
Infection Reduces B Lymphopoiesis in Bone Marrow and Truncates Compensatory Splenic Lymphopoiesis through Transitional B-Cell Apoptosis
African trypanosomes of the Trypanosoma brucei species are extracellular protozoan parasites that cause the deadly disease African trypanosomiasis in humans and contribute to the animal counterpart, Nagana. Trypanosome clearance from the bloodstream is mediated by antibodies specific for their Variant Surface Glycoprotein (VSG) coat antigens. However, T. brucei infection induces polyclonal B cell activation, B cell clonal exhaustion, sustained depletion of mature splenic Marginal Zone B (MZB) and Follicular B (FoB) cells, and destruction of the B-cell memory compartment. To determine how trypanosome infection compromises the humoral immune defense system we used a C57BL/6 T. brucei AnTat 1.1 mouse model and multicolor flow cytometry to document B cell development and maturation during infection. Our results show a more than 95% reduction in B cell precursor numbers from the CLP, pre-pro-B, pro-B, pre-B and immature B cell stages in the bone marrow. In the spleen, T. brucei induces extramedullary B lymphopoiesis as evidenced by significant increases in HSC-LMPP, CLP, pre-pro-B, pro-B and pre-B cell populations. However, final B cell maturation is abrogated by infection-induced apoptosis of transitional B cells of both the T1 and T2 populations which is not uniquely dependent on TNF-, Fas-, or prostaglandin-dependent death pathways. Results obtained from ex vivo co-cultures of living bloodstream form trypanosomes and splenocytes demonstrate that trypanosome surface coat-dependent contact with T1/2 B cells triggers their deletion. We conclude that infection-induced and possibly parasite-contact dependent deletion of transitional B cells prevents replenishment of mature B cell compartments during infection thus contributing to a loss of the host's capacity to sustain antibody responses against recurring parasitemic waves.
Published in the journal:
. PLoS Pathog 7(6): e32767. doi:10.1371/journal.ppat.1002089
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002089
Summary
African trypanosomes of the Trypanosoma brucei species are extracellular protozoan parasites that cause the deadly disease African trypanosomiasis in humans and contribute to the animal counterpart, Nagana. Trypanosome clearance from the bloodstream is mediated by antibodies specific for their Variant Surface Glycoprotein (VSG) coat antigens. However, T. brucei infection induces polyclonal B cell activation, B cell clonal exhaustion, sustained depletion of mature splenic Marginal Zone B (MZB) and Follicular B (FoB) cells, and destruction of the B-cell memory compartment. To determine how trypanosome infection compromises the humoral immune defense system we used a C57BL/6 T. brucei AnTat 1.1 mouse model and multicolor flow cytometry to document B cell development and maturation during infection. Our results show a more than 95% reduction in B cell precursor numbers from the CLP, pre-pro-B, pro-B, pre-B and immature B cell stages in the bone marrow. In the spleen, T. brucei induces extramedullary B lymphopoiesis as evidenced by significant increases in HSC-LMPP, CLP, pre-pro-B, pro-B and pre-B cell populations. However, final B cell maturation is abrogated by infection-induced apoptosis of transitional B cells of both the T1 and T2 populations which is not uniquely dependent on TNF-, Fas-, or prostaglandin-dependent death pathways. Results obtained from ex vivo co-cultures of living bloodstream form trypanosomes and splenocytes demonstrate that trypanosome surface coat-dependent contact with T1/2 B cells triggers their deletion. We conclude that infection-induced and possibly parasite-contact dependent deletion of transitional B cells prevents replenishment of mature B cell compartments during infection thus contributing to a loss of the host's capacity to sustain antibody responses against recurring parasitemic waves.
Introduction
Trypanosoma brucei is a highly antigenically variable uniflagellate protozoan of which the subspecies T. b. gambiense and T. b. rhodesiense cause Human African Trypanosomiasis (HAT), also called Sleeping Sickness. In addition the parasite infects domestic animals, contributing to Nagana, which is a fatal disease of livestock in sub-Saharan Africa. T. brucei is transmitted in tsetse fly saliva and lives and replicates in blood, lymph and interstitial fluids of its mammal hosts protected from lytic plasma components by a coat of variable surface glycoprotein (VSG). The surface coat of a T. brucei parasite consists of 107 identical densely packed VSG molecules which can be varied among a possibly unlimited repertoire of coat types via a mechanism called antigenic variation [1]–[5]. Clearance of T. brucei and other African trypanosomes from the host blood stream is mainly mediated by VSG specific antibodies [6]–[8]. T. brucei parasites have been shown to (i) deplete marginal zone and follicular B cells from the spleen [9] , (ii) induce non-specific, polyclonal B cell activation leading to clonal exhaustion [10]–[12], and (iii) cause a general decrease in bone marrow cells [13] consistent with a negative impact on lymphopoiesis and erythropoiesis. Infection of trypanosomiasis-susceptible hosts with African trypanosomes has been shown to compromise host humoral immune competence resulting in the loss of B cell responsiveness to new antigens and of recall responses to previously encountered antigens, including trypanosome VSGs and vaccines [9]. Hence, vaccination against trypanosomiasis has so far never been successful in a natural infection setting.
B2 B cell lineage development under normal conditions occurs via a series of bone marrow (BM) stromal cell facilitated processes that begin within the hematopoietic stem cell pool and proceed in hierarchical steps of lineage commitment [14], [15]. Hematopoietic stem cells (HSC), which can self renew, give rise to multi lineage progenitors (MLP) and lymphocyte primed multi lineage progenitors (LMPP) that no longer self renew. LMPP, in turn, give rise to common lymphoid progenitors (CLP), which have been shown to sustain both T and B lymphopoiesis, although these lineages may diverge within the CLP. CLP give rise to several types of precursor cells, including pre-pro-B cells [16], [17]. B lymphopoiesis then proceeds in the bone marrow yielding several developmental stages of pre-pro-B, pro-B, pre-B and eventually immature B cells, which show a high expression of the IgM form of the antigen receptor and low or no expression of the IgD maturation marker [18], [19]. To complete their development, immature B cells migrate through the periphery, however only 10% reaches the spleen as transitional B cells of the T1 type. Important is the fact that under inflammatory immune conditions, BM lymphopoiesis is often severely reduced, and is compensated for by a splenic cell differentiation process that involves the same B-cell differentiation steps, referred to as extramedullary lymphopoiesis [20], [21]. Once the transitional T1 stage has been reached, B cells develop further into T2 transitional B cells that in turn can mature into either Marginal Zone B (MZB) cells or Follicular B (FoB) cells [22]. T2 cells can also give rise to T3 transitional B cells, but the latter don't give rise to mature B cells, due to being hyper-responsive to stimulation through their BCR [23]. Each of these populations is distinguished by a unique set of cell surface antigens, allowing monoclonal antibody (mAb) phenotyping by multicolor flow cytometry [24]–[26]. Using this approach, Radwanska et al. reported that the splenic MZB and FoB cell populations become rapidly depleted in T. brucei-infected mice and do not recover [9]. Since these findings suggest an impaired replacement of mature B-cells during infection, we have now investigated B cell development, maturation and cell death of various B-cell populations in T. brucei-infected mice.
