doi:10.1371/journal.pgen.1003645
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003645
Summary
Azoospermia is one of the major reproductive disorders which cause male infertility in humans; however, the etiology of this disease is largely unknown. In the present study, six missense mutations of WT1 gene were detected in 529 human patients with non-obstructive azoospermia (NOA), indicating a strong association between WT1 mutation and NOA. The Wilms tumor gene, Wt1, is specifically expressed in Sertoli cells (SCs) which support spermatogenesis. To examine the functions of this gene in spermatogenesis, Wt1 was deleted in adult testis using Wt1flox and Cre-ERTM mice strains. We found that inactivation of Wt1 resulted in massive germ cell death and only SCs were present in most of the seminiferous tubules which was very similar to NOA in humans. In investigating the potential mechanism for this, histological studies revealed that the blood–testis barrier (BTB) was disrupted in Wt1 deficient testes. In vitro studies demonstrated that Wt1 was essential for cell polarity maintenance in SCs. Further studies found that the expression of cell polarity associated genes (Par6b and E-cadherin) and Wnt signaling genes (Wnt4, Wnt11) were downregulated in Wt1 deficient SCs, and that the expression of Par6b and E-cadherin was regulated by Wnt4. Our findings suggest that Wt1 is important in spermatogenesis by regulating the polarity of SCs via Wnt signaling pathway and that WT1 mutation is one of the genetic causes of NOA in humans.
Introduction
Infertility is a common health problem which affects about 15–20% of couples. Among infertile couples, about 50% are related to male infertility [1], a major cause of which is azoospermia. Genetic causes of azoospermia include autosomal chromosome abnormalities, Y chromosome microdeletions, and single gene mutations. Several genes have been reported to play a role in azoospermia, including PRM1, SPATA16, AURKC, and KLHL10 [2]–[5].
As support cells, Sertoli cell (SCs) play central roles in testis development and spermatogenesis. During mouse embryogenesis, SCs emerge at E10.5 and are involved in seminiferous cord formation and prevent germ-cell entry into meiosis [6]. Mullerian duct regression in males is induced by anti-Müllerian hormone (AMH) which is secreted by immature SCs [6], [7]. At puberty, SCs undergo terminal differentiation and develop complex morphological interactions with each other and with adjacent germ cells. Mammalian spermatogenesis is dependent on the proper functioning of SCs which provide structural support and nutrition to developing germ cells, phagocytose degenerating germ cells and residual bodies, release spermatids at spermiation, and produce a host of proteins that regulate or respond to pituitary hormone [8]–[10].
Our previous studies have demonstrated that Wt1, which encodes a nuclear transcription factor, is important for testes development with inactivation of Wt1 in SCs between E12.5–E14.5, resulting in testicular cord disruption and testes dysgenesis [11]. However, the functional significance of Wt1 in adult testis has been unclear, in part due to the gonadal agenesis of Wt1−/− [12] and Wt1R394W/R394W [13] mice. Rao et al reported that knock-down of Wt1 using siRNA in postnatal SCs caused reduced sperm count [14], suggesting that Wt1 plays a role in spermatogenesis. However, the exact function of Wt1 in spermatogenesis and underlying mechanism by which it plays a role are still largely unknown.
In this study, we demonstrated that inactivation of Wt1 in adult SCs resulted in massive germ cell death with only SCs surviving in the seminiferous tubules. Six WT1 missense mutations were detected in 529 NOA patients by mutational analysis, indicating a strong association between WT1 mutation and spermatogenic defects in human. We further demonstrated that Wt1 is critical for maintaining the polarity of SCs, likely via Wnt signaling pathways. Inactivation of Wt1 resulted in loss of polarity in SCs and abnormal tight junction assembly which in turn caused germ cell death.
Results
Inactivation of Wt1 in adult testis results in massive cell death in seminiferous tubules
Wt1−/flox; Cre-ERTM and control mice (Wt1−/flox, Wt1+/flox; Cre-ERTM) were obtained by intercrossing of Wt1flox/flox and Wt1+/−; Cre-ERTM mice. The growth of Wt1−/flox; Cre-ERTM mice were indistinguishable from that of control mice and the morphology and histology of Wt1−/flox; Cre-ERTM testes were completely normal (data not shown). To induce Cre activity, Wt1−/flox; Cre-ERTM and littermate control mice were injected with 9 mg/40 g (body weight) Tamoxifen for two consecutive days at 8 weeks of age. The testes were collected at 1, 2, and 3 weeks after Tamoxifen injection. The efficiency of Tamoxifen induced Cre recombination was examined by Wt1 Real-time PCR (Figure S2G) and western blot (Figure S3). Compared to control testis Wt1 mRNA was reduced about 50% in Wt1−/flox; Cre-ERTM testis, indicating that Wt1 was deleted in about 50% of SCs. Because the Cre activation results in the in-frame deletion of exons 8 and 9, the Wt1Δ allele results in a truncated protein. Our previous work indicated that this truncation has the same phenotypic effect as Wt1 deletion [11]. As shown in Figure S3, approximately the same amount of wild type and truncated Wt1 protein was observed in Wt1−/flox; Cre-ERTM testes 1 week after Tamoxifen induction, indicating that Wt1 function was lost in approximately 50% of Sertoli cells. This was consistent with real time PCR results. The size of testes from Wt1−/flox; Cre-ERTM mice was dramatically reduced 3 weeks after Tamoxifen treatment (Figure 1C). Histological analysis results revealed that the seminiferous tubules were grossly normal in Wt1−/flox; Cre-ERTM testis 1 week after Tamoxifen induction (Figure 1G), although vacuolization was first noted in a small number of tubules. Severe epithelial vacuolization (Figure 1H, arrows) was noted in mutant testis 2 weeks after Tamoxifen induction. At 3 weeks post-Tamoxifen, atrophic seminiferous tubules with massive cell loss were evident in mutant testes (Figure 1I), whereas the control testis was completely normal (Figure 1F). In addition to germ cell loss, inactivation of Wt1 in adult mice also resulted in multiple phenotypes, including accumulation of ascitic fluid (Figure S1A), atropic spleen (Figure S1B), abnormal pancreas (Fig. S1C), and renal failure (Figure S1D), consistent with a previous report [15]. In this study, however, the defect in spermatogenesis was not observed, likely because mice were treated with Tamoxifen for 5 consecutive days and, died earlier in our model system,

Wt1 is essential for germ cell survival
Apoptotic cells (Figure S4C, white arrows) were noted by TUNEL assay at the peripheral region of seminiferous tubules in Wt1−/flox; Cre-ERTM testis at 1 week after Tamoxifen induction, and the number of TUNEL positive cells (Figure S4D, white arrows) was dramatically increased at 2 weeks. In contrast, very few apoptotic cells were detected in control testis at both 1 (Figure S4A) and 2 weeks (Figure S4B). The results of statistical analysis showed that the number of apoptotic cells was significantly increased in Wt1−/flox; Cre-ERTM mice at both 1 and 2 weeks after Tamoxifen treatment compared to control testes (Figure S4E).
