
FUS Transgenic Rats Develop the Phenotypes of Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration
Fused in Sarcoma (FUS) proteinopathy is a feature of frontotemporal lobar dementia (FTLD), and mutation of the fus gene segregates with FTLD and amyotrophic lateral sclerosis (ALS). To study the consequences of mutation in the fus gene, we created transgenic rats expressing the human fus gene with or without mutation. Overexpression of a mutant (R521C substitution), but not normal, human FUS induced progressive paralysis resembling ALS. Mutant FUS transgenic rats developed progressive paralysis secondary to degeneration of motor axons and displayed a substantial loss of neurons in the cortex and hippocampus. This neuronal loss was accompanied by ubiquitin aggregation and glial reaction. While transgenic rats that overexpressed the wild-type human FUS were asymptomatic at young ages, they showed a deficit in spatial learning and memory and a significant loss of cortical and hippocampal neurons at advanced ages. These results suggest that mutant FUS is more toxic to neurons than normal FUS and that increased expression of normal FUS is sufficient to induce neuron death. Our FUS transgenic rats reproduced some phenotypes of ALS and FTLD and will provide a useful model for mechanistic studies of FUS–related diseases.
Published in the journal:
. PLoS Genet 7(3): e32767. doi:10.1371/journal.pgen.1002011
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002011
Summary
Fused in Sarcoma (FUS) proteinopathy is a feature of frontotemporal lobar dementia (FTLD), and mutation of the fus gene segregates with FTLD and amyotrophic lateral sclerosis (ALS). To study the consequences of mutation in the fus gene, we created transgenic rats expressing the human fus gene with or without mutation. Overexpression of a mutant (R521C substitution), but not normal, human FUS induced progressive paralysis resembling ALS. Mutant FUS transgenic rats developed progressive paralysis secondary to degeneration of motor axons and displayed a substantial loss of neurons in the cortex and hippocampus. This neuronal loss was accompanied by ubiquitin aggregation and glial reaction. While transgenic rats that overexpressed the wild-type human FUS were asymptomatic at young ages, they showed a deficit in spatial learning and memory and a significant loss of cortical and hippocampal neurons at advanced ages. These results suggest that mutant FUS is more toxic to neurons than normal FUS and that increased expression of normal FUS is sufficient to induce neuron death. Our FUS transgenic rats reproduced some phenotypes of ALS and FTLD and will provide a useful model for mechanistic studies of FUS–related diseases.
Introduction
Amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD) are two common neurodegenerative diseases [1], [2]. ALS is characterized by degeneration of motor neurons, denervation atrophy of skeletal muscles, and progressive paralysis of limbs [3], [4]. FTLD mainly affects cortical neurons and causes cortical dementia [5]. ALS patients may develop cortical dementia that overlaps with FTLD in pathology [2], [6]. ALS and FTLD share a common feature of pathology—ubiquitin-positive inclusion [7]–[10]. Although selective groups of neurons are primarily affected in each disease condition [2], increasing evidence suggests that ALS and FTLD may fall the same disease spectrum.
Fused in Sarcoma (FUS) has recently been linked to both ALS and FTLD [11], [12]. FUS is a highly conserved ribonucleoprotein that mainly resides in the nucleus while shuttling between the cytoplasm and the nucleus [13]–[15]. Fus was initially reported to translocate and fuse with one of several genes to form chimeric oncogenes in leukemia and liposarcoma [16], [17]. The N-terminus of the FUS protein is rich in glutamine, serine, and tyrosine residues, and may be responsible for transactivation activity of FUS oncogenic fusion [18], [19]. The C-terminal part of the FUS protein contains several structural motifs important for nucleic acid binding [18], [20], [21]. FUS may also play an important role in regulating mRNA [14], [22], [23]. Deletion of the fus gene results in chromosomal instability and perinatal death in inbred mice [24], but causes only male sterility in outbred mice [25]. FUS-positive inclusion is considered a hallmark of some sporadic FTLD [9], [26]. FUS, Tau, and TDP-43 are the important components of ubiquitinated proteins in FTLD, but exclude one another in ubiquitin-positive inclusion [8]–[10], [27]. Mutations in the fus gene segregate with ALS and FTLD [11], [12], [28], [29], implying a pathogenic role of FUS in these diseases.