B-lymphocyte cell death in the context of inflammation and infection has been attributed in the past to several major mechanisms that include TNF-TNFR1 and Fas/Fas-L induced apoptosis, prostaglandin triggered cell death and BCR-cross-linking in the absence of proper T-cell help [20], [27]–[32]. With respect to African Trypanosomiasis, none of these aspects have been addressed so far, despite the crucial need for an effective B-cell compartment for parasitemia control. In contrast, their role and modulation during intracellular Trypanosoma cruzi infections is better documented, showing that: (i) in the absence of TNF-TNFR1 signaling, susceptibility to infection increases and coincides with abnormal B-cell differentiation in secondary lymphoid tissues [33]. (ii) CD95/FasL interaction between B cells can mediate the fratricide of IgG+ B lymphocytes [27], and (iii) myeloid cell-derived prostaglandins contribute to infection-associated apoptosis of immature B cells in a Fas-FasL independent manner [28].
Programmed cell death can be induced by a number of death factors, including Fas-FasL interaction [34], [35] and the TNF-TNF-R1 apoptosis pathway [36], [37]. With respect to the latter, it has been well established that (i) T. brucei infections induce severe inflammatory disease in susceptible hosts leading to the excessive production of pro-inflammatory factors including TNF and prostaglandins [38]–[42], and (ii) that excess induction of TNF negatively affects various lymphoid compartments [20]. In contrast to TNF, to date no information on the role of Fas has been reported in a T. brucei infection setting. The Fas apoptosis pathway is normally important in both the regulation of the immune response as well as T and B lymphocyte homeostasis [35]. Therefore, Fas-FasL mediated apoptosis plays a critical role in the mechanism for negative selection of B cells [43]–[45] and for the establishment of the B cell repertoire in the memory compartment [46]. Following activation, B cells can rapidly upregulate both Fas and FasL expression [47], [48], but the control of B lymphocyte expansion appears mainly to be regulated by FasL-expressing T cells [47].
Here we investigate the contributions of different mechanisms to T. brucei-induced abrogation of B cell development and infection-associated B cell apoptosis. Our results show that following T. brucei infection, B lymphopoiesis is truncated in the bone marrow and compensatory extramedullary B lymphopoiesis is induced (but not completed) in the spleen. Splenic B lymphopoiesis up to the stage of immature B cells was triggered but final development was severely limited by apoptosis of transitional B cells, thus preventing replenishment of mature B2 B cells. Despite the pro-inflammatory immune environment induced by experimental T. brucei infections, these events occurred independent from TNF-TNF-R1, Fas-Fas-L and prostaglandin-mediated pathways. Interestingly, in an ex vivo setting in which naïve or infection-derived splenocytes were co-cultured with living bloodstream form trypanosomes, transitional B-cell apoptosis was only observed when cell-cell contact between lymphocytes and parasites occurred. This observation corroborates the previous findings from Radwanska et al. that showed that trypanosomes can induce contact dependent cell death in anti-VSG hybridoma B-cells [49]. Using a Trans-well co-culture system, or a VSG specific Nanobody, i.e. a variable heavy chain fragment of a single chain camelid antibody devoid of its Fc part [50], we now show that preventing direct contact between the trypanosome surface coat and transitional B-cells results in an abrogation of infection-induced apoptosis in the ex vivo setting.
Results
Impaired B lymphopoiesis in T. brucei infected mice
T. brucei infections in mice have been shown to compromise host humoral immune competence and to induce the loss of specific mature B cell populations in the spleen [9]. However, little is known about how B lymphopoiesis in the bone marrow is affected during T. brucei infection. Here, B lymphopoiesis has been examined using a C57BL/6 mouse T. brucei AnTat 1.1E infection model, which is characterized by successive waves of parasitemia and a median infection survival time of 35 days [51]. Bone marrow and spleen cells were isolated at different time points of infection and prepared for cellular characterization by multicolor flow cytometry as described in Table 1 and shown in Figure S1, S2 and S3. As presented in Figure 1, the number of very early progenitors, i.e., HSC-LMPP was minimally affected, showing only a transient reduction on day 20 p.i. Subsequent bone marrow B lymphopoiesis was severely affected in T. brucei infected mice, with major declines in all bone marrow B cell developmental stages starting with the CLP fraction. A drop in the number of CLP progenitors was detected on day 10 p.i. and this cell population remained severely depleted thereafter. The pre-pro-B cell population showed a 50% reduction by day 20 p.i., while the subsequent B-cell maturation stage i.e. the pro-B, pre-B and immature B cell populations reached more than 95% depletion by day 10 p.i. and failed to recover throughout the further course of infection. Combined, these results show that at the end stage of differentiation in the bone marrow, B cell numbers are severely depleted.


T.brucei infection induces extramedullary lymphopoiesis in the spleen
Inflammation in general has been described to induce mobilization of immature bone marrow lymphocytes [20]. Because T. brucei infection is characterized by a strong type 1 inflammatory immune response, mobilization of bone marrow precursors may result in the appearance of developing B cells in the spleen. Cells were harvested from the spleen at different time points of infection and a multicolor flow-cytometric analysis was performed according to Table 1. As shown in Figure 2, T. brucei infection induced an increase in HSC-LMPP fractions in the spleen by day 10 p.i. In addition, there was a significant rise in CLP, pre-pro-B, pro-B and pre-B cell numbers in the spleen on day 10 p.i., coinciding with the drastic losses of B cell precursors from the bone marrow. While pre-pro-B, pro-B and pre-B cell numbers remained significantly elevated in the spleen by day 20 p.i., these populations returned to pre-infection levels towards the end of infection (day 30 p.i.). In contrast, while there was no early stage decrease in immature B cells in the spleen, there was a significant loss of this population towards the end of infection.

T. brucei infection causes depletion of transitional B cells in the spleen through induction of apoptosis
In contrast to the elevation in early B cell developmental stages observed in the spleen, Radwanska et al. [9] have reported depletion of mature marginal zone and follicular B cells suggesting impaired replacement. Therefore, B cell development at the transitional B cell stage in the spleen was examined here, as these cells provide the link between the immature B cell stage and the mature marginal zone and follicular B cell stages. Flow cytometric analysis of the T1, T2 and T3 transitional B cell populations of T. brucei infected mice (Figure 3) revealed a transient increase in transitional B cells numbers that however rapidly faded towards day 10 p.i. On days 20 and 30 p.i. the number of transitional T2 and T3 B cells was significantly decreased compared to uninfected control mice.

To address whether apoptosis is contributing to the depletion of transitional B cells in the spleen, the amount of active caspases-1, −3, −4, −5, −6, −7, −8 and −9 inside the transitional T1, T2 and T3 B cells was measured at different time points of infection by flow cytometry (Figure 4A). Although infection did induce apoptosis in both the T1 and T2 transitional B cell population, the T3 population only showed a temporary increase of caspase activation after the first week of infection (Figure 4B). The elevation in levels of caspases in transitional B cells coincided with the contraction of these B cell populations between days 6 and 10 p.i. (Figure 3A), i.e., immediately following peak parasitemia (8×107 T. brucei/ml blood). In the model used here, infections are initiated with 5×103 T. brucei AnTat 1.1 and remission of the first parasitemic wave occurs between 6 and 7 days p.i. [41]. It is noteworthy that when the infection was initiated with 108 T. brucei AnTat 1.1, peak parasitemia and wave remission occurred at 4 days p.i. (data not shown) and transitional B-cell apoptosis was observed as early as 3 days p.i. (Figure 5). These observations suggest a direct link between levels of parasitemia and the kinetics of induction of splenic T1/2 B-cell apoptosis. Combined, these data indicate the induction of apoptosis in both T1 and T2 transitional B cell populations occurs at, or close to, peak parasitemia. The transitional B cells would, under normal conditions, give rise to the continuous replenishment of marginal zone and follicular B cell populations, but clearly are unable to do so in the infected mice.