To identify the cells which survived in Wt1-deficient testes, immunohistochemistry analyses were carried out. Because the truncated WT1 protein encoded by the recombined Wt1Δ allele is recognized by the Wt1 antibody, Wt1 antibody could be employed to identify Sertoli cells. Wt1 positive Sertoli cells were localized at the periphery of the seminiferous tubules in both control (Figure 2A, arrows) and Wt1−/flox; Cre-ERTM (Figure 2B, arrows) testes. Multiple layers of GCNA1-positive germ cells were noted in seminiferous tubules of control testes (Figure 2C). However, very few GCNA1 positive germ cells were present in Wt1−/flox; Cre-ERTM testis (Figure 2D, arrows), and some tubules were complete devoid of germ cells (Figure 2D, asterisks). The cauda epididymes of control mice were filled with normal mature sperm (Figure 2E, arrows). In contrast, only cellular debris and round, prematurely released spermatocytes were noted in Wt1−/flox; Cre-ERTM mice (Figure 2F, arrow heads). These results indicate that Wt1 is essential for SCs function such that Wt1-deficient SCs cannot support the development of germ cells, eventually resulting in germ cell death.

WT1 mutation is associated with azoospermia in humans
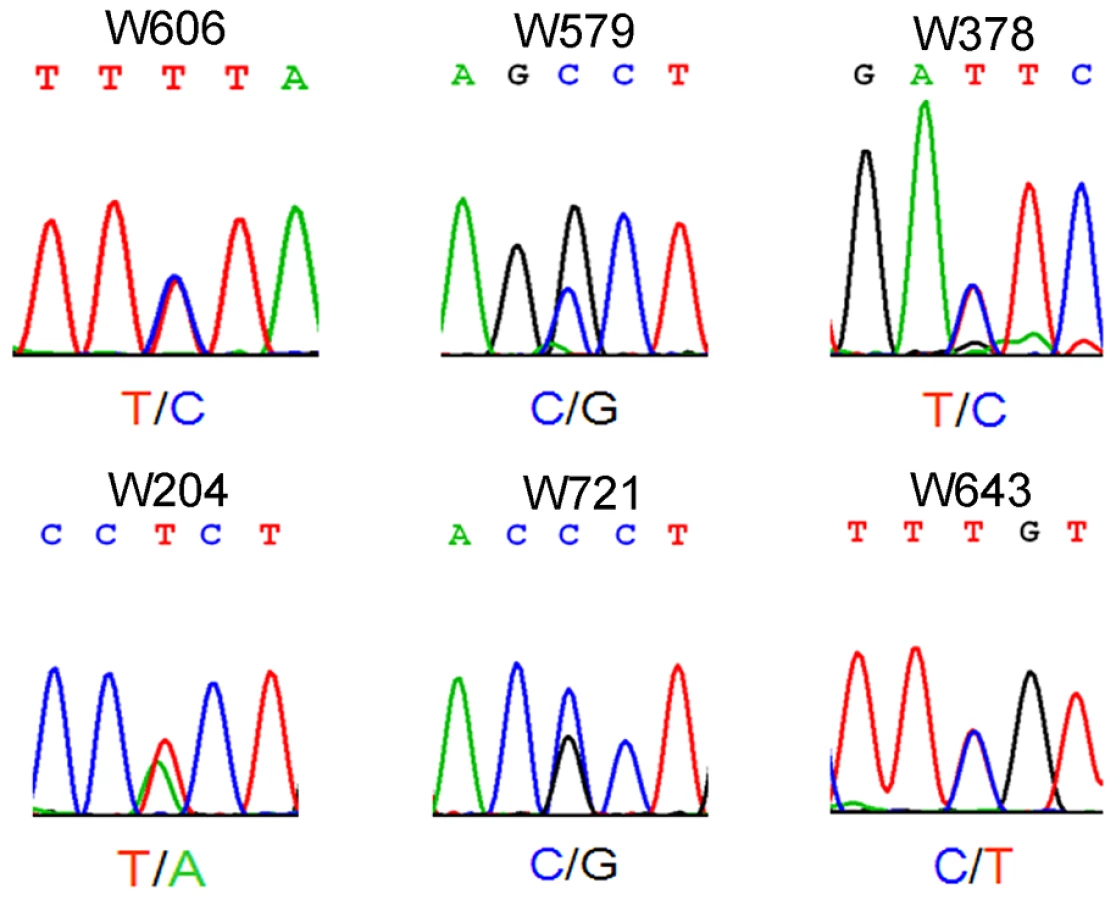
Azoospermia is one of the major causes of male infertility in human. The testes histology of human non-obstructive azoospermia (NOA) was very similar to Wt1-deficient testes. As shown in Figure S6, only Sertoli cells (black arrows) were presented in the seminiferous tubules (asterisks) of human NOA patients. To determine whether WT1 mutation is associated with spermatogenic defects in humans, following exonic capture we sequenced (mean depth of 43×) the exons of WT1 in 529 non-obstructive azoospermia (NOA) patients and 709 men with proven fertility. On average per sample, 96% of target bases were covered at least once, and 78% were covered sufficiently for variant calling (Table S1). The first coding exon was poorly captured due to its extremely high GC content. As shown in Figure 3 and Table S2, 6 WT1 missense mutations were detected in 6 patients while no mutations were found in the control group. By Chi-square analysis, there was a statistically significant (p = 0.004) difference in WT1 status between the NOA population and the control group. The WT1 variants present in the NOA patients have not been detected as SNPs in the 1000 Genomes Project, further indicating the significance of the observation. Two mutations were in the two zinc finger domains (encoded by exons 8 and 9) which are most important for DNA-protein interaction [16]. The other four mutations were localized to the transcription regulatory domain (Exons 3, 4, 6). The functional significance of the NOA-associated mutations was predicted using a combination of several approaches as previously described [17]. All 6 mutations were predicted to be deleterious by several complementary nsSNV scoring algorithms (Table S2), including SIFT [18], PolyPhen2 [19], PhastCons [20] and GERP scores [21] (Table S2). The observation of these predicted deleterious mutations specifically in the NOA population strongly indicates that WT1 is also important for spermatogenesis in human and that WT1 mutation plays an etiologic role in azoospermia.