Given the importance of FUS in human diseases, the consequences of mutation in the fus gene must be examined. Here we show that overexpression of a mutant, but not normal, human FUS in rats induced progressive paralysis resembling ALS. Mutant FUS transgenic rats developed severe axonopathy of motor neurons, denervation atrophy of skeletal muscles, and a substantial loss of cortical and hippocampal neurons. At advanced ages, normal FUS transgenic rats displayed deficits in spatial learning and memory, and a loss of cortical and hippocampal neurons. Neuronal loss was accompanied by ubiquitin aggregation and glial reaction. Our FUS transgenic rats recapitulated some features of ALS and FTLD.
Results
Overexpression of a mutant, but not normal, human fus gene causes progressive paralysis in transgenic rats
To study the consequences of mutation in the fus gene, we created transgenic rats expressing the human fus gene with or without mutation (Table S1). Most mutations in the fus gene are a single amino acid alteration, as exemplified by the substitution of arginine for cysteine at residue 521 (R521C) that is identified in geographically unrelated patients [11], [12], [30]. We therefore chose R521C as an example of fus mutation for our transgenic studies. The ribonucleoproteins FUS and TDP-43 are both linked to ALS and FTLD [11], . FUS and TDP-43 are robustly and ubiquitously expressed in rodents during development [33], implying an important role for these genes in development. Constitutive expression of a mutant TDP-43 causes early death to transgenic founder rats [34], preventing transgenic lines from establishment. To overcome this potential difficulty, we used a tetracycline-inducible system to express human fus transgenes in a controlled manner [34]. From 26 transgenic founders carrying the normal (12 rats) or the mutant (14 rats) fus transgene, we established four transgenic lines (line number corresponding to copy number of the transgenes) that expressed human FUS, under tight control by Doxycycline (Dox), at substantial levels (Figure 1A, Figure S1, and Table S1). FUS transgenic rats were crossed with a CAG-tTA transgenic line to produce double transgenic offspring that expressed human FUS transgene in the absence of Dox [35]. Breeding female rats were given Dox in their drinking water until delivery such that expression of the fus transgenes would be recovered in the offspring after Dox withdrawal (Figures S1 and S2).

Immunoreactivity to human FUS was detected in the brain and spinal cord (gray and white matter) of FUS transgenic rats (Figure 1B, 1D, and 1E), but not in tissues of nontransgenic rats (Figure 1C and 1F). While transgenic rats of lines 16, 20, and 22 expressed human FUS at comparable levels (Figure 1A), only the mutant FUS transgenic rats (lines 16 and 22) developed paralysis resembling ALS (Figure 1G-1J and Videos S1 and S2). Similar disease phenotypes were observed in two independent lines of mutant FUS transgenic rats (Figure 1G–1J), suggesting that the disease phenotypes resulted from expression of the mutant fus gene.
Axonopathy of the motor neurons contributes to paralysis in mutant FUS transgenic rats
Pathological analysis revealed that few motor neurons in the spinal cord were undergoing degeneration (Figure 2A–2E). Degenerating axons were detected in the dorsal corticospinal tracts (Figure 2G), the ventral roots (Figure 2I and 2M), and the dorsal roots (Figure 2K) of mutant FUS transgenic rats at paralysis stages. As a result of motor axon degeneration, groups of skeletal muscle cells were atrophied (Figure 2O), although there were some perimysial cells with small nuclei suggestive of inflammation. These pathological changes were not observed in nontransgenic rats (Figure 2A) and also not observed in age-matched, wild-type FUS transgenic rats (Figure 2B, 2D, 2F, 2H, 2J, 2L, and 2N) expressing human FUS at comparable levels (Figure 1A). Collectively, these findings suggest that mutation of the fus gene is pathogenic. Electromyography of the gastrocnemius muscle revealed fibrillation potential, a characteristic of denervation atrophy (Figure 2P). Confocal microscopy showed that a substantial number of neuromuscular junctions were denervated in paralyzed FUS transgenic rats (Figure 2Q and 2R). Through stereological cell counting, we estimated the number of spinal motor neurons and did not detect a significant loss of motor neurons, although a trend of neuron loss was observed in the mutant FUS rats at paralysis stages (Figure 2S). Our results suggest that degeneration of motor axons contributed to paralysis in the mutant FUS transgenic rats.