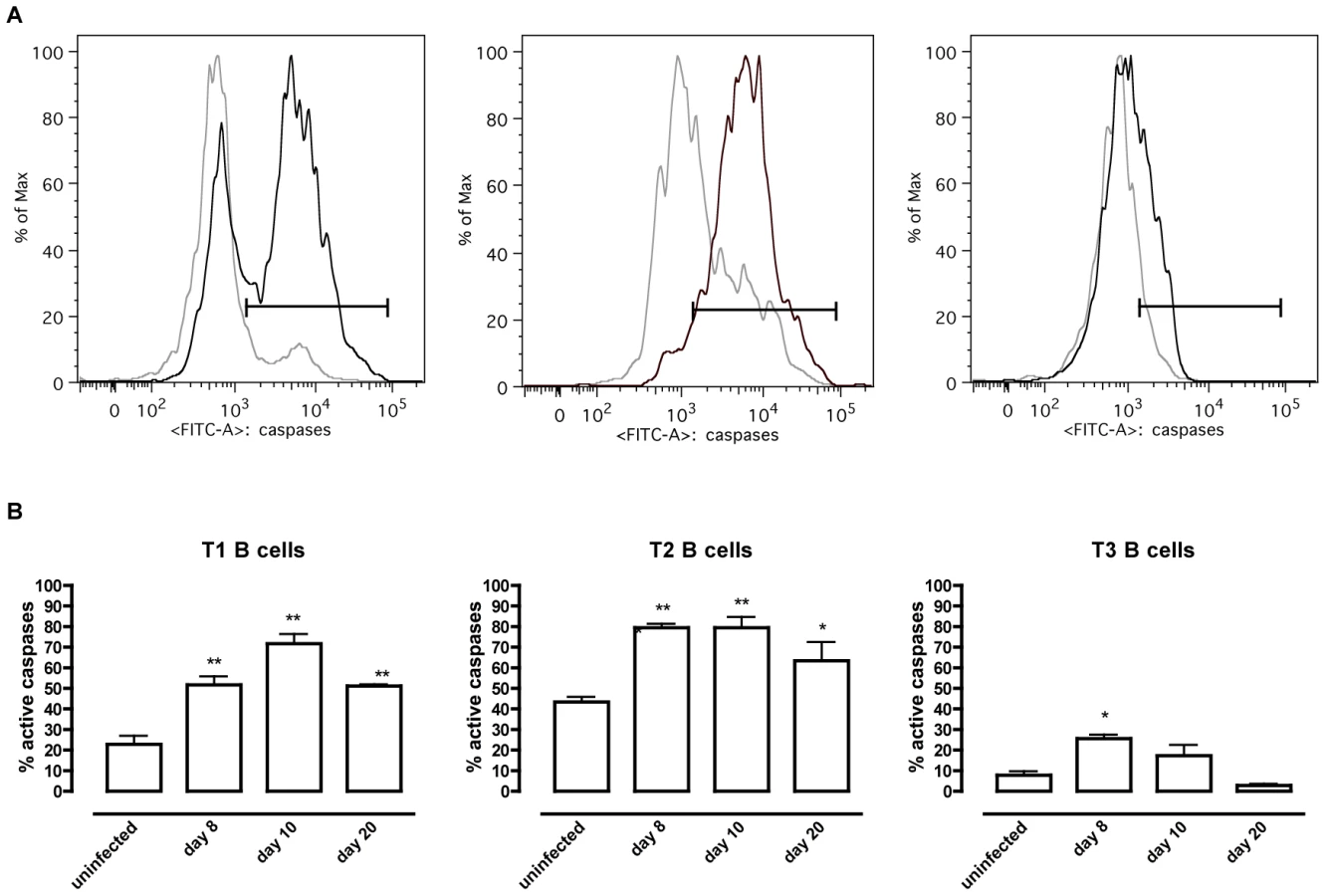

T. brucei infection-induced apoptosis of transitional B cells occurs independently of TNF and Fas
Experimental T. brucei infections induce high circulating levels of TNF [38], a host cytokine that has been reported to be involved in the induction of immunopathology during T. brucei infections [38]–[42]. Furthermore, TNF is known to be a potent inducer of apoptosis through the TNF-R1 signal pathway. Therefore, to examine the possibility of TNF-mediated apoptosis of T1 and T2 transitional B cells, TNF−/− mice were infected with T. brucei and the number of T1 and T2 transitional B cells in the spleen was examined at different time points of infection. Figure 6 illustrates that on day 10 p.i. WT mice and TNF−/− mice both suffered from similar levels of transitional T1 and T2 B cell depletion. While on day 20 p.i. a temporary recovery of T1 transitional B cells occurred only in the TNF−/− mice, the same final level of 75% reduction in both T1 and T2 transitional B cells was observed in WT as well as TNF−/− mice towards the end of infection. As an additional control, apoptosis of transitional B cells was recorded using the poly-capase activation FACS analysis outlined above. Here, the percentage of T1 and T2 B cells undergoing apoptosis in T. brucei-infected TNF−/− as well as TNF-R1−/− mice equaled the results reported for WT mice (data not shown).

Next, the expression of the death receptor Fas (CD95) on T1 and T2 transitional B cells, and the potential involvement of the Fas-FasL apoptosis pathway were analyzed at different time points of infection. Control T1 and T2 transitional B cells express low, but detectable levels of Fas on their surface. During infection however, there was a strong increase in the level of Fas expression on the surface of both T1 and T2 transitional B cells, as shown here for days 8, 10, 14 and 20 p.i., (Figures 7A and B). The increase in surface Fas expression coincided with elevated caspases activity (Figure 4) and with the loss of transitional B cells from the spleen (Figure 6). Interestingly, also in TNF−/− mice as well as TNF-R1−/− mice, the increase in Fas expression on both T1 and T2 transitional B-cells preceded the rapid loss of these cells from the spleen further suggesting a correlation between the up-regulation of surface-expressed Fas and the trypanosomiasis-associated destruction of the transitional B-cell compartment (Figure S4).

If apoptosis of transitional B cells in T. brucei-infected mice is mediated through Fas, then the loss of the cells would be expected to be reduced in mice with constitutively low Fas expression. However, this proved not to be the case as the extent and kinetics of transitional B cell loss following infection of lpr mice with T. brucei AnTat 1.1 was the same as in wild type mice (data not shown). In addition, if apoptosis were to be mediated through Fas in wild type mice, it would be expected to be ameliorated by blocking the activation of Fas by its ligand, FasL, through administration of neutralizing anti-FasL antibody [27]. However, this also proved not to be the case. Indeed, mice infected with T. brucei and treated by i.v. injection with 100 µg of anti-FasL antibody on days 4, 5 and 6 p.i. did not manifest a measurable alteration in transitional B cell loss from the spleen (Figure 8a), nor did the remaining transitional B-cells in these mice exhibit a change in their caspase activity profile (Figure 8b) relative to mice receiving a control immunoglobulin treatment. Thus, while the occurrence of transitional B cell apoptosis after infection with T. brucei coincides with an increased expression of the death receptor Fas on the surface, apoptosis of transitional B cells could not be prevented by the anti-FasL antibody treatment used here.