The integrity of the blood-testis barrier (BTB) is damaged in Wt1-deficient testes
One of the major functions of SCs is to maintain the integrity of the BTB, which, when disrupted, results in germ cell death and spermatogenic defects [22], [23]. To test whether the integrity of the BTB was damaged in Wt1-deficient testes, surface biotinylated reagent was injected into the testicular interstitium of both control and Wt1−/flox; Cre-ERTM testes at 1 week after Tamoxifen treatment and before obvious histological changes were observed. In control mice, biotin tracer was restricted to the testicular interstitium and the basal compartment of the seminiferous tubules with no tracer being observed in the tubular lumen (Figure 4A, Figure S8A and C). However in Tamoxifen treated Wt1−/flox; Cre-ERTM testes, biotin tracer was also present along the SCs plasma membrane from the basement membrane to the lumen in about 30% of seminiferous tubules (Figure 4B, Figure S8B and D, asterisks). This difference was significant (Figure S8E). These results indicated that the integrity of BTB in Wt1−/flox; Cre-ERTM testes was disrupted after Wt1 inactivation such that it was permeable to the biotin tracer.

The BTB structure in Wt1-deficient testes was further examined by transmission electron microscope (TEM). As shown in Figure 4E, a normal BTB (bracket) structure with F-actin bundles (black arrows) was observed in control testes. In contrast, in Wt1-deficient testes, the BTB was aberrant with intermittent loss of F-actin bundles (Figure 4F, black arrows) and the occurrence of vacuoles, likely formed as a result of loss of interaction between SCs (Figure 4F, asterisks and black arrowheads). We also found that the structure of apical ES (Ectoplasmic Specialization) was damaged in Wt1−/flox; Cre-ERTM testes at 1 week after Tamoxifen treatment. In control testes, the apical ES between Sertoli cell and elongated sperm was well organized with F-actin bundles (Figure 4C, arrows), whereas the apical ES structure was disrupted and there was a loss of F-actin bundles in Wt1-deficient testes (Figure 4D, arrows). The expression of BTB components was examined by immunofluorescence at 1 week after Tamoxifen treatment. As shown in Figure S7, tight junction protein Claudin11 (A, B), adhesion junction proteins N-cadherin (C, D) and β-catenin (E, F), and gap junction protein CX43 (G, H) were detected at the peripheral region of seminiferous tubules where tight junctions are formed in control testes (A, C, E, G). In contrast, the expression of these genes was significantly reduced in Wt1−/flox; Cre-ERTM testes (B, D, F, H), indicating the BTB structure was disrupted in Wt1-deficient testes at 1 week after Tamoxifen induction.
Loss of epithelial cell morphology and tight junction formation in Wt1-deficient SCs
To explore the physiological function of Wt1 in spermatogenesis, control and Wt1−/flox; Cre-ERTM SCs were isolated from 2 months old mice and cultured in vitro. Wt1 was deleted by addition of 4-OH-tamoxifen to the culture medium. As shown in Figure 5A, the control SCs had a cuboidal epithelial morphology as visualized by F-actin staining (Figure 5A). In contrast, SCs assumed a mesenchymal-like morphology when Wt1 was inactivated by Tamoxifen induction (Figure 5C). The quantitative results showed that the number of mesenchymal-like cells was dramatically increased after Wt1 inactivation, and this difference was significant (Figure 5E). Tight junctions were well established in control SCs after a few days culture indicating by ZO-1 staining (Figure 5B, white arrows). However, in Wt1-deficient SCs, ZO-1 protein was diffused in the cytosol and no obvious staining was noted at the cell junctions (Figure 5D). The tight junction formation by SCs was further examined by Paracellular FITC-dextran Flux assay. As shown in Figure 5F, the permeability of Wt1-deficient SCs was significantly increased compared to control cells. This result further confirmed that the tight junction formation of SCs is disrupted when Wt1 is inactivated, consistent with the Zo-1 staining data.

The expression of SCs specific genes is not changed in Tamoxifen treated Wt1−/flox; Cre-ERTM testes and Wt1-deficient SCs
Our previous study found that deletion of Wt1 in SCs during early stage of embryonic development resulted in downregulation of Sox9 and AMH [11]. To test whether deletion of Wt1 in adult testes also affects the expression of SC specific genes, immunohistochemistry and real time PCR were conducted. The results of immunohistochemistry showed that the expression of Sox9 (Figure S2A, B), AR (Figure S2C, D), and Gata4 (Figure S2E, F) was not changed in Wt1−/flox; Cre-ERTM testes (Figure S2B, D, F) 3 weeks after Tamoxifen treatment. Real time PCR further confirmed these results (Figure S2G). The expression of SC-specific genes was also examined in Wt1-deficient SCs by immunofluorescence and real time PCR. As shown in Figure S9, inactivation of Wt1 in cultured primary SCs did not affect the expression of SC specific genes, such as Sox9, Dmrt1, Gata4, AR, Gata1, and Nr5a1.
Molecular characterization of spermatogenic defect in Wt1-deficient testis
To explore the molecular mechanism of the spermatogenic defect in Wt1-deficient testes, RNA-Seq analysis were performed using mRNA from control and Wt1-deficient SCs. A total of 710 differentially (p-value<0.05) expressed genes were identified (456 upregulated and 254 downregulated genes), the raw data has been uploaded to http://www.ncbi.nlm.nih.gov/geo/, the accession number is GSE46664. As listed in Table S3, the genes were differentially expressed in multiple pathways based on the pathway term analysis. However, no specific pathway was significantly altered in Wt1-deficient SCs. In the present study, we found that the morphology of SCs was transformed from epithelium into mesenchyme with a concurrent loss tight junction formation after Wt1 inactivation. It has been reported previously that Wt1 is involved in EMT process during kidney and heart development [24], [25]. Therefore, EMT and cell polarity related genes were selected for further analysis. RNA-Seq analysis revealed that cell polarity-associated genes, such as E-cadherin, Par6b, were significantly decreased in Wt1-deficient SCs. In contrast, the expression of the EMT related genes was not significantly changed. We also found that Wnt signaling gene, Wnt11 was downregulated in Wt1-deficient SCs. The expression of Wnt4 was also decreased in Wt1-deficient SCs, but this was not statistically significant.