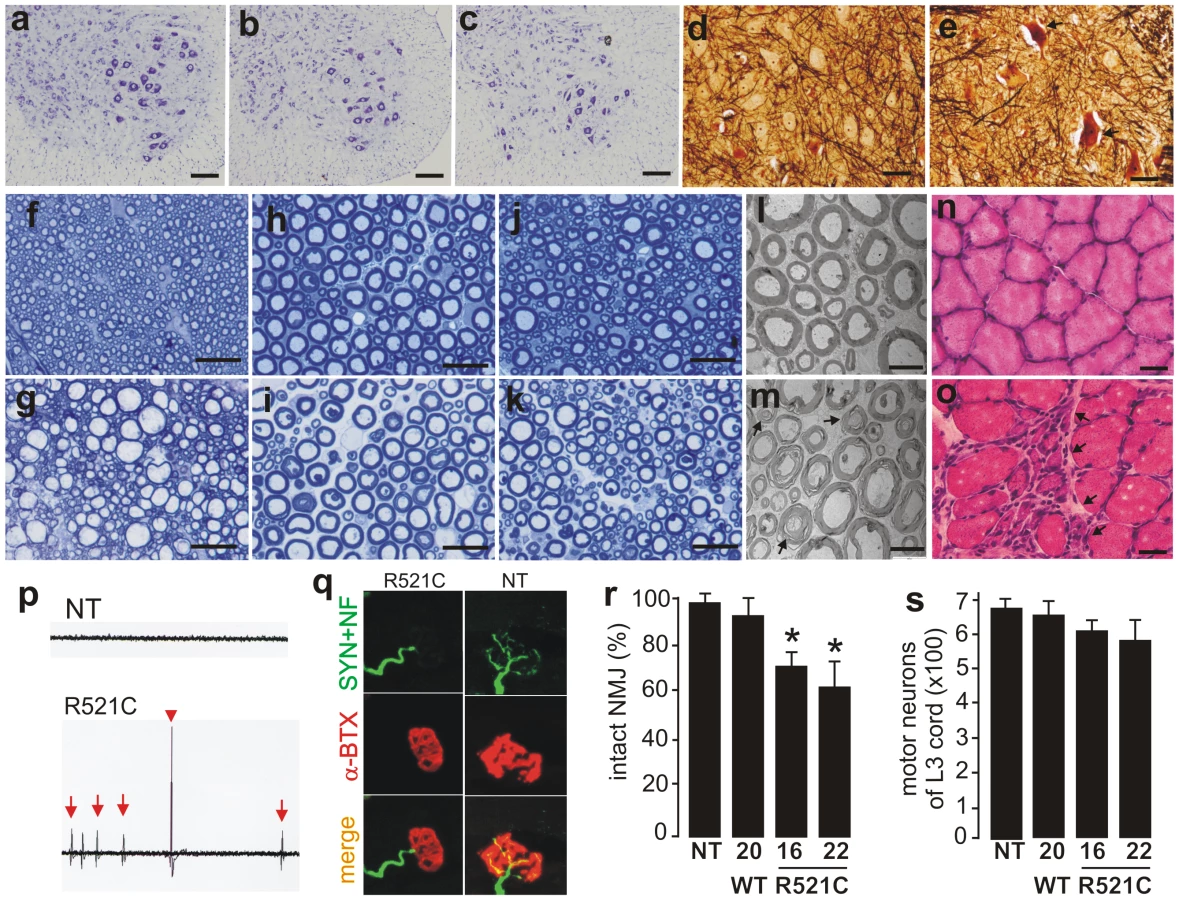
Overexpression of mutant human FUS causes a substantial loss of neurons in the brains of FUS transgenic rats
ALS and FTLD somewhat overlap in pathology [2], and mutation of the fus gene is linked to both ALS and FTLD [28], [29]. We therefore examined the pathology in the brains of mutant FUS transgenic rats. Through stereological cell counting (Figure S3), we detected a significant loss of neurons in the frontal cortex and dentate gyrus of mutant FUS transgenic rats at paralysis stages (Figure 3). This neuronal loss was not observed in age-matched, normal FUS transgenic rats of line 20, although they expressed human FUS at comparable levels (Figure 1A and Figure 3). While cortical neurons are the primary targets of degeneration in FTLD, hippocampal neurons could be affected particularly at advanced disease stages [36], [37]. Our results show that overexpression of mutant FUS induced a substantial loss of cortical and hippocampal neurons in FUS transgenic rats, a phenotype of FTLD in rat models.