Induction of transitional B cell apoptosis occurs independently from the cyclooxygenase pathway
Besides TNF and Fas, also prostaglandins have been implicated in B-cell apoptosis, in particular in a (intracellular) T. cruzi infection setting [28]. To examine the possible contributions of cyclo-oxygenase (COX) products to T. brucei-induced transitional B cell death in vivo, indomethacin (a nonsteroidal anti-inflammatory drug that inhibits COX activity), or control physiological buffer, was administered daily to infected mice by i.p. injection using a previously described protocol [52]. The indomethacin treated mice infected with T. brucei did not differ from control infected mice with respect to transitional B cell apoptosis (Figure S5). In a second set-up T. brucei-infected mice received indomethacin in the drinking water at a concentration of 14 µg/ml [53]. Here again, no difference was observed in the percentage of transitional B cells undergoing apoptosis between treated mice and untreated mice (data not shown).
In addition to the in vivo analysis of prostaglandin contribution to the induction of transitional B cell apoptosis, an in vitro assay was designed to further examine the contribution of cyclooxygenase products to transitional B cell apoptosis. Here, spleen cells from uninfected mice were co-cultured in a Trans-well system [29] with spleen cells from uninfected mice or from mice that had been infected for varying times with T. brucei Antat 1.1. Culture conditions included incubations in the presence or absence of trypanosomes and of indomethacin. However, in none of the experimental conditions was there a difference in percentage of transitional B cells undergoing apoptosis compared to the controls (representative data are presented in Figure S6), leading us to conclude that prostaglandins (either host or parasite derived) are not a major contributor to transitional B-cell apoptosis during T. brucei infections.
Apoptosis of transitional B cells is blocked when cell-cell contact between trypanosomes and lymphocytes is prevented
When performing the Trans-well experiments described above to address the potential role of prostaglandins in transitional B cell apoptosis, control conditions included co-cultures in which parasites and splenocytes (either from uninfected or day 5 T. brucei AnTat 1.1 infected mice) were not separated by a physical barrier. In line with previous results obtained in co-cultures of bloodstream form trypanosomes and B-cell hybridoma cells raised against VSG [49], here the T1/2 transitional B-cell population survival was significantly impaired, which was not observed when parasites and B-cells were separated by a 0,4 µm polycarbonate transmembrane. Indeed, Figure 9A (columns 1 and 2) shows that incubation of spleen cells from uninfected mice with freshly isolated living trypanosomes (10 trypanosome/spleen cell) resulted in 75% lower survival of T1 and T2 B cells relative to incubation in medium. In contrast, there was no difference in the numbers of viable IgM−ve B220−ve leukocytes recovered from splenocytes cultured in the presence or absence of trypanosomes (data not shown).

Similarly, when spleen cells from mice infected 5 days earlier with T. brucei were incubated with trypanosomes, there was a 50% reduction in viable T1 and T2 B cells relative to cells cultured in medium (Figure 9A columns 3 and 4) and again no reduction in the recovery of IgM−ve B220−ve leukocytes (data not shown). However, when cultured cells and trypanosomes were separated by a 0.4 µm polycarbonate transmembrane there was no decrease in the number of viable transitional B cells in the culture (Figure 9A column 5) indicating that B cell loss does not result from diffusible trypanosome products or from consumption of essential medium components by living trypanosomes, but from the direct contact between the cells and the parasites. Culturing splenocytes from 5 day-infected mice with parasite lysate, or purified soluble VSG (sVSG) from the parasites, rather than living parasites, also did not result in the loss of transitional B cells (Figure 9B, columns 1–3). To further examine whether contact with VSG on living T. brucei is required for depletion of transitional B cells in vitro, T. brucei AnTat 1.1 trypanosomes were pre-incubated with a VSG-specific Nanobody (Nb, a dromedary heavy chain antibody fragment, devoid of its Fc part, having no detrimental effect on parasite survival [50]), prior to addition to spleen cells from 5 day-infected mice. A comparison of Figure 9B column 4 (control) and column 5 (Nb) shows that the pre-treatment of trypanosomes with nanobodies prevented the killing of B cells. Thus, these results corroborate the obtained Trans-well results indicating that VSG dependent direct contact between intact living trypanosomes and transitional B-cells can trigger cell death.
Discussion
Infection with African trypanosomes causes mice and other trypanosomiasis-susceptible mammals to develop non-specific hypergammaglobulinemia and polyclonal activation that in the end leads to B cell clonal exhaustion [10]–[12]. While the mechanisms underlying B cell clonal exhaustion have yet to be resolved, results by Radwanska et al. showed that the mature marginal zone and follicular B cell populations rapidly disappear during experimental trypanosome infections and that vaccine-induced B-cell memory is destroyed in a non-specific manner [9]. Additional results suggested that while trypanosome infection initially leads to rapid immune activation and buildup of trypanosome/VSG-specific immunity against trypanosomes in the initial parasitemic wave, humoral immune responsiveness is rapidly lost. Impairment in the replacement and recruitment of naïve B-cells to the spleen apparently prevents the efficient activation of specific immunity later on in infection [47], [48]. In order to gain insight into the mechanisms of infection-induced B-cell dysfunction, we have examined how T. brucei infection affects B cell development in the bone marrow and the survival and maturation of B cells in the periphery. Our studies show that BM lymphopoiesis is shut down during infection and that compensatory extramedullary B lymphopoiesis is truncated by apoptosis of transitional B cells thus preventing replenishment of mature marginal zone and follicular B cell compartments.
Mice infected with T. brucei exhibit reduced numbers of B cells at all developmental stages in the bone marrow and transitional stages in the spleen. Within the bone marrow, loss of CLP preceded that of other progenitor populations raising the possibility that downstream losses resulted from depletion of this precursor population. However, if the loss of CLP was solely responsible for the downstream disrupted B cell development, then the ratio of a subsequent B cell developmental population to its immediate upstream progeny and downstream precursors should remain unaffected, which was not the case. Hence, additional processes besides progenitor depletion must be operating to limit B cell development in T. brucei infected mice.
Although aberrant differentiation, apoptosis and expulsion from the bone marrow may singly or jointly contribute to loss of B cell precursors from the bone marrow, we favor the latter mechanisms based on the following arguments: (1) during infection we could not measure any increase in B cell precursor apoptosis in the bone marrow (Figure S7), (2) alterations in the expression of essential B cell development-specific transcription factors in the BM like Icaros, PU.1, EBF and E2A and the IL-7 growth factor have never been reported and were not observed in our laboratory either (data not shown), (3) a reduction in bone marrow CXCL12 expression was the only parameter found by us to correlate with the observed loss of developing B cells during infection (Figure S8). The importance of this result is underlined by the knowledge that inflammation-induced reductions in stromal bone marrow CXCL12 expression indeed have been reported by others to correlate with a premature lymphocyte efflux [20]; (4) there are elevated numbers of HSC-LMPP and CLP as well as pre-pro-B, pro-B, pre-B and immature B cells in the spleen, suggesting the possibility of an increased influx of these cells. In addition, during the early stage of infection small numbers of HSC-LMPP, CLP, pre-pro-B, pro-B, pre-B and immature B cells were also found in the blood, the liver, peritoneum and lungs and small numbers of pro-B, pre-B and immature B cells were found in the lymph nodes. Taking these findings and arguments together, we hypothesize that during early T. brucei infections, B cell precursors prematurely migrate out of the bone marrow as a result of the initiation of inflammation, and at least in part home to the spleen, allowing transient extramedullary B lymphopoiesis to take place.