To further verify the RNA-Seq results, the expression of genes related to EMT and cell polarity was analyzed by real time PCR. As shown in Figure 6A, the expression of Par6b, E-cadherin, Wnt11, and Wnt4 was significantly decreased in Wt1-deficient SCs. Western blot analysis also showed that the expression of E-cadherin and Par6b was dramatically reduced in Wt1-deficient SCs, however, ZO-1 and N-cadherin expression was not changed (Figure 6B). In contrast, Snail1, Snail2, Twist, Zeb, Sip, Mmp2 were not differentially expressed between control and Wt1-deficient SCs (Figure S10). The expression of cell polarity related genes in Wt1-deficient testes was also examined by immunofluorescence and real time PCR. The results of immunofluorescence (Figure S5A–D) showed that the localization of E-cadherin and Par6b was disorganized in Wt1−/flox; Cre-ERTM testes at 1 week after Tamoxifen induction. Real time PCR results showed that the mRNA level of Par6b and Wnt4 was significantly reduced in Wt1−/flox; Cre-ERTM testes at 1 week after Tamoxifen treatment. mRNA level of E-cadherin and Wnt11 was also reduced, but this was not statistically significant (Figure S5E). These results were consistent with the results from the in vitro study. Notably, when Tamoxifen treated Wt1−/flox; Cre-ERTM SCs were transfected with Wt1 expressing adenovirus, the mRNA level of Par6b, E-cadherin, Wnt4, and Wnt11 was completely rescued, indicating that the expression of these genes was regulated by Wt1 directly or indirectly (Figure 6C).

The expression of E-cadherin and Par6b is regulated by Wnt4 in SCs
To examine whether Wt1's role in regulating SCs polarity is mediated by Wnt signaling, Wnt4 was knocked down in cultured SCs using siRNA. As shown in Figure 6F, compared to treatment with scrambled siRNA, the mRNA level of Wnt4 was reduced about 70% after transfection with Wnt4-specific siRNA, and the expression of E-cadherin and Par6b was also reduced about 30–50% respectively (Figure 6F), suggesting that the expression of E-cadherin and Par6b is regulated by Wnt4 in SCs. To further confirm these results, Tamoxifen treated Wt1−/flox; Cre-ERTM SCs were transfected with Wnt4 - and Wnt11 - expressing adenovirus. We found that the expression of Par6b was dramatically increased upon over-expression of both Wnt4 and Wnt11 in Wt1-deficient SCs (Figure 6D). However, the expression of E-cadherin was not rescued (Figure 6E), suggesting that the expression of E-cadherin was also directly regulated by Wt1.
The mutations detected in NOA patient results in WT1 protein loss of function
Although the WT1 mutations detected in NOA patients were predicted to be deleterious by several complementary nsSNV scoring algorithms (Table S2), the effect of mutations on Wt1 function was further assessed by in vitro functional analyses. Two Wt1 expressing adenovirus carrying the zinc finger domain mutations detected in patient W643 and W606 were generated by site-directed mutagenesis and designated as Wt1R362Q and Wt1K386R. We found that both of these mutations did not affect the nuclear localization of Wt1 protein (Figure S11). However, functional analysis showed that both these mutated Wt1 could not induced the expression of E-cadherin, Par6b, Wnt4, and Wnt11 in Wt1-deficient SCs (Figure 7A), indicating that these mutations caused Wt1 protein loss of function. To examine whether these mutation affect the ability of Wt1 protein binding to the promoter of a Wt1 targeting gene, a ChIP assay was performed using HepG2 cells. As shown in Figure 7B, C, a 211 bp DNA fragment in the Wnt4 promoter region was pulled down by Wt1, but not Wt1R362Q and Wt1K386R. These results indicated that these mutations affected the interaction between Wt1 protein and DNA.

Discussion
Wt1 is specifically expressed in SCs of the testes and our previous study has demonstrated that it is critical for testicular development [11], [12]. However, the functional significance of Wt1 in adult testes is largely unknown. In the present study, we have demonstrated for the first time in vivo, using a conditional Wt1 knockout mice strain, that Wt1 plays an important role in maintaining SC polarity and that inactivation of Wt1 leads to the loss of epithelial characteristics of SCs which in turn results in germ cell death.
In adult mammalian testes, SCs are polarized epithelial cells which, as nurse cells, are essential for germ cell division and differentiation [26], providing structural support and creating an immunological barrier from the systemic circulation [26], [27]. The latter function is conferred by the blood-testes barrier (BTB), which is formed by tight junctions (TJs), basal ectoplasmic specialization (ES), and desmosome-like junctions between SCs [23], [28]. The integrity of the BTB structure is very important for spermatogenesis, and abnormal assembly of BTB is known to cause germ cell loss and male infertility [29].
In this study, we have demonstrated, both by biotin tracer injection experiments (Figure 4B) and TEM (Figure 4F), that loss of Wt1 disrupted the normal BTB structure. While Wt1 is a transcriptional regulator, the expression of genes known to be important in spermatogenesis, AR [30], Dmrt1 [31], Gata4 [32], Connexin43 [33], Claudin11 [34], was not changed in Wt1-deficient testes, suggesting that the spermatogenic defect of Wt1-deficient mice was mechanistically different.
Analysis of cultured SCs revealed that upon Wt1 deletion, cells underwent a morphologic transformation from cuboidal epithelium into mesenchyme-like cells with a concomitant loss of tight junctions and a significant reduction in E-cadherin expression, both of which are normal features of epithelial cells. These results indicate that Wt1 is critical for the maintenance of epithelial characteristics of SCs and that the loss of these characteristics results in germ cell death.
A recent study has shown that Wt1 is essential for epithelial to mesenchymal transition (EMT) in epicardial cells by controlling the expression of Snai1 [25]. However, we found that the expression of the EMT-related genes (such as Snail, Twist, Zeb, Sip, et al) was not changed in Wt1-deficient SCs. Instead, genes important for cell polarity maintenance (such as Par6b, Cdc42ep5) and Wnt pathway signaling genes (Wnt4 and Wnt11) were significantly decreased in SCs when Wt1 gene was deleted. These results suggest that the function of Wt1 is different in different cell types which is probably dependent on the interaction with different cofactors.
PAR6b directly binds to PAR3, aPKC, and CDC42 [35]. This complex is involved in tight junction formation in epithelial cells [35], and inhibiting the expression of Par3, Par6, or aPKC blocks tight junction assembly in MDCK cells [35], [36]. The PAR3/PAR6b/aPKC complex is also involved in regulating apical ectoplasmic specialization (ES) and blood testes barrier (BTB) restructuring in the testis. Knockdown of Par6b or Par3 results in defective tight junction formation in cultured SCs [37]. These data suggest that the spermatogenesis defect we observed following Wt1 ablation is due to the loss of SC polarity.