Overexpression of normal FUS is sufficient to induce neurodegeneration in transgenic rats
FUS proteinopathy is a hallmark of some sporadic FTLD cases [9], [26]. How normal FUS is related to neurodegeneration in the disease remains to be examined. Our wild-type (line 20) and mutant (line 16) FUS transgenic rats expressed human FUS at comparable levels (Figure 1A), but only the mutant FUS transgenic rats developed paralysis at an early age (Figure 1G–1I). We further examined the normal FUS transgenic rats at advanced ages (Figure 4). Although the normal FUS transgenic rats were asymptomatic by the age of 1 year, they displayed a deficit in spatial learning and memory at the advanced age (Figure 4J and 4K). By stereological cell counting, we detected a moderate, but significant, loss of neurons in the frontal cortex and dentate gyrus of the normal FUS transgenic rats at advanced ages (Figure 4L and 4M). These findings suggest that increased expression of normal FUS is sufficient to induce neurodegeneration and that mutant FUS is more toxic to neurons than is normal FUS.

Neuron death is accompanied by ubiquitin aggregation and glial reaction
Ubiquitin-positive inclusion is a hallmark of ALS and FTLD [8]–[27]. Accumulated ubiquitin was detected in the cortex (Figure 5D–5F) and spinal cord (Figure 5J–5L) of mutant FUS transgenic rats at paralysis stages, but was not detected in the tissues of age-matched normal FUS transgenic rats (Figure 5A–5C and 5G–5I). In the normal FUS transgenic rats, ubiquitin aggregates were observed only when neuronal loss was detected at advanced ages (Figure 4), suggesting that ubiquitin aggregation accompanied neurodegeneration. Ubiquitin inclusions were detected only in FUS-expressing cells, but were not colocalized with FUS (Figure 4G–4I and Figure 5). Ubiquitinated aggregates were positive for the mitochondrial marker COXIV (Figure S4), suggesting that damaged mitochondria may be ubiquitinated for degradation. No typical FUS inclusion was detected in FUS transgenic rats (Figure 1B and 1E, and Figure 5). FUS mainly resided in the nucleus, but was also diffusely located in the cytoplasm (Figure 1E). The C-terminus of FUS contains a nuclear localization signal that is necessary for the nuclear import of FUS. Most mutations occur within the C-terminus of FUS and disrupt this nuclear localization signal [38], leading to cytoplasmic accumulation of FUS. The R521C mutation tested in our transgenic studies affects FUS distribution to a minimal extent [38], and may be less potent in eliciting redistribution and aggregation of FUS in transgenic rats. Glial cells are key players in neurodegeneration [39]. Here we found that astrocytes and microglia proliferated in the brain (Figure 6A–6F) and spinal cord (Figure 6H–6K) of FUS transgenic rats at paralysis stages. Our results indicate that neurodegeneration was accompanied by ubiquitin aggregation and glial reaction.


Discussion
ALS and FTLD are two related neurodegenerative diseases [2], [6] and may fall within the same disease spectrum. While a subset of FTLD patients develop motor neuron disease [40], ALS patients may develop the symptoms and pathology of FTLD [41]–[43]. FUS and TDP-43 are two ribonucleoproteins and their mutant forms are linked to both ALS and FTLD [7]–[29]. We obtained two FUS transgenic lines expressing a mutant or normal human fus transgene at comparable levels. Transgenic rats expressing a mutant FUS developed progressive paralysis secondary to axonal degeneration and displayed a substantial loss of neurons in the cortex and hippocampus, reproducing some phenotypes of ALS and FTLD. While the mutant FUS transgenic rats developed some phenotypes of ALS and FTLD, the age-matched normal FUS transgenic rats were asymptomatic. Our findings in FUS transgenic rats confirm that mutation of the fus gene is related to these two diseases and suggest that mutation of the fus gene is pathogenic.
FUS proteinopathy characterizes a subset of sporadic FTLD, in which ubiquitin-positive inclusions are negative for TDP-43 and tau but positive for FUS protein [27], [44]. However, it is not known how normal FUS is related to neurodegeneration in these diseases. While overexpression of mutant FUS induced severe phenotypes in young animals, overexpression of the normal FUS also induced neuron death as well as learning and memory deficits in aged rats. Mutated FUS appeared more toxic in transgenic rats, but an increase in the expression or function of the fus gene may elicit neurotoxicity. The effects of gene mutation include gain-of-function, loss-of-function, and dominant-negative effects. Overexpression of either the mutant or wild-type FUS induced disease phenotypes in transgenic rats, suggesting that mutation of the fus gene may cause the disease by a gain of toxic properties. Since gain-of-function and dominant-negative mutations can induce similar effects in transgenic models, more sophisticated genetic approaches, such as gene knockin, may be required for determining the nature of FUS mutations.