Despite the initiation of extramedullary B lymphopoiesis, infection-induced loss of mature B-cells marks progressing trypanosomiasis in experimental infections. Various factors, in particular the induction of systemic inflammation, could contribute to this. Hence, we addressed the potential involvement of three likely participants, i.e. TNF, Fas and prostaglandins, all previously shown to be potentially involved in immune and B-cell malfunctioning. Paradoxically, despite the presence of high systemic TNF levels during infection [38] and the clear evidence shown here that transitional B cell loss through apoptosis coincides with elevation of Fas expression on both T1 and T2 B cells, our results showed through the use of knock-out mice as well as anti-FasL antibodies that neither the TNF - nor the Fas - death pathways acting alone are responsible for transitional B cell apoptosis in a T. brucei infection setting. In addition, despite the reported production of prostaglandins by trypanosomes [29] and macrophages in infected mice [54] inhibition of prostaglandin/cyclooxygenase activity in T. brucei infected mice by administration of indomethacin did not rescue transitional B cells, contrasting with results obtained in T. cruzi infections [28]. However, as each of the death pathways could be redundant in a multi-factorial complex event such as infection-induced apoptosis, their in vivo individual contribution to transitional B cell apoptosis in the T. brucei infection model should not be formally excluded.
The absence of a clear role for the Fas apoptosis pathway in T. brucei induced transitional B-cell death is particularly surprising, taken that (i) Fas upregulation on these cells is reported here to be a clear hallmark of progressing infection, and (ii) that Fas-FasL B cell killing in the context of an infectious disease has previously been described in the case of Trypansosoma cruzi infections, which induce Fas-mediated fratricide of IgG+ B lymphocytes specific for parasite antigens but not self antigens [27]. In addition, Fas-mediated cell death is known to be important in the regulation of the immune response and T and B lymphocyte homeostasis [35], where for instance Fas-FasL mediated apoptosis plays a critical role in the mechanism for negative selection of B cells [44], [45] and the establishment of the B cell repertoire in the memory compartment [46]. However, despite the reported role for Fas in parasite-induced fratricide, its contribution to the killing of virus-infected, damaged or excess cells and its implication in various immunopathological disorders [43], [44], [47], our study did not provide any evidence functionally linking Fas upregulation and B cell apoptosis. Indeed, treating infected mice three times (with daily interval) with 100 µg of anti-FasL antibody just prior to peak infection, in order to block in vivo the activation of the Fas death cascade did not alter the kinetics at which the transitional B cell population underwent apoptosis. It could be argued that the doses of anti-FasL antibody used in this experiment do not functionally block the activation of the Fas apoptosis pathway, however, when Lpr mice are infected with T. brucei, transitional B cells are lost from the spleen to the same extend as in the wild type mice (data not shown), arguing once again against a major involvement of the Fas-apoptosis pathway.
Having shown that T. brucei driven transitional B-cell apoptosis occurs in a TNF-, Fas - and prostaglandin-independent manner, our study next focused on a model system that could help to functionally unravel infection-induced B-cell apoptosis. Interestingly, since B cell apoptosis only occurs when living parasites are administered to the mice, and not when mice are treated on a daily basis by the injection of high doses of trypanosome lysate (results not shown), the presence of a toxin or a super-antigen-like activity by trypanosome molecules appears to be excluded. These results are in sharp contrast to the superantigen-mediated death of mature B cells in the case of Staphylococcus aureus, where injection with the virulence factor protein A alone is enough to mimic massive Fas and TNF-independent bacterial induced B cell death, and cause a ‘hole’ in the immune repertoire recognizing the pathogen [55], [56]. Also, it is worth recalling that the in vivo experiments presented in this study show that initiation of transitional B cell apoptosis depends on the timing at which peak levels of parasitemia are reached, which is a function of the number of living bloodstream form parasites used for initiation of the infection. This observation suggests a link between the presence of high numbers of living parasites and the induction of parasite-induced transitional B-cell apoptosis consistent with the possibility that B cell hyper-stimulation, through multiple VSG (variable surface glycoprotein - attached to the parasite surface)/BCR (B cell antigen specific receptor - attached to the B-cell surface) interactions, could be a major contributor to this process, as might exposure of B cells to short-lived parasite products which would be active only when directly delivered to the target cell. With respect to BCR signaling, cross-linking of the BCR has been well described to trigger apoptosis of T1 and T2 transitional B cells, which can be ameliorated in the case of T2 B cells by anti-apototic signaling through the BlyS receptor BR3. Transitional B cells of the T2 type can also be rescued from BCR crosslinking-induced apoptosis by T cell help [30]–[32], [57], [58]. However, T cell suppression is one of the hallmarks of T. brucei infection [54], making it likely that T2 transitional B cells in infected mice are exposed to the parasites in the absence of proper survival-stimulating T cell help. Of crucial importance is the notion that membrane-bound antigens that can extensively engage BCRs trigger rapid BCR-mediated apoptosis in a Fas-independent manner [59]. Hence, in the context of trypanosome-B cell interaction, the presence of 10 million identical VSG molecules densely packed on the surface of the parasite could be responsible for causing BCR receptor clustering on the surface of the transitional B cells, leading to hyper-stimulation of the B cell and TNF/Fas-independent cell death in the absence of proper T-cell signaling. Avidity, due to multiple VSG/BCR interactions, in this case would have a much higher impact than actual BCR affinity/specificity for a given VSG.
Using an in vitro co-culture system of splenocytes and live bloodstream form trypanosomes, we showed that direct contact between the living parasites and host B cells can indeed trigger the deletion of the latter. Depletion of T1 and T2 transitional B cells was maximally induced with 10 trypanosomes per spleen cell in culture but was observed with as few as 0.1 trypanosomes/spleen cell in culture, with 50% depletion occurring at a ratio of between 0.1 and 0.5 trypanosome/spleen cell (Figure S9). This falls within the physiologic range in vivo for infections with T. brucei AnTat 1.1, where the ratio of trypanosomes to viable nucleated cells in the spleen is 0.25 : 1 at peak parasitemia. In co-cultures of trypanosomes and splenocytes in which contact between the two is prevented using a Trans-well system or a pre-incubation of the parasite with a VSG-specific Nanobody, abrogation of transitional B cell deletion is observed. This result mirrors a previous observation by Radwanska et al. where cell death was triggered in IgM expressing hybridoma cells when cultured in the presence of living trypanosomes [49]. Again, in the co-culture system neither lysate, nor purified parasite VSG could mimic the apoptosis-inducing effect of living parasites (nor could anti-FasL or prostaglandin inhibition block the detrimental effect of living trypanosomes). Together, these observations strengthen the hypothesis that the direct interaction of B cells with epitopes on T. brucei causes transitional B cell death. Unfortunately we cannot evaluate Nanobody blockage of transitional B cell apoptosis in vivo as these antibody fragments are very rapidly cleared from the circulation by the kidneys [60]. Thus, although the mechanism of T. brucei-induced transitional B cell depletion in vivo remains to be fully elucidated, we did observe that living trypanosomes induce cell death in transitional B cells in vitro through a contact-dependent mechanism. Micro-array analysis of material from both hybridoma co-culture assays and spleen-derived B-cell co-cultures with trypanosomes are now underway in order to shed light on the signal cascades involved in this contact triggered apoptosis.