Non-canonical Wnt signaling is known to play an important role in regulating cell polarity and motility [38]. In vertebrates, Wnt4, Wnt5a and Wnt11 encode ligands that activate the Wnt signaling pathway [39]–[41] that is critical for regulating epithelial versus mesenchymal cell characteristics. Wnt4 is necessary and sufficient for MET during kidney development [42], [43], and Wt1 directly activates Wnt4 expression in developing kidney. Interestingly, by recruiting different cofactors, Wt1 represses Wnt4 expression and induces an epithelial to mesenchymal transition in epicardium [24]. In this study, we found that the expression of Wnt4 and Wnt11 was activated by Wt1 in SCs and that deletion of Wt1 resulted in downregulation of these genes and loss of epithelial cell polarity. Wnt4 RNAi knockdown experiments showed that the expression of Par6b and E-cadherin was regulated by Wnt4 in SCs. On the other hand, overexpression of Wnt4 and Wnt11 in Wt1-deficient SCs induced Par6b expression. These data suggest that Wt1 controls SCs polarity indirectly through Wnt4. However, studies in 3T3 cells suggest that Wt1 can directly regulates E-cadherin expression [44], and our data cannot exclude this possibility in SCs.
WT1 germline mutation is known to result in a predisposition to Wilms tumors, male sex differentiation disorder, and early-onset renal failure [45]–[51]. These WT1 mutations are truncating mutations or missense mutations that are predicted, based on structural studies, to alter the ability of zinc finger domains to bind to DNA. We have now identified 6 novel missense mutations in NOA patients. These data strongly suggest that WT1 is also important for spermatogenesis in humans and that WT1 gene mutations play an etiologic role in azoospermia. Four of these mutations occur in the regulatory domain of the protein. Two occur in exons encoding zinc finger domains known to be critical for DNA binding, and functional studies revealed that they affected the ability of Wt1 to bind to Wnt4 promoter and result in a failure to induce targeting gene expression. Of not, these Wt1 mutants did not act in a dominant negative manner.
Interestingly, neither aberrant sex determination nor renal failure was noted in the NOA patients carrying WT1 mutations. We speculate that the NOA-associated mutations represent a functionally new type of WT1 alteration which probably also affects the ability to bind other transcription factors and/or chromatin-remodeling factors critical for its regulatory role.
In summary, our data strongly support a model by which loss of Wt1 in SCs results in downregulation of non-canonical Wnt signaling genes (Wnt4 and Wnt11) and cell polarity genes (E-cadherin and Par6b). This altered expression subsequently leads to loss of the epithelial characteristic and BTB integrity in SCs and concomitant germ cell loss. Such disruption of the BTB is also known to result in germ cell loss and male infertility in humans, and our observation of WT1 mutations in NOA patients strongly suggests that altered WT1 function constitutes one genetic cause of azoospermia in humans. Wt1 mutational analysis will be potentially useful clinically for characterizing NOA patients.
Materials and Methods
Patient samples
Peripheral blood samples from 529 patients with non-obstructive azoospermia (NOA) and 709 men with proven fertility were collected from Peking University Shenzhen Hospital and the Center of Reproductive Medicine, Tongji Medical College, Huazhong University of Science and Technology. The inclusion criteria for NOA patients included: 1) no sperm detected in the pellets of semen samples on three different occasions, 2) no inflammation and injury of the reproductive system or pelvic cavity, 3) no endocrinological defect, and 4) no karyotypic abnormality nor Y chromosome microdeletion. Testicular biopsy and histological analysis were conducted for azoospermic men wherever possible. All the control men had fathered at least one child. This study was approved by the ethical committees of Peking University Shenzhen Hospital and Tongji Medical College, and all participants signed the consent form permitting the collection and use of their blood samples in the study.
Selective exon capture and massively parallel sequencing
Genomic DNA was isolated from the blood samples using QIAamp DNA Mini Kits (QIAGEN). The exons of WT1 and other 600 genes were selectively captured by NimbleGen custom arrays (Roche NimbleGen, Inc, USA) and sequenced following the standard Illumina-based resequencing procedures as described [52]. Reads were mapped to the UCSC human reference genome build hg19 by SOAPaligner [53]. Mutation analysis for all the exons of WT1 were performed with SOAPsnp [54]. Novel coding mutations in WT1 were defined as those variants that had not been annotated in dbSNP nor in the publicly available dataset from the 1000 Genome Project. To further refine those novel mutations that may be associated with NOA, all the genetic variants detected in the fertile men were also eliminated for subsequent analysis.
Validation of novel mutations by Sanger sequencing
To validate the novel mutations identified in the WT1 gene by the massively parallel sequencing, primers flanking the point mutations were designed with Primer3 software (primer sequences are given in Table S5). PCR was performed using the following conditions: 94°C for 7 minutes; 30 cycles of denaturation at 94°C for 30 seconds, annealing at 57–60°C for 45 seconds, extension at 72°C for 1 minute, and a final extension at 72°C for 7 minutes. PCR products were checked on 1.5% agarose gel. The amplification product was directly sequenced using the 3730 DNA analyzer (Applied Biosystems).
Mice
All animal work was carried out in accordance with institutional animal care and use committee (IACUC) regulations. All the mice were maintained in a C57BL/6;129/SvEv mixed background. Wt1+/flox [11] mice were mated with mice carrying the Wt1-null allele (Wt1+/−) [12] and Cre-ERTM [55] transgenic mice to produce Wt1−/flox; Cre-ERTM offspring. DNA isolated from tail biopsies was used for genotyping. Genotyping was performed by PCR as described previously [11], [56].
Tamoxifen injection
Tamoxifen (Sigma) was dissolved in corn oil at a final concentration of 20 mg/ml. Two month old control (Wt1+/flox; Cre-ERTM, Wt1−/flox, and Wt1+/flox) and Wt1−/flox; Cre - ERTM males were injected intraperitoneally with 9 mg/40 g body weight for 2 consecutive days.
Tissue collection and histological analysis
Testes were dissected from mutant and control mice immediately after euthanasia and fixed in 4% paraformaldehyde for up to 24 hr, stored in 70% ethanol, and embedded in paraffin. Five-micrometer-thick sections were cut and mounted on glass slides. After deparaffinization, slides were stained with H&E for histological analyses.
Immunohistochemistry analysis
IHC analysis of tissues from at least three mice for each genotype was performed using a Vectastain ABC (avidin–biotin–peroxidase) kit (Vector Laboratories, Burlingame, CA) as recommended and using antibodies to WT1 (Santa Cruz, sc-192) and GCNA1 (gift of Dr. George Enders). The IHC procedure was performed as described previously [13]. Stained slides were examined with a Leica DMR Epifluorescence Microscope, and images were captured by a Hamamatsu CCD camera.