FUS and TDP-43 show a similarity in disease induction. Mutant forms of these genes are more toxic than the normal genes [34], and increased expression of the normal genes is sufficient to induce neurodegeneration [45], [46]. Both FUS and TDP-43 are ribonucleoproteins and may have overlapping functions. Indeed, FUS and TDP-43 are found in one protein complex regulating HDAC6 mRNA [47]. Like TDP-43, FUS predominantly resides in the nucleus, but also shuttles between the nucleus and the cytoplasm to perform multiple functions [13]. Similar to results for TDP-43 [34], we found that FUS was diffusely located in the cytoplasm in transgenic rats. Possibly, redistribution of FUS may alter the functions of this multifunctional protein, incurring cellular toxicity.
In summary, our results suggest that mutant FUS is more toxic to neurons than normal FUS and that increased expression of normal FUS is sufficient to induce neuron death. Our FUS transgenic rats reproduced some phenotypes of ALS and FTLD. The establishment of these FUS transgenic rat lines will allow for more detailed studies of FUS-related diseases.
Material and Methods
Ethics statement
Animal use followed NIH guidelines. The animal use protocol was approved by the Institutional Animal Care and Use Committees (IACUC) at Thomas Jefferson University.
Transgenic rat production
The open reading frame (ORF) of the normal human fus gene was PCR-amplified from a human cDNA pool (Invitrogen) and the mutation was introduced by site-directed mutagenesis (Stratagene). The normal and mutant human FUS ORF were inserted downstream of the TRE promoter as described previously [34]. Linearized transgenic DNA was purified from agar gel and injected into the pronuclei of fertilized eggs of Sprague-Dawley (SD) rats to produce transgenic founder rats [34], [35]. Transgenes were maintained on the SD genomic background and were identified by PCR analysis of rat's tail DNA.
Animal behavior tests
Grip strength of the rat's fore and hind paws was measured twice a week (Columbus Instruments) and used for determining disease onset and progression. Disease onset was defined as an unrecoverable reduction in the grip strength of fore or hind paws. Disease end-stage was defined as paralysis in two or more legs or as a 30% reduction in body weight.
Spatial learning and memory tasks were examined with a Barnes Maze (Med Associates). Compared to Morris Water Maze or Radial Arm Maze, the Barnes Maze not only avoids dietary restriction and intense stress, but also gives comparable results on rodent's spatial learning and memory tasks [48], [49]. The Barnes Maze consists of a white, acrylic, circular disk (122 cm diameter) with 18 holes (9.5 cm diameter) spaced every 20° and a high stand (140 cm height) supporting the disk that is designed to discourage animals from jumping to the floor. Rats were given one training session and four test sessions for 5 consecutive days. During training or testing sessions, rats were placed in the same initial orientation inside a transparent cylinder (start box) that was located at the center of the maze disk and the rats remained in the start box for 1 minute such that a standard starting context was ensured. When a lamp above the maze was turned on to make the surface of the maze aversive, the start box was removed to allow the animal to escape the maze surface by locating and crawling through the correct hole under which a black safe box was located. When the animal entered the safe box, the light was turned off and the safe box was covered with a black sheet. The animal was allowed to stay in the safe box for 1 minute before it was placed back to its home cage. Before training, each rat was given 2 minutes to explore the maze and then placed inside the safe box for 1 minute for habituation. During training, each rat was guided to the safe box twice and then given two trials to locate the safe box by itself. During the test, rats were placed inside the start box for 1 minute to locate the fixed safe box. The number of incorrect hole pokes (error) and the latency to locate the safe box were recorded. An incorrect hole poke was indicated when an animal closely approached and visually inspected a wrong hole. Latency to locate the safe box was calculated from the time testing started to the time when the animal entered, or its four paws touched, the safe box. The maze was wiped clean with 70% ethanol and then with dry paper towel after each test to prevent animals following odor trails.
Antibody production
An antibody to human FUS was produced by immunizing rabbits with a synthetic peptide (Genemed): (N-terminal)-SYGQPQSGSYSQQPS. Antiserum was affinity-purified with a peptide-affinity column (Pierce).