Combined, our study provides evidence for trypanosomiasis-induced apoptosis of transitional B cells in the spleen and it proposes a mechanism for T. brucei-induced B cell clonal exhaustion and loss of humoral immune competence in trypanosomaisis-susceptible hosts. Under normal conditions the production of high-affinity, antigen-specific, class-switched, antibodies takes up to 10 days after immunization [61], [62]. Here, in our infection model, about 90% of the transitional B cells are undergoing apoptosis by day 8 of infection, making the replenishment of the mature marginal zone and follicular B cell populations impossible and therefore obstructing efficient germinal center reaction and the renewal of the plasma B cell pool. Since parasite-specific antibodies are essential for parasite control, inhibition of B cell maturation at the transitional stage is an efficient evasive mechanism developed by the parasite to interfere with the protective antibody responses of the host and establish a sustained infection. It is important to stress here that our studies are based on a mouse model system for African trypanosomiasis, which manifests certain limitations. However, the results obtained in this model provide guidelines for analysis of B cell pathology in more relevant hosts including susceptible livestock species such as cattle and goat, or the retention of immune function in natural trypanotolerant animals such as the Cape Buffalo, which have a sustained capacity to generate effective protective antibody responses against T. brucei and other African trypanosomes during chronic infection, thus, maintaining cryptic parasitemia with few or no signs of disease [7]. Based on our findings and the earlier data reported by Radwanska et al. [9], it would also be crucial to investigate whether a similar B cell pathology occurs in T. brucei infected humans. In this context, it would be interesting to compare B cell pathology between T. brucei gambiense (manifesting a chronic infection in humans with prolonged parasitemia control) and T. brucei rhodesiense (manifesting an acute infection whereby parasitemia control is lost very early) infected patients, where in the latter it could be that rapid destruction of the B-cell compartment is the underlying reason for failure of parasitemia control, the rapid induction of systemic inflammation, and the subsequent passage of the parasite through the blood-brain barrier.
Materials and Methods
Ethics statement
The study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and Guidelines for the Use of Laboratory Animals in Research, Teaching and Testing of the International Council for Laboratory Animal Science. All animal work was approved by the appropriate committee at the University of Massachusetts (IACUC protocol #s 26-09-09/27-09-09 and 2010-0028) and at the Vrije Universiteit Brussel (ethics committee protocol # 09-220-1).
Parasites and infection in mice
All mice were housed under barrier conditions. Male C57BL/6 wild type (Taconic, Germantown, NY), Lpr and TNF−/− C57BL/6 mice (provided by VUB, Belgium) (7–9 week old) were infected by intraperitoneal (i.p.) injection of 5000 exponentially growing pleomorphic Trypanosoma brucei Antat 1.1 (EATRO 1125 stock) [51]. Parasitemia was assessed in blood collected from the tail vein during infection. Blood was diluted in RPMI (Gibco, Grand Island, NY, USA) and the number of trypanosomes present in the blood was estimated using a hemocytometer and a light microscope.
Cell isolation and flowcytometric analysis
B cell populations were analyzed by flowcytometry. Both spleen and bone marrow from femur and tibia were harvested from non-infected control and infected mice at different time points of infection. Cell suspensions were prepared in FACS buffer (1.0% BSA [Sigma, St. Louis, MO] in DPBS) and red blood cells were lysed using ACK lysis buffer (0.15M NH4Cl, 1.0 mM KHCO3, 0.1 mM Na2-EDTA). Non-specific binding sites were blocked using Fc block (CD16/CD32 Fcγ III/II, BD biosciences, San Jose, CA) for 30 minutes at 4°C. Cells were washed twice with FACS buffer and stained with biotin - or fluorochrome-conjugated primary antibodies (see following section) for 30 minutes at 4°C. After washing twice, cell suspensions stained with biotin-conjugated antibodies were incubated with streptavidin-conjugated fluorochromes (listed in the text), which detects cell bound biotinylated antibodies, and incubated for an additional 30 minutes at 4°C. Finally, cells were resuspended in FACS buffer with 1 µg of 7-amino-actinomycin D (7AAD), a fluorescent DNA dye that binds to membrane permeable dead or dying cells, (BD biosciences). Analyses were performed using a FACS Canto II flow cytometer (BD Biosciences) and data were processed using FLOWJO software (Tree Star Inc., Ashland, OR). The total number of cells in each population was determined by multiplying the percentages of subsets within a series of marker negative or positive gates by the total cell number determined for each tissue.
Antibodies and detection reagents
The following antibodies were added to 100 µl aliquots of 106 Fc-blocked leukocytes prepared as described above: 0.5 µg anti-IL7rα-FITC (clone A7R34), 0.2 µg anti-IgM-PE (clone II/41), 0.2 µg anti-IgM PE-Cy7 (clone II/41), 0,25 µg hamster IgG2 κ isotype control (clone B81-3), 0.5 µg anti-CD11b-FITC (clone M1/70; 0.5 mg/ml), 0.5 µg anti-CD23-FITC (clone B3B4), 0.5 µg anti-CD45R (B220)-FITC (clone RA3-6B2), 0.2 µg of anti-CD45R (B220)-PE-Cy7 (clone RA3-6B2), 0.2 µg anti-CD93–PE (clone AA4.1), 0.2 µg of anti-CD93-APC (clone AA4.1), 0.5 µg anti-CD95 –FITC (clone Jo2), 0.2 µg of anti-CD117 (ckit)-APC (clone 2B8), purchased from eBioscience (San Diego, CA); 0.2 µg anti-CD1d-PE (clone 1b1), 0.2 µg of anti-CD19-APC-Cy7 (clone 1D3), 0.2 µg of anti-CD43-PE (clone 1B11), 0.2 µg of anti-CD45R (B220)-APC-Cy7 (clone RA3-6B2), 0.2 µg of streptavidin-PerCP, 0.2 µg of streptavidin-PE-Texas Red, purchased from BD Biosciences (Erembodegem, Belgium); 2 µg of each of the following antibodies: CD3ε, CD11b (Mac-1), Gr-1 (Ly-6G and Ly-6C) and Ter-119 (Ly-76) from the Biotin-conjugated Mouse Lineage Panel (BD Biosciences, San Jose, CA).
Flow cytometric analyses of apoptosis
Cells were stained as described in the previous section with antibodies. For the polycaspases-based apoptosis assay, labeled cells were further reacted with the FLICATM fluorescent inhibitor of caspase-1, −3, −4, −5, −6, −7, −8 and −9, using the FAM Poly Caspases Assay Kit for flow-cytometric analysis (Molecular probes, Invitrogen, Leiden, the Netherlands).
In vivo anti-FasL treatment
Mice infected with T. brucei where treated i.v. with 100 µg of purified anti-FasL antibody (clone MFL3, purchased from BD Biosciences) on days 4, 5 and 6 of infection and on day 7 of infection total spleen cells were isolated from treated mice and untreated controls and prepared for flowcytometric analysis as described above.