RNA-Seq analysis
Total RNA was prepared from cultured SCs isolated from 2 month old control (Wt1+/flox; Cre-ERTM, Wt1−/flox, and Wt1+/flox) and Wt1−/flox; Cre-ERTM mice after Tamoxifen treatment using an RNeasy Minikit (Ambion, Austin, TX). The main reagents and instruments used for RNA library construction and deep sequencing were the Illumina Gene Expression Sample Prep Kit, Solexa Sequencing Chip (flowcell), Illumina Cluster Station and Illumina HiSeq 2000 System. Sequence tags were prepared using the Illumina Digital Gene Expression Tag Profiling Kit, according to the manufacturer's protocol. The RNA-Seq was performed as described by Zhang et al [57]. In brief, raw data was filtered to remove adaptor tags, low quality tags and tags with a single copy number. Clean tags were classified according to their copy number and the saturation of the library was analyzed. All clean tags were mapped to the reference sequences, filtered and the remainder of the clean tags was designated as unambiguous clean tags. The number of unambiguous clean tags for each gene was calculated and normalized to the number of transcripts per million clean tags (TPM). To identify DEGs between control and Wt1-deficient Sertoli cells, the number of raw clean tags in each library was normalized to the TPM to obtain the normalized gene expression level. DEGs was identified as previously described [58] using a false discovery rate (FDR)≤0.001 and a threshold absolute log2-fold change ≥1 for the sequence counts across the libraries.
Primary Sertoli cell isolation
A modified method was used to isolate primary Sertoli cells from the testes of 6-week-old mice [59]. Testes were decapsulated under the dissection microscope. The seminiferous tubules were pooled and washed with phosphate-buffered saline (PBS) three times. The tubules were incubated with 2 mg/ml collagenase I (Sigma) and 0.5 mg/ml DNase I (sigma) in DMEM for 30 minutes at 37°C on a shaker, then washed twice with DMEM and further digested with 2 mg/ml collagenase I, 0.5 mg/ml DNase I and 1 mg/ml hyaluronidase type III (Sigma) for 20–30 minutes at 37°C. The tubules were allowed to settle and were then washed twice with DMEM before being digested with 2 mg/ml collagenase I, 0.5 mg/ml DNase I, 2 mg/ml hyaluronidase, and 1 mg/ml trypsin for 40–60 minutes at 37°C. This final digestion step resulted in a cell suspension containing primarily Sertoli cells and type A spermatogonia. The dispersed cells were then washed twice with DMEM and placed into culture dishes in DMEM containing 10% fetal calf serum and incubated at 37°C and 5% CO2. Spermatogonia were unable to attach to the dish and were removed after the medium change on the next day. 4-OH-Tamoxifen (Sigma, H7904) was dissolved in ethanol to generate a 1 mM stock solution and further diluted to appropriate concentrations prior to use. Recombination was initiated by adding 4-OH-TM to cultured Sertoli cells at a final concentration of 1 µM. After 3 days culture, total RNA and protein were extracted as described below.
Nucleic acid isolation and quantitative reverse transcription-PCR
Total RNA was extracted from cultured Sertoli cells or testes using a Qiagen RNeasy kit in accordance with the manufacturer's instructions. To quantify gene expression, real-time SybrGreen assay was performed with the isolated RNA. Gene expression was quantified relative to the expression of the gene for Gapdh (glyceraldehyde-3-phosphate dehydrogenase). Primers used for the RT-PCR are listed in sTable 4.
Western blotting
Cells were lysed in radioimmune precipitation assay lysis buffer (RIPA) containing complete Mini protease-inhibitor cocktail tablets (Roche, Mannheim, Germany). The protein concentration in the supernatants was estimated using a Bradford assay (Bio-Rad Laboratories, Hercules, CA). The proteins were electrophoresed under reducing conditions in 12% SDS-PAGE gels and transferred to nitrocellulose membranes. Blots were incubated overnight at 4°C with primary antibody and followed by 1 h of incubation at room temperature with HRP-labeled secondary antibody. Specific signals were detected using the ECL Western blotting detection system.
Biotin tracer
The permeability of the BTB was assessed by using a biotin tracer ENREF-37 [22]. Two-month-old control and Wt1−/flox; Cre - ERTM animals were injected with 9 mg/40 g body weight of tamoxifen. A week later they were anesthetized with avertin, and 50 µl of 10 mg/ml EZ-Link Sulfo-NHS-LC-Biotin (Pierce Chemical Co.) freshly diluted in PBS containing 1 mM CaCl2 was injected into the interstitium of one testis and the other testis was injected with 50 µl of 1 mM CaCl2 in PBS as an internal control. The animals were euthanized 30 min later, and the testes were removed immediately and embedded with OCT. Cryosections were prepared for further staining.
Electron microscopy assay
Testes were collected from control and Wt1−/flox; Cre-ERTM males 1 week after Tamoxifen treatment and fixed overnight in 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4). They were then washed in phosphate buffer (two changes), postfixed with 1.0% osmium tetroxide, dehydrated in a graded series of ethanol, and embedded in EPON/Araldite resin. Thin sections were cut, mounted on 200-mesh grids, and stained with uranyl acetate and lead citrate.
Site-directed mutagenesis and generation of adenovirus
Mutant Wt1 cDNA was generated using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). Mouse Wt1 cDNA (-KTS) was cloned by our lab previously. The WT1 mutations detected in patients W643 and W606 caused amino acid changed from Arg to Gln and Lys to Arg respectively (Table S2). We generated the same mutations with mouse Wt1 cDNA using the following primers (Wt1R362Q: forward, 5′CTTCAAGGACTGCGAGCAAAGGTTTTCTCGCTCAG3′, reverse, 5′CTGAGCGAGAAAACCTGTTCTCGCAGTCCTTGAAG3′; Wt1K386R: forward, 5′ CATTCCAGTGTAGAACTTGTCAGCG3′, reverse, 5′ CGCTGACAAGTTCTACACTGGAATG3′). The adenovirus containing wild type Wt1, Wt1R362Q, and Wt1K386R cDNA were generated using the Gateway Expression System (Invitrogen). The candidate genes were amplified by PCR and inserted into the pEntr 3C vector (Invitrogen). The resulting plasmids were then generated by homologous L/R recombination. Viral constructs were transduced into a 293A cell line, and high titer (108 IU/ml) viral particles were obtained by 4 rounds of amplification. The titer of virus was determinated as previous described [60].