Histology and immunostaining
Anesthetized rats were transcardially perfused with 4% paraformaldehyde (PFA) dissolved in 1X PBS buffer and tissues were dissected after perfusion. Tissues were cryopreserved in 40% sucrose and cut into sections on a Cryostat. Tissue sections of 12 µm were immunostained with the following antibodies: rabbit polyclonal antibody to human FUS (made in-house), chicken antibody to ubiquitin (Sigma), mouse monoclonal antibodies against Iba-1 (Wako Chemical) or GFAP (Millipore), and mouse monoclonal antibody against NeuN (Millipore). For histochemistry, immunostained sections were visualized with an ABC kit in combination with diaminobenzidine (Vector) and counterstained with hematoxylin to display nuclei. For immunofluorescent staining, tissue sections were incubated first with specific primary antibodies and then with secondary antibodies labeled with fluorescent dyes (Jackson Immunoresearch). Primary antibodies were diluted at 1∶1000 and secondary antibodies diluted at 1∶200. The primary antibodies were incubated overnight at 4°C and the secondary antibodies were incubated for 2 hours at room temperature. For detection of degenerating neurons, paraffin-embedded spinal cords were cut into transverse sections of 10 µm and stained using a protocol for Bielschowsky silver staining [34].
As described in a previous publication [34], neuromuscular junctions (NMJ) were visualized by immunofluorescent staining and confocal microscopy. PFA-fixed gastrocnemius muscles were cut into sections of 100 µm on a Cryostat. Muscle sections were incubated with α-bungarotoxin (Invitrogen) for 30 minutes at room temperature and subsequently immunostained with mouse monoclonal antibodies to neurofilament (Sigma) and synaptophysin (Millipore). Both primary and secondary antibodies were diluted at 1∶1000. The primary antibodies were incubated overnight at 4°C and the secondary antibodies were incubated for 2 hours at room temperature. NMJ images were captured with a Zeiss LSM510 META confocal system and the NMJ was reconstructed through z-stack projections from serial scanning every 1 µm.
Toluidine blue staining and electron microscopy
As described previously [34], anesthetized rats were perfused with a mixture of 4% PFA plus 2% glutaraldehyde. Cervical spinal cords and L3 ventral and dorsal roots were dissected and post-fixed in the same fixative at 4°C overnight. Fixed tissues were embedded in Epon 812 (Electron Microscopic Sciences, PA) and cut into semithin and thin sections. Semithin sections (1 µm) were stained with 1% toluidine blue and visualized under a light microscope. Thin sections (80 nm) were stained with uranyl acetate and lead citrate and observed under a transmission electron microscope (Hitachi H7500-I).
Electromyography (EMG)
Anesthetized rats were examined by EMG. Fibrillation and fasciculation potentials of gastrocnemius muscles were recorded with an EMG machine (CMS6600; COTEC Inc.) as previously described [34].
Stereological cell counting
Motor neurons in the ventral horn of the L3 lumbar cord were stereologically counted as previously described [34]. Neurons larger than 25 µm in diameter were counted in the ventral horns on both sides. For estimation of neurons in the frontal cortex and dentate gyrus, one hemisphere of the brain was used for cell counting. The forebrain was cut into coronal sections of 30 µm between the apical rostral part of the brain and the first occurrence of hippocampus, and every 12th section (a total of 15 to 18 sections) was counted for neurons in the defined frontal cortex (Figure S3). The portion of the brain containing the dentate gyrus was cut into consecutive sections (20 µm) and every 12th section (a total of 16 to 21 sections) was counted for neurons in the dentate gyrus. Tissue sections were stained with Cresyl violet and mounted in sequential order (rostral-caudal). The number of targeted neurons was estimated using a fractionator-based unbiased stereology software program (Stereologer) run on a PC computer that was attached to a Nikon 80i microscope fitted with a motorized XYZ stage (Prior). At low magnification, the targeting area was outlined and a random sampling grid was created. At high magnification, an optical dissector probe in the designated area was randomly generated by the program. The presence of clearly definable neurons was noted according to defined inclusion and exclusion limits of the dissector. This process was repeated on all selected sections. The total number of defined neurons was calculated by the software according to the result of random counts as previously described [34].