Co-culture experiments with total spleen cells and live trypanosomes
On day 5 of infection mice were sacrificed to collect blood. In addition, total spleen cells were isolated from the infected mice and uninfected control mice and prepared for cell culture as described above. Trypanosomes were purified from the blood by anion exchange chromatography according to the method of Lanham & Godfrey [63]. Then, 106 total spleen cells were put in co-culture with or without 107 (or fewer) live bloodstream form parasites at 37°C, 5% CO2 and 95% humidity in RPMI1640 medium containing 10% FBS, 2 mM pyruvate, 0.2 mM 2-mercaptoethanol, and penicillin/streptomycin under different experimental conditions and 20 h later, the cells were stained for flowcytometric analysis. Parasite lysate was prepared by 3 repeated cycles of freezing at −80°C and thawing and used at indicated concentration. Soluble VSG (sVSG) was prepared through heat-shock treatment of a purified trypanosome suspension, which forces them to release their VSG molecules, followed by anion exchange chromatography, and used at indicated concentrations. Nanobodies against soluble trypanosome VSG were prepared as described in [50], starting from an immune library of VHH fragments of heavy chain dromedary antibodies, obtained after multiple vaccination with T. brucei AnTat 1.1 VSG. Trypanosomes were pre-incubated with Nanobody BankIt1413802 Nb_An05–04 HQ680968 (monovalent and devoid of an Fc chain; 15 µg/ml medium ) at the indicated concentrations 1 hour prior to addition to the splenocytes, and the antibody was included in culture medium during subsequent incubations. Under these assay conditions, binding of the Nanobody onto the trypanosome surface did not result in altered parasite survival.
Statistical analysis
Statistical comparisons were performed by ANOVA and means were compared using Tukey and Dunnett's post test when p≤0.05 (GraphPad Prism v.4.0, GraphPad Software Inc. San Diego, CA).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. VickermanK 1989 Trypanosome sociology and antigenic variation. Parasitology 99Suppl.S37 47
2. VickermanK 1978 Antigenic variation in trypanosomes. Nature 273 613 617
3. PaysE 2006 The variant surface glycoprotein as a tool for adaptation in African trypanosomes. Microbes Infect 8 930 937
4. BorstP 2002 Antigenic variation and allelic exclusion. Cell 109 5 8
5. PaysE 1995 Antigenic variation and the problem of vaccines against African trypanosomes. Bull Mem Acad R Med Belg 150 : 123-131; discussion 131-125
6. LevineRFMansfieldJM 1984 Genetics of resistance to the African trypanosomes. III. Variant Specific antibody responses of H-2-compatible resistant and susceptible mice. J immunol 133 1564 1569
7. GuirnaldaPMurphyNBNolanDBlackSJ 2007 Anti-Trypanosoma brucei activity in Cape buffalo serum during the cryptic phase of parasitemia is mediated by antibodies. Int J Parasitol 37 1391 1399
8. RadwanskaMMagezSMichelAStijlemansBGeuskensM 2000 Comparative analysis of antibody responses against HSP60, invariant surface glycoprotein 70, and variant surface glycoprotein reveals a complex antigen specific pattern of immunoglobulin isotype switching during infection by Trypanosoma brucei. Infect Immun 68 848 860
9. RadwanskaMGuirnaldaPDe TrezCRyffelBBlackSJ 2008 Trypanosomiasis-induced B cell apoptosis results in loss of protective anti-parasite antibody responses and abolishment of vaccine-induced memory responses. PLoS Pathog 4 e1000078 doi:10.1371/journal.ppat.1000078
10. CorsiniACClaytonCAskonasBAOgilvieBM 1977 Suppressor cells and loss of B-cell potential in mice infected with Trypanosoma brucei. Clin Exp Immunol 29 122 131
11. Mayor-WitheyKSClaytonCERoelantsGEAskonasBA 1978 Trypanosomiasis leads to extensive proliferation of B, T and null cells in spleen and bone marrow. Clin Exp Immunol 34 359 363
12. DiffleyP 1983 Trypanosomal surface coat variant antigen causes polyclonal lymphocyte activation. J Immunol 131 1983 1986
13. ClaytonCESelkikrMECorsiniCAOgilvieBMAskonasBA 1980 Murine trypanosomiasis: cellular proliferation and functional depletion in the blood, peritoneum, and spleen related to changes in bone marrow stem cells. Infect Immun 28 824 831
14. MaQJonesDSpringerTA 1999 The chemokine receptor CXCR4 is required for the retention of B lineage and granulocytic precursors within the bone marrow microenvironment. Immunity 10 463 471
15. NagasawaT 2006 Microenvironmental niches in the bone marrow required for B cell development. Nat Rev Immunol 6 107 116
16. KondoMSchererDCMiyamotoTKingAGAkashiK 2000 Cell-fate conversion of lymphoid-commited progenitors by instructive actions of cytokines. Nature 407 383 386
17. Montecino-RodriguezEDorshkindK 2003 To T or not to T: reassessing the common lymphoid progenitor. Nat Immunol 4 100 101
18. VitettaESUhrJW 1975 Immunoglobulin receptors revised. Science 189 964 969
19. AbneyERCooperMDKearneyJFLawtonARParkhouseRME 1978 Sequential expression of immunoglobulin on developing mouse B lymphocytes: a systematic survey that suggests a model for the generation of immunoglobulin isotype diversity. J. Immunol. 120 2041 2049
20. UedaYYangKFosterSJKondoMKelsoeG 2004 Inflammation controls B lymphopoiesis by regulating chemokine CXCL12 expression. J Exp Med 199 47 57
21. CainDKondoMChenHKelsoeG 2009 Effects of acute and chronic inflammation on B-cell development and differentiation. J Invest Dermatol 129 266 277
22. SrivastavaBLindsleyRCNikbakhtNAllmanD 2005 Models for peripheral B cell development and homeostasis. Semin Immunol 17 175 182
23. TeagueBNPanYMuddPANakkenBZhangQ 2007 Cutting edge: Transitional T3 B cells do not give rise to mature B cells, have undergone selection, and are reduced in murine lupus. J Immunol 178 7511 7515
24. Labrie JE3rdSahAPAllmanDMCancroMPGersteinRM 2004 Bone marrow microenvironmental changes underlie reduced RAG-mediated recombination and B cell generation in aged mice. J Exp Med 200 411 423
25. AllmanDLindsleyRCDeMuthWRuddKShintonSA 2001 Resolution of three nonproliferative immature splenic B cell subsets reveals multiple selection points during peripheral B cell maturation. J Immunol 167 6834 40
26. GorelikLCutlerAHThillGMiklaszSDSheaDE 2004 Cutting Edge: BAFF regulates CD21/35 and CD23 expression independent of its B cell survival function. J Immunol 172 762 766
27. ZunigaEMotranCCMontesCLYagitaHGruppiA 2002 Trypanosoma cruzi infection selectively renders parasite-specific IgG+ B lymphocytes susceptible to Fas/Fas ligand-mediated fratricide. J Immunol 168 3965 3973