ChIP assay
Chromatin immunoprecipitation (ChIP) assays were performed according to the protocol provided by Upstate Biotechnology (Charlottesville, VA). In brief, HepG2 cells were transiently transfected with Wt1, Wt1 R362Q, and Wt1K386R expressing adenovirus. 72 hr after transfection, cells were crosslinked with 1% formaldehyde in medium at 37°C for 15 min. Cells were then washed in ice-cold phosphate-buffered saline (PBS) and resuspended in 200 µl of SDS lysis buffer containing protease inhibitor mixture. The suspension was sonicated on ice and pre-cleared with protein A-agarose beads blocked with sonicated salmon sperm DNA (Upstate Biotechnology) for 30 min at 4°C. The beads were removed, and the chromatin solution was immunoprecipitated with anti-Wt1 antibody at 4°C, followed by incubation with protein A-agarose beads for an additional 1 h at 4°C. Normal rabbit IgG was used as a negative control, anti-Histone antibody was used as a positive control. The immune complexes were eluted with 100 µl of elution buffer (1% SDS and 0.1 M NaHCO3), and formaldehyde cross-links were reversed by heating at 65°C for 6 h. Proteinase K was added to the reaction mixtures and incubated at 45°C for 1 h. DNA of the immunoprecipitates and control input DNA were purified and then analyzed by standard PCR using mouse Wnt4 promoter specific primers (Forward, 5′ ATAGCAAGCTCATGTGGTGTGCAG3′, reverse, 5′ ATATAGGCCGCCGCACTTATCAGA3′).
Statistical analysis
Experiments were repeated at least three times. The data were evaluated for statistical differences using student T-test. A p-value<0.05 was considered significant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. FerlinA, ArrediB, ForestaC (2006) Genetic causes of male infertility. Reprod Toxicol 22 : 133–141.
2. OlivaR (2006) Protamines and male infertility. Hum Reprod Update 12 : 417–435.
3. DamAH, KoscinskiI, KremerJA, MoutouC, JaegerAS, et al. (2007) Homozygous mutation in SPATA16 is associated with male infertility in human globozoospermia. Am J Hum Genet 81 : 813–820.
4. DieterichK, Soto RifoR, FaureAK, HennebicqS, Ben AmarB, et al. (2007) Homozygous mutation of AURKC yields large-headed polyploid spermatozoa and causes male infertility. Nat Genet 39 : 661–665.
5. YatsenkoAN, RoyA, ChenR, MaL, MurthyLJ, et al. (2006) Non-invasive genetic diagnosis of male infertility using spermatozoal RNA: KLHL10 mutations in oligozoospermic patients impair homodimerization. Hum Mol Genet 15 : 3411–3419.
6. MackayS (2000) Gonadal development in mammals at the cellular and molecular levels. Int Rev Cytol 200 : 47–99.
7. JossoN, di ClementeN, GouedardL (2001) Anti-Mullerian hormone and its receptors. Mol Cell Endocrinol 179 : 25–32.
8. DymM, RajHG (1977) Response of adult rat Sertoli cells and Leydig cells to depletion of luteinizing hormone and testosterone. Biol Reprod 17 : 676–696.
9. FeigLA, BellveAR, EricksonNH, KlagsbrunM (1980) Sertoli cells contain a mitogenic polypeptide. Proc Natl Acad Sci U S A 77 : 4774–4778.
10. JohnsonL, VarnerDD, ThompsonDLJr (1991) Effect of age and season on the establishment of spermatogenesis in the horse. J Reprod Fertil Suppl 44 : 87–97.
11. GaoF, MaitiS, AlamN, ZhangZ, DengJM, et al. (2006) The Wilms tumor gene, Wt1, is required for Sox9 expression and maintenance of tubular architecture in the developing testis. Proc Natl Acad Sci U S A 103 : 11987–11992.
12. KreidbergJA, SariolaH, LoringJM, MaedaM, PelletierJ, et al. (1993) WT-1 is required for early kidney development. Cell 74 : 679–691.
13. GaoF, MaitiS, SunG, OrdonezNG, UdthaM, et al. (2004) The Wt1+/R394W mouse displays glomerulosclerosis and early-onset renal failure characteristic of human Denys-Drash syndrome. Mol Cell Biol 24 : 9899–9910.
14. RaoMK, PhamJ, ImamJS, MacLeanJA, MuraliD, et al. (2006) Tissue-specific RNAi reveals that WT1 expression in nurse cells controls germ cell survival and spermatogenesis. Genes Dev 20 : 147–152.
15. ChauYY, BrownsteinD, MjosengH, LeeWC, Buza-VidasN, et al. (2011) Acute multiple organ failure in adult mice deleted for the developmental regulator Wt1. PLoS Genet 7: e1002404.
16. StollR, LeeBM, DeblerEW, LaityJH, WilsonIA, et al. (2007) Structure of the Wilms tumor suppressor protein zinc finger domain bound to DNA. J Mol Biol 372 : 1227–1245.
17. LyonGJ, JiangT, Van WijkR, WangW, BodilyPM, et al. (2011) Exome sequencing and unrelated findings in the context of complex disease research: ethical and clinical implications. Discov Med 12 : 41–55.
18. NgPC, HenikoffS (2003) SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31 : 3812–3814.
19. AdzhubeiIA, SchmidtS, PeshkinL, RamenskyVE, GerasimovaA, et al. (2010) A method and server for predicting damaging missense mutations. Nat Methods 7 : 248–249.
20. BlanchetteM, KentWJ, RiemerC, ElnitskiL, SmitAF, et al. (2004) Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res 14 : 708–715.
21. CooperGM, GoodeDL, NgSB, SidowA, BamshadMJ, et al. (2010) Single-nucleotide evolutionary constraint scores highlight disease-causing mutations. Nat Methods 7 : 250–251.
22. MengJ, HoldcraftRW, ShimaJE, GriswoldMD, BraunRE (2005) Androgens regulate the permeability of the blood-testis barrier. Proc Natl Acad Sci U S A 102 : 16696–16700.
23. WongCH, ChengCY (2005) The blood-testis barrier: its biology, regulation, and physiological role in spermatogenesis. Curr Top Dev Biol 71 : 263–296.
24. EssafiA, WebbA, BerryRL, SlightJ, BurnSF, et al. (2011) A wt1-controlled chromatin switching mechanism underpins tissue-specific wnt4 activation and repression. Dev Cell 21 : 559–574.
25. Martinez-EstradaOM, LetticeLA, EssafiA, GuadixJA, SlightJ, et al. (2010) Wt1 is required for cardiovascular progenitor cell formation through transcriptional control of Snail and E-cadherin. Nat Genet 42 : 89–93.
26. MrukDD, ChengCY (2004) Sertoli-Sertoli and Sertoli-germ cell interactions and their significance in germ cell movement in the seminiferous epithelium during spermatogenesis. Endocr Rev 25 : 747–806.
27. JegouB (1993) The Sertoli-germ cell communication network in mammals. Int Rev Cytol 147 : 25–96.
28. RussellLD, PetersonRN (1985) Sertoli cell junctions: morphological and functional correlates. Int Rev Cytol 94 : 177–211.
29. GowA, SouthwoodCM, LiJS, ParialiM, RiordanGP, et al. (1999) CNS myelin and sertoli cell tight junction strands are absent in Osp/claudin-11 null mice. Cell 99 : 649–659.