Statistical analysis
The number of defined neurons in the defined region was statistically compared between groups of transgenic rats and comparison among experimental groups was performed by one-way ANOVA followed by Tukey's post-hoc test. The null hypothesis was rejected at the level of 0.05.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. WangJSluntHGonzalesVFromholtDCoonfieldM 2003 Copper-binding-site-null SOD1 causes ALS in transgenic mice: aggregates of non-native SOD1 delineate a common feature. Hum Mol Genet 12 2753 2764
2. LilloPHodgesJR 2009 Frontotemporal dementia and motor neurone disease: Overlapping clinic-pathological disorders. Journal of Clinical Neuroscience 16 1131 1135
3. YamanakaKBoilleeSRobertsEAGarciaMLMcAlonis-DownesM 2008 Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proc Natl Acad Sci U S A 105 7594 7599
4. BoilleeS 2006 Onset and Progression in Inherited ALS Determined by Motor Neurons and Microglia. Science 312 1389 1392
5. FormanMSFarmerJJohnsonJKClarkCMArnoldSE 2006 Frontotemporal dementia: Clinicopathological correlations. Annals of Neurology 59 952 962
6. BenajibaLLe BerICamuzatALacosteMThomas-AnterionC 2009 TARDBPmutations in motoneuron disease with frontotemporal lobar degeneration. Annals of Neurology 65 470 473
7. DengHXZhaiHBigioEHYanJFectoF 2010 FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann Neurol 67 739 748
8. NeumannMSampathuDMKwongLKTruaxACMicsenyiMC 2006 Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314 130 133
9. UrwinHJosephsKARohrerJDMackenzieIRNeumannM 2010 FUS pathology defines the majority of tau - and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathologica 120 33 41
10. Van DammePRobberechtW 2009 Recent advances in motor neuron disease. Current Opinion in Neurology 22 486 492
11. KwiatkowskiTJJrBoscoDALeclercALTamrazianEVanderburgCR 2009 Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323 1205 1208
12. VanceCRogeljBHortobagyiTDe VosKJNishimuraAL 2009 Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323 1208 1211
13. FujiiROkabeSUrushidoTInoueKYoshimuraA 2005 The RNA Binding Protein TLS Is Translocated to Dendritic Spines by mGluR5 Activation and Regulates Spine Morphology. Current Biology 15 587 593
14. FujiiR 2005 TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. Journal of Cell Science 118 5755 5765
15. WangXAraiSSongXReichartDDuK 2008 Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 454 126 130
16. Perez-LosadaJPintadoBGutierrez-AdanAFloresTBanares-GonzalezB 2000 The chimeric FUS/TLS-CHOP fusion protein specifically induces liposarcomas in transgenic mice. Oncogene 19 2413 2422
17. RabbittsTHForsterALarsonRNathanP 1993 Fusion of the dominant negative transcription regulator CHOP with a novel gene FUS by translocation t(12;16) in malignant liposarcoma. Nat Genet 4 175 180
18. ZinsznerHImmanuelDYinYLiangFXRonD 1997 A topogenic role for the oncogenic N-terminus of TLS: nucleolar localization when transcription is inhibited. Oncogene 14 451 461
19. CrozatAAmanPMandahlNRonD 1993 Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 363 640 644
20. YangLEmbreeLJTsaiSHicksteinDD 1998 Oncoprotein TLS interacts with serine-arginine proteins involved in RNA splicing. J Biol Chem 273 27761 27764
21. ZinsznerHSokJImmanuelDYinYRonD 1997 TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J Cell Sci 110 1741 1750
22. BellyAMoreaugachelinFSadoulRGoldbergY 2005 Delocalization of the multifunctional RNA splicing factor TLS/FUS in hippocampal neurones: exclusion from the nucleus and accumulation in dendritic granules and spine heads. Neuroscience Letters 379 152 157
23. YoshimuraAFujiiRWatanabeYOkabeSFukuiK 2006 Myosin-Va Facilitates the Accumulation of mRNA/Protein Complex in Dendritic Spines. Current Biology 16 2345 2351
24. HicksGGSinghNNashabiAMaiSBozekG 2000 Fus deficiency in mice results in defective B-lymphocyte development and activation, high levels of chromosomal instability and perinatal death. Nat Genet 24 175 179
25. KurodaMSokJWebbLBaechtoldHUranoF 2000 Male sterility and enhanced radiation sensitivity in TLS(-/-) mice. EMBO J 19 453 462
26. SeelaarHKlijnsmaKYKoningILugtAChiuWZ 2009 Frequency of ubiquitin and FUS-positive, TDP-43-negative frontotemporal lobar degeneration. Journal of Neurology 257 747 753