28. ZunigaEMotranCCMontesCLYagitaHGruppiA 2002 Trypanosoma cruzi infection selectively renders parasite-specific IgG+ B lymphocytes susceptible to Fas/Fas ligand-mediated fratricide. J Immunol 168 3965 3973
29. FigarellaKRawerMUzcateguiNLKubataBKLauberK 2005 Prostaglandin D2 induced programmed cell death in Trypanosoma brucei bloodstream form. Cell Death Differ 12 335 346
30. MonroeJGBannishGFuentes-PananaEMKingLBSandelPC 2003 Positive and negative selection during B cell development. Immunologic research 27 427 442
31. MelamedDBenschopRJCambierJCNemazeeD 1998 Developmental regulation of B lymphocyte immune tolerance compartementalizes clonal selection from receptor selection. Cell 92 173 182
32. SaterRASandelPCMonroeJG 1998 B cell receptor-induced apoptosis in primary transitional murine B cells: signaling requirements and modulation by T cell help. Int Immunol 10 1673 1682
33. Castanos-VelezEMaerlanSOsorioLMBiberfeldPOmARottenbergME 1998 Trypanosoma cruzi infection in tumor receptor p55-deficient mice. Infect. Immun. 66 2960 2968
34. NagataS 1997 Apoptosis by death factor. Cell 88 355 365
35. KrammerPH 2000 CD95's deadly mission in the immune system. Nature 407 789 795
36. AshkenaziADixitVM 1999 Apoptosis control by death and decoy receptors. Curr Opin Cell Biol 11 255 260
37. MicheauOTschoppJ 2003 Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114 181 190
38. MagezSTruyensCMerimiMRadwanskaMStijlemansB 2004 P75 tumor necrosis factor-receptor shedding occurs as a protective host response during African trypanosomiasis. J Infect Dis 189 527 539
39. DrennanMBStijlemansBVan den AbbeeleJQuesniauxVJBarkhuizenM 2005 The induction of a type 1 immune response following a Trypanosoma brucei infection is MyD88 dependent. J Immunol 175 2501 2509
40. LucasRMagezSSongaBDarjiAHamersR 1993 A role for TNF during African trypanosomiasis: involvement in parasite control, immunosuppression and pathology. Res Immunol 144 370 376
41. MagezSRadwanskaMBeschinASekikawaKDe BaetselierP 1999 Tumor necrosis factor alpha is a key mediator in the regulation of experimental Trypanosoma brucei infections. Infect Immun 67 3128 3132
42. MagezSStijlemansBBaralTDe BaetselierP 2002 VSG-GPI anchors of African trypanosomes: their role in macrophage activation and induction of infection-associated immunopathology. Microbes Infect 4 999 1006
43. MoriTAndoKTanakaKIkedaYKogaY 1997 Fas-mediated apoptosis of hematopoietic progenitor cells in mice infected with murine cytomegalovirus. Blood 89 3565 3573
44. CohenPLEisenbergRA 1991 Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annu Rev Immunol 9 243 269
45. PasqualettoVVasseurFZavalaFSchneiderEEzineS 2005 Fas receptor signaling is requisite for B cell differentiation. J Leukoc Biol 78 1106 1117
46. TakahashiYOhtaHTakemoriT 2001 Fas is required for colonal selection in germinal centers and the subsequent establishment of the memory B cell repertoire. Immunity 14 181 192
47. TannerJEAlfieriC 1999 Epstein-Barr virus induces Fas (CD95) in Tcells and Fas ligand in B cells leading to T-cell apoptosis. Blood 94 3439 3447
48. HahneMRennoTSchroeterMIrmlerMFrenchL 1996 Activated B cells express functional Fas ligand. Eur J Immunol 26 721 724
49. RadwanskaMBockstalVBrombacherFBlackSMagezS 2010 Parasite-induced B-cell apoptosis results in loss of specific protective anti-trypanosome antibody responses, and abolishment of vaccine induced protective memory responses. XII International Congress of Parasitology – ICOPA.In press
50. StijlemansBConrathKCortez-RetamozoVVan XongHWynsL 2004 Efficient targeting of conserved cryptic epitopes of infectious agents by single domain antibodies. African trypanosomes as paradigm. J Biol Chem 279 1256 1261
51. Van MeirvenneNMagnusEBusherP 1995 Evaluation of variant specific trypanolysis tests for serodiagnosis of human infections with Trypanosoma brucei gambiense. Acta Trop 60 189 199
52. BlahaMDLeonLR 2008 Effects of indomethacin and buprenorphine analgesia on the postoperative recovery of mice. J Am Assoc Lab Anim Sci 47 8 19
53. MontineKSMontineTJMorrowJDFreiBMilatovicD 2002 Mouse cerebral prostaglandins, but not oxidative damage, change with age and are responsive to indomethacin treatment. Brain Res 930 75 82
54. SchleiferKWMansfieldJM 1993 Suppressor macrophages in African Trypanosomiasis inhibit T cell proliferative responses by nitric oxide and prostaglandins. J Immunol 151 5492 5503
55. SilvermanGJGoodyearCS 2006 Confounding B-cell defenses: lessons from a staphylococcal superantigen. Nat Rev Immunol 6 465 475
56. GoodyearCSCorrMSugiyamaFBoyleDLSilvermanGJ 2007 Cutting Edge: Bim is required for superantigen-mediated B cell death. J Immunol 178 2636 2640
57. PetroJBGersteinRMLoweJCarterRSShinnersN 2002 Transitional type 1 and 2 B lymphocyte subsets are differentially responsive to antigen receptor signaling. J Biol Chem 277 48009 48019
58. SuTTRawlingsDJ 2002 Transitional B lymphocyte subsets operate as distinct checkpoints in murine splenic B cell development. J Immunol 168 2101 2110
59. YoshidaTHiguchiTHagiyamaHStrasserANishiokaK 2000 Rapid B cell apoptosis induced by antigen receptor ligation does not require Fas (CD95/Apo-1), the adaptor protein FADD/MORT1 or CrmA-sensitive caspases but is defective in both MRL-+/+ and MRL-lpr/lpr mice. Int Immunol 12 517 526
60. De GroeveKDeschachtNDe KoninckCCaveliersVLahoutteT 2010 Nanobodies as tools for in vivo imaging of specific immune cell types. J Nucl Med 51 782 789
61. MagezSSchwegmannAAtkinsonRClaesFDrennanM 2008 The role of B cells and IgM antibodies in parasitemia, anemia and VSG switching in Trypanosoma brucei-infected mice. PLoS Pathog 4 e1000122
62. MagezSRadwanskaM 2009 African trypanosomiasis and antibodies: implications for vaccination, therapy and diagnosis. Future Microbiol 4 1075 1087
63. LanhamSMGodfreyDG 1970 Isolation of salivarian trypanosomes from man and other mammals using DEAE-cellulose. Exp Parasitol 28 521 534
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 6
- Farmakovigilanční studie perorálních antivirotik indikovaných v léčbě COVID-19
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Měli bychom postcovidový syndrom léčit antidepresivy?
- 10 bodů k očkování proti COVID-19: stanovisko České společnosti alergologie a klinické imunologie ČLS JEP
Nejčtenější v tomto čísle
- High Affinity Nanobodies against the VSG Are Potent Trypanolytic Agents that Block Endocytosis
- Structural and Mechanistic Studies of Measles Virus Illuminate Paramyxovirus Entry
- Sporangiospore Size Dimorphism Is Linked to Virulence of
- The Binding of Triclosan to SmeT, the Repressor of the Multidrug Efflux Pump SmeDEF, Induces Antibiotic Resistance in