30. WangRS, YehS, TzengCR, ChangC (2009) Androgen receptor roles in spermatogenesis and fertility: lessons from testicular cell-specific androgen receptor knockout mice. Endocr Rev 30 : 119–132.
31. MatsonCK, MurphyMW, SarverAL, GriswoldMD, BardwellVJ, et al. (2011) DMRT1 prevents female reprogramming in the postnatal mammalian testis. Nature 476 : 101–104.
32. KyronlahtiA, EulerR, BielinskaM, SchoellerEL, MoleyKH, et al. (2011) GATA4 regulates Sertoli cell function and fertility in adult male mice. Mol Cell Endocrinol 333 : 85–95.
33. BrehmR, ZeilerM, RuttingerC, HerdeK, KibschullM, et al. (2007) A sertoli cell-specific knockout of connexin43 prevents initiation of spermatogenesis. Am J Pathol 171 : 19–31.
34. Mazaud-GuittotS, MeugnierE, PesentiS, WuX, VidalH, et al. Claudin 11 deficiency in mice results in loss of the Sertoli cell epithelial phenotype in the testis. Biol Reprod 82 : 202–213.
35. JobertyG, PetersenC, GaoL, MacaraIG (2000) The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat Cell Biol 2 : 531–539.
36. GaoL, JobertyG, MacaraIG (2002) Assembly of epithelial tight junctions is negatively regulated by Par6. Curr Biol 12 : 221–225.
37. WongEW, MrukDD, LeeWM, ChengCY (2008) Par3/Par6 polarity complex coordinates apical ectoplasmic specialization and blood-testis barrier restructuring during spermatogenesis. Proc Natl Acad Sci U S A 105 : 9657–9662.
38. ChenWS, AnticD, MatisM, LoganCY, PovelonesM, et al. (2008) Asymmetric homotypic interactions of the atypical cadherin flamingo mediate intercellular polarity signaling. Cell 133 : 1093–1105.
39. RauchGJ, HammerschmidtM, BladerP, SchauerteHE, StrahleU, et al. (1997) Wnt5 is required for tail formation in the zebrafish embryo. Cold Spring Harb Symp Quant Biol 62 : 227–234.
40. HeisenbergCP, TadaM, RauchGJ, SaudeL, ConchaML, et al. (2000) Silberblick/Wnt11 mediates convergent extension movements during zebrafish gastrulation. Nature 405 : 76–81.
41. QianD, JonesC, RzadzinskaA, MarkS, ZhangX, et al. (2007) Wnt5a functions in planar cell polarity regulation in mice. Dev Biol 306 : 121–133.
42. KispertA, VainioS, McMahonAP (1998) Wnt-4 is a mesenchymal signal for epithelial transformation of metanephric mesenchyme in the developing kidney. Development 125 : 4225–4234.
43. StarkK, VainioS, VassilevaG, McMahonAP (1994) Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nature 372 : 679–683.
44. HosonoS, GrossI, EnglishMA, HajraKM, FearonER, et al. (2000) E-cadherin is a WT1 target gene. J Biol Chem 275 : 10943–10953.
45. BrueningW, BardeesyN, SilvermanBL, CohnRA, MachinGA, et al. (1992) Germline intronic and exonic mutations in the Wilms' tumour gene (WT1) affecting urogenital development. Nat Genet 1 : 144–148.
46. GesslerM, PoustkaA, CaveneeW, NeveRL, OrkinSH, et al. (1990) Homozygous deletion in Wilms tumours of a zinc-finger gene identified by chromosome jumping. Nature 343 : 774–778.
47. PelletierJ, BrueningW, LiFP, HaberDA, GlaserT, et al. (1991) WT1 mutations contribute to abnormal genital system development and hereditary Wilms' tumour. Nature 353 : 431–434.
48. PfeifferDC, VoglAW (1991) Evidence that vinculin is co-distributed with actin bundles in ectoplasmic (“junctional”) specializations of mammalian Sertoli cells. Anat Rec 231 : 89–100.
49. CallKM, GlaserT, ItoCY, BucklerAJ, PelletierJ, et al. (1990) Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms' tumor locus. Cell 60 : 509–520.
50. HuffV, MiwaH, HaberDA, CallKM, HousmanD, et al. (1991) Evidence for WT1 as a Wilms tumor (WT) gene: intragenic germinal deletion in bilateral WT. Am J Hum Genet 48 : 997–1003.
51. Royer-PokoraB, BeierM, HenzlerM, AlamR, SchumacherV, et al. (2004) Twenty-four new cases of WT1 germline mutations and review of the literature: genotype/phenotype correlations for Wilms tumor development. Am J Med Genet A 127A: 249–257.
52. GuiY, GuoG, HuangY, HuX, TangA, et al. (2011) Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet 43 : 875–878.
53. LiR, YuC, LiY, LamTW, YiuSM, et al. (2009) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25 : 1966–1967.
54. LiR, LiY, FangX, YangH, WangJ, et al. (2009) SNP detection for massively parallel whole-genome resequencing. Genome Res 19 : 1124–1132.
55. HayashiS, McMahonAP (2002) Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol 244 : 305–318.
56. HaradaN, TamaiY, IshikawaT, SauerB, TakakuK, et al. (1999) Intestinal polyposis in mice with a dominant stable mutation of the beta-catenin gene. EMBO J 18 : 5931–5942.
57. ZhangX, HaoL, MengL, LiuM, ZhaoL, et al. (2013) Digital gene expression tag profiling analysis of the gene expression patterns regulating the early stage of mouse spermatogenesis. PLoS One 8: e58680.
58. AudicS, ClaverieJM (1997) The significance of digital gene expression profiles. Genome Res 7 : 986–995.
59. van der WeeKS, JohnsonEW, DiramiG, DymTM, HofmannMC (2001) Immunomagnetic isolation and long-term culture of mouse type A spermatogonia. J Androl 22 : 696–704.
60. MengF, ChenS, MiaoQ, ZhouK, LaoQ, et al. (2012) Induction of fibroblasts to neurons through adenoviral gene delivery. Cell Res 22 : 436–440.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 8
Nejčtenější v tomto čísle
- Chromosomal Copy Number Variation, Selection and Uneven Rates of Recombination Reveal Cryptic Genome Diversity Linked to Pathogenicity
- Genome-Wide DNA Methylation Analysis of Systemic Lupus Erythematosus Reveals Persistent Hypomethylation of Interferon Genes and Compositional Changes to CD4+ T-cell Populations
- Associations of Mitochondrial Haplogroups B4 and E with Biliary Atresia and Differential Susceptibility to Hydrophobic Bile Acid
- A Role for CF1A 3′ End Processing Complex in Promoter-Associated Transcription