27. NeumannMRademakersRRoeberSBakerMKretzschmarHA 2009 A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132 : 2922-2931. Epub 2009 Aug 2911
28. Van LangenhoveTvan der ZeeJSleegersKEngelborghsSVandenbergheR 2010 Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology 74 366 371
29. YanJDengHXSiddiqueNFectoFChenW 2010 Frameshift and novel mutations in FUS in familial amyotrophic lateral sclerosis and ALS/dementia. Neurology 75 807 814
30. DrepperCHerrmannTWessigCBeckMSendtnerM 2009 C-terminal FUS/TLS mutations in familial and sporadic ALS in Germany. Neurobiol Aging 15 15
31. SreedharanJBlairIPTripathiVBHuXVanceC 2008 TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319 1668 1672
32. YokosekiAShigaATanCFTagawaAKanekoH 2008 TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol 63 538 542
33. HuangCXiaPYZhouH 2010 Sustained expression of TDP-43 and FUS in motor neurons in rodent's lifetime. Int J Biol Sci 6 396 406
34. ZhouHHuangCChenHWangDLandelCP 2010 transgenic rat model of neurodegeneration caused by mutation in the TDP gene. PLoS Genet 6 e1000887 doi:10.1371/journal.pgen.1000887
35. ZhouHHuangCYangMLandelCPXiaPY 2009 Developing tTA Transgenic Rats for Inducible and Reversible Gene Expression. Int J Biol Sci 2 171 181
36. van de PolLA 2006 Hippocampal atrophy on MRI in frontotemporal lobar degeneration and Alzheimer's disease. Journal of Neurology, Neurosurgery & Psychiatry 77 439 442
37. MackenzieIRAFotiDWoulfeJHurwitzTA 2007 Atypical frontotemporal lobar degeneration with ubiquitin-positive, TDP-43-negative neuronal inclusions. Brain 131 1282 1293
38. DormannDRoddeREdbauerDBentmannEFischerI 2010 ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. The EMBO Journal
39. YangYGozenOVidenskySRobinsonMBRothsteinJD 2010 Epigenetic regulation of neuron-dependent induction of astroglial synaptic protein GLT1. Glia 58 277 286
40. MitsuyamaYInoueT 2009 Clinical entity of frontotemporal dementia with motor neuron disease. Neuropathology 29 649 654
41. NishihiraYTanCFHoshiYIwanagaKYamadaM 2009 Sporadic amyotrophic lateral sclerosis of long duration is associated with relatively mild TDP-43 pathology. Acta Neuropathol 117 45 53
42. MachidaYTsuchiyaKAnnoMHagaCItoT 1999 Sporadic amyotrophic lateral sclerosis with multiple system degeneration: a report of an autopsy case without respirator administration. Acta Neuropathol 98 512 515
43. TsuchiyaKSanoMShiotsuHAkiyamaHWatabikiS 2004 Sporadic amyotrophic lateral sclerosis of long duration mimicking spinal progressive muscular atrophy exists: additional autopsy case with a clinical course of 19 years. Neuropathology 24 228 235
44. MunozDGNeumannMKusakaHYokotaOIshiharaK 2009 FUS pathology in basophilic inclusion body disease. Acta Neuropathologica 118 617 627
45. TsaiKJYangCHFangYHChoKHChienWL 2010 Elevated expression of TDP-43 in the forebrain of mice is sufficient to cause neurological and pathological phenotypes mimicking FTLD-U. Journal of Experimental Medicine 207 1661 1673
46. WilsHKleinbergerGJanssensJPeresonSJorisG 2010 TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proceedings of the National Academy of Sciences 107 3858 3863
47. KimSHShanwareNBowlerMJTibbettsRS 2010 ALS-associated proteins TDP-43 and FUS/TLS function in a common biochemical complex to coregulate HDAC6 mRNA. J Biol Chem
48. HarrisonFEReisererRSTomarkenAJMcDonaldMP 2006 Spatial and nonspatial escape strategies in the Barnes maze. Learn Mem 13 809 819
49. BaytanSHAlkanatMOkuyanMEkinciMGedikliE 2008 Simvastatin impairs spatial memory in rats at a specific dose level. Tohoku J Exp Med 214 341 349
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 3
Nejčtenější v tomto čísle
- Whole-Exome Re-Sequencing in a Family Quartet Identifies Mutations As the Cause of a Novel Skeletal Dysplasia
- Origin-Dependent Inverted-Repeat Amplification: A Replication-Based Model for Generating Palindromic Amplicons
- FUS Transgenic Rats Develop the Phenotypes of Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration
- Limited dCTP Availability Accounts for Mitochondrial DNA Depletion in Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE)