
The Genomes of the Fungal Plant Pathogens and Reveal Adaptation to Different Hosts and Lifestyles But Also Signatures of Common Ancestry
We sequenced and compared the genomes of the Dothideomycete fungal plant pathogens Cladosporium fulvum (Cfu) (syn. Passalora fulva) and Dothistroma septosporum (Dse) that are closely related phylogenetically, but have different lifestyles and hosts. Although both fungi grow extracellularly in close contact with host mesophyll cells, Cfu is a biotroph infecting tomato, while Dse is a hemibiotroph infecting pine. The genomes of these fungi have a similar set of genes (70% of gene content in both genomes are homologs), but differ significantly in size (Cfu >61.1-Mb; Dse 31.2-Mb), which is mainly due to the difference in repeat content (47.2% in Cfu versus 3.2% in Dse). Recent adaptation to different lifestyles and hosts is suggested by diverged sets of genes. Cfu contains an α-tomatinase gene that we predict might be required for detoxification of tomatine, while this gene is absent in Dse. Many genes encoding secreted proteins are unique to each species and the repeat-rich areas in Cfu are enriched for these species-specific genes. In contrast, conserved genes suggest common host ancestry. Homologs of Cfu effector genes, including Ecp2 and Avr4, are present in Dse and induce a Cf-Ecp2 - and Cf-4-mediated hypersensitive response, respectively. Strikingly, genes involved in production of the toxin dothistromin, a likely virulence factor for Dse, are conserved in Cfu, but their expression differs markedly with essentially no expression by Cfu in planta. Likewise, Cfu has a carbohydrate-degrading enzyme catalog that is more similar to that of necrotrophs or hemibiotrophs and a larger pectinolytic gene arsenal than Dse, but many of these genes are not expressed in planta or are pseudogenized. Overall, comparison of their genomes suggests that these closely related plant pathogens had a common ancestral host but since adapted to different hosts and lifestyles by a combination of differentiated gene content, pseudogenization, and gene regulation.
Published in the journal:
. PLoS Genet 8(11): e32767. doi:10.1371/journal.pgen.1003088
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003088
Summary
We sequenced and compared the genomes of the Dothideomycete fungal plant pathogens Cladosporium fulvum (Cfu) (syn. Passalora fulva) and Dothistroma septosporum (Dse) that are closely related phylogenetically, but have different lifestyles and hosts. Although both fungi grow extracellularly in close contact with host mesophyll cells, Cfu is a biotroph infecting tomato, while Dse is a hemibiotroph infecting pine. The genomes of these fungi have a similar set of genes (70% of gene content in both genomes are homologs), but differ significantly in size (Cfu >61.1-Mb; Dse 31.2-Mb), which is mainly due to the difference in repeat content (47.2% in Cfu versus 3.2% in Dse). Recent adaptation to different lifestyles and hosts is suggested by diverged sets of genes. Cfu contains an α-tomatinase gene that we predict might be required for detoxification of tomatine, while this gene is absent in Dse. Many genes encoding secreted proteins are unique to each species and the repeat-rich areas in Cfu are enriched for these species-specific genes. In contrast, conserved genes suggest common host ancestry. Homologs of Cfu effector genes, including Ecp2 and Avr4, are present in Dse and induce a Cf-Ecp2 - and Cf-4-mediated hypersensitive response, respectively. Strikingly, genes involved in production of the toxin dothistromin, a likely virulence factor for Dse, are conserved in Cfu, but their expression differs markedly with essentially no expression by Cfu in planta. Likewise, Cfu has a carbohydrate-degrading enzyme catalog that is more similar to that of necrotrophs or hemibiotrophs and a larger pectinolytic gene arsenal than Dse, but many of these genes are not expressed in planta or are pseudogenized. Overall, comparison of their genomes suggests that these closely related plant pathogens had a common ancestral host but since adapted to different hosts and lifestyles by a combination of differentiated gene content, pseudogenization, and gene regulation.
Introduction
Cladosporium fulvum and Dothistroma septosporum are two related fungal species belonging to the class Dothideomycetes. C. fulvum is a biotrophic pathogen of tomato that has served as a model system for plant-microbe interactions since its first effector gene, Avr9, was cloned in 1991 [1]. It is not related to species in the genus Cladosporium sensu strictu, and has recently been renamed Passalora fulva [2]. However, to be consistent with past literature it will be referred to here as C. fulvum. Phylogenetic analyses based on sequences of the internal transcribed spacer (ITS) region of the ribosomal DNA revealed that C. fulvum is closely related to D. septosporum and other Dothideomycete fungi such as species of Mycosphaerella isolated from eucalyptus [3]. D. septosporum is an economically important hemibiotrophic pathogen of pine species that is well known for its production of an aflatoxin-like toxin, dothistromin [4]. A taxonomic revision also occurred for this species: prior to 2004 the name Dothistroma pini (syn. D. septosporum syn. D. septospora) was widely used. The revision involved a split into two species: the best-studied and most widespread species was named D. septosporum, and a less common species retained the name of D. pini [5].
The disease caused by C. fulvum, leaf mold of tomato, likely originates from South America, the center of origin of tomato [6]. The first outbreak of the disease was reported in South Carolina, USA, in the late 1800s [7]. Since then, disease outbreaks have occurred worldwide in moderate temperature zones with high relative humidity. The disease was of high economic importance during the first half of the 20th century, but its importance waned after introgression of Cf (for C. fulvum) resistance genes by breeders into tomato cultivars began providing effective control [8]. However, recent outbreaks have been reported in countries where tomato cultivars lacking Cf resistance genes are grown, and in areas where intensive year-round cultivation of resistant tomato plants led to fungal strains overcoming Cf genes [9], [10].
In contrast, the foliar forest pathogen D. septosporum (Dorog.) Morelet has a relatively recent history and has been less intensively studied than C. fulvum. D. septosporum infects over 70 species of pine, as well as several minor hosts including some Picea species [11]. During the 1960s–1980s, Dothistroma needle blight (DNB) was largely a problem of Southern hemisphere pine plantations, where primary control was achieved by fungicide applications or planting of resistant species (reviewed in [12]). Since the early 1990s DNB incidence has increased greatly in the Northern hemisphere, with some epidemics causing unprecedented levels of mortality [13], [14]. In northwest British Columbia, disease outbreaks are correlated with summer rainfall levels, suggesting that climate change could have unpredictable and severe effects on DNB outbreaks in forests [15].
Infection in both the C. fulvum-tomato and D. septosporum-pine pathosystems starts with conidia that germinate on the leaf surface and produce runner hyphae that enter the host through open stomata. Subsequently, the fungi colonize the apoplastic space between mesophyll cells. In the case of C. fulvum, conidiophores emerge from stomata 10–14 days later producing massive amounts of conidia that can re-infect tomato [16], [17], [18] (Figure 1A–1D). D. septosporum produces conidia, several weeks after infection, on conidiomata that erupt through the needle epidermis where they can be spread to other pines by rain splash [19], [20] (Figure 1E–1H). Whilst C. fulvum is considered a biotroph, D. septosporum is assumed to be a hemibiotroph based on similarities of its lifecycle to other Dothideomycete fungi.

There is no evidence that C. fulvum has an active sexual cycle, although both mating type idiomorphs occur in its global population [21]. Although D. septosporum also has a predominantly asexual lifestyle, it is known to be sexually active in some parts of the world. The sexual stage Mycosphaerella pini Rostr. (syn Scirrhia pini Funk & Parker) has been reported in some forests in Europe and North America but has not yet been found in other regions, such as South Africa or the United Kingdom, even though both mating types are known to be present [22]. The rare sightings of the sexual stage are due partly to difficulties in identification, but also reflect findings from population studies that show mixed modes of reproduction with a significant clonal component [23], [24]. So far, attempts to induce a sexual cycle between opposite mating types of D. septosporum in culture in our laboratory or others (Brown A, unpublished data) have failed. Further research is required to determine environmental conditions conducive to sexual reproduction. The D. septosporum isolate whose genome was sequenced is derived from a clonal population with a single mating type that was introduced into New Zealand in the 1960s [22], [25].
The C. fulvum-tomato interaction complies with the gene-for-gene model [2], [26]. During infection C. fulvum secretes effector proteins into the apoplast of tomato leaves which function not only as virulence factors, but also as avirulence (Avr) factors when recognized by corresponding tomato Cf resistance proteins. This recognition leads to Cf-mediated resistance that often involves a hypersensitive response (HR) preventing further ingress of the fungus into its host plant tomato [8]. To date many cysteine-rich effectors have been cloned from C. fulvum, including Avr2, Avr4, Avr4E and Avr9, that can trigger Cf-2-, Cf-4-, Cf-4E-, and Cf-9-mediated resistance, respectively, and Ecps (extracellular proteins) like Ecp1, Ecp2, Ecp4, Ecp5 and Ecp6 that trigger Cf-Ecp-mediated resistance [27], [28], [29]. Specific functions for some C. fulvum effectors have been determined: Avr4 is a chitin-binding protein that protects fungi against the deleterious effects of plant chitinases [30], [31], Ecp2 is a virulence factor that occurs in many fungi [32], [33] and Ecp6 sequesters chitin fragments released from fungal cell walls by chitinases during infection thereby dampening their potential to induce pathogen-associated molecular pattern (PAMP)-triggered immunity [28]. Initially, the Avr and Ecp effectors seemed unique to C. fulvum, but in recent years homologs of Avr4, Ecp2 and Ecp6 with functions in virulence have been found in other fungal genomes, including members of the Dothideomycetes [27], [28], [33].
Whilst most studies of C. fulvum have focused on effectors and their interactions with components in both resistant and susceptible plants, studies of D. septosporum have instead focused on dothistromin, a toxin produced by the fungus that accumulates in infected pine needles. Dothistromin is a broad-spectrum toxin with structural resemblance to a precursor of the highly toxic and carcinogenic fungal metabolite, aflatoxin [34]. Although dothistromin is not essential for pathogenicity [35], recent observations suggest it to be a virulence factor, affecting lesion size and spore production (Kabir MS and Bradshaw RE, unpublished data). Some dothistromin biosynthetic genes were identified in D. septosporum but unexpectedly they were in several mini-clusters rather than in one co-regulated cluster of genes as reported for aflatoxin-producing species of Aspergillus [36], [37], [38]. The similarity of dothistromin to aflatoxin enabled predictions to be made about other D. septosporum genes involved in dothistromin production [39]; the complete set of dothistromin genes will help us understand the evolution of dothistromin and aflatoxin gene clusters.
Here we report the sequence and comparison of the genomes of C. fulvum and D. septosporum, which have very similar gene contents but differ significantly in genome size as a result of different repeat contents. We found unexpectedly high levels of similarity in genes previously studied in one or other of these fungi, including those encoding Avr and Ecp effectors of C. fulvum, and dothistromin toxin genes of D. septosporum. Surprisingly, compared to D. septosporum, C. fulvum has higher numbers of genes normally associated with a necrotrophic or hemibiotrophic lifestyle such as genes for carbohydrate-degrading enzymes and secondary metabolite biosynthesis. However, in C. fulvum some of these genes were lowly or not expressed in planta and others were pseudogenized. Other C. fulvum genes that are absent in D. septosporum are putatively involved in virulence on its host plant tomato, such as the α-tomatinase gene. We suggest that regulation of gene expression and pseudogenization, in addition to evolution of new genes, are important traits associated with adaptation to different hosts and lifestyles of the two fungi that, however, also retained some signatures of their common ancestral host.
Results/Discussion
C. fulvum and D. septosporum are closely related species with very different genome sizes
The 30.2-Mb genome of D. septosporum (http://genome.jgi.doe.gov/Dotse1/Dotse1.home.html; GenBank AIEN00000000) was sequenced at 34-fold coverage (Table S1) and then assembled into 20 scaffolds (>2-kb), 14 of which were 407-kb or larger, have telomere sequences at one or both ends (Table S2) and mostly match chromosome sizes estimated from pulsed-field gel electrophoresis [36]. The six smallest scaffolds ranged from 2.3 - to 5.2-kb in size so are not significant parts of the genome. The excellent assembly of the D. septosporum genome was facilitated by its very low repeat content of only 3.2% (Table 1; Table 2; Protocol S1). In contrast, the repeat-rich genome of C. fulvum (http://genome.jgi-psf.org/Clafu1/Clafu1.home.html; GenBank number AMRR00000000) was very difficult to assemble. Fourteen 2-kb paired-end or shotgun 454 sequencing runs for C. fulvum resulted in a 21-fold coverage of the 61.1-Mb assembly in 2664 scaffolds >2-kb (Table 1) with a total repeat content of 47.2% (Table 2). The sequencing strategy was initially based on the assumption of a genome size of around 40-Mb, but soon it appeared that the C. fulvum genome was much larger due to the high repeat content. Problems with the assembly are not caused by the sequencing coverage of C. fulvum because it is estimated to be sufficiently high for good coverage of the gene-encoding areas. Instead, they are a consequence of its high repeat content. An estimated additional 26-Mb of C. fulvum DNA reads could not be assembled as they were predominantly repeat sequences (Figure 2). In the remainder of the manuscript we refer to chromosomes (1 to 14) for D. septosporum and scaffolds for C. fulvum. Summary statistics for the two genomes are shown in Table 1 and at the Joint Genome Institute (JGI) Genome portal (jgi.doe.gov/fungi) [40]. The C. fulvum and D. septosporum genomes are predicted to encode approximately 14 and 12.5 thousand gene models, respectively. Nevertheless, the C. fulvum and D. septosporum genomes share more than 6,000 homologous gene models with at least 80% similarity at the predicted amino acid level, whereas this number drops to 3,000 gene models this similar when comparing C. fulvum or D. septosporum with other closely related Dothideomycete species such as Mycosphaerella graminicola and M. fijiensis (Figure 2, Figure S1). Similarly, most introner-like element clusters found in C. fulvum and D. septosporum are closely related, more than to elements in other Dothideomycetes [41].



Phylogenetic analysis of C. fulvum and D. septosporum genomes in the context of nine other Dothideomycetes [42] confirms that these two species are the most closely related of the sampled species (Figure 2), as was inferred earlier from ITS [3] and mating type sequences [21]. This gives us two very closely related genomes with drastically different genome sizes mostly due to the greatly increased repeat content of C. fulvum.
C. fulvum and D. septosporum differ in content and classes of repeats that are affected by repeat-induced point mutation
The massive increase in repetitive elements in C. fulvum might result from expansion of one or more repeat families that are also present in D. septosporum. Therefore, we classified the different repeat families in D. septosporum and compared them with those in C. fulvum. This revealed that some of the repetitive element families present in D. septosporum have expanded in C. fulvum (Table 2). This is most remarkable for the Class I retrotransposons which comprise over 90% of the repetitive fraction in C. fulvum and together account for over 26-Mb of the assembled genome. Retrotransposons are also highly abundant in the large repeat-rich genome of the hemibiotrophic sexual pathogen Mycosphaerella fijiensis (Dhillon B, Goodwin SB and Kema GHJ, unpublished data). Both Copia and Gypsy LTR retroelements are expanded in C. fulvum compared to the D. septosporum genome, whereas LINEs are detected only in C. fulvum (Table 2). Some other fungal species that are closely related to each other, but have a different lifestyle, also differ in repeat content, such as the Leotiomycetes of which Botrytis cinerea (<1% repeats) and Sclerotinia sclerotiorum (7% repeats) are necrotrophs, while Blumeria graminis f. sp. hordei (64% repeats) is an obligate biotroph [43], [44]. The latter species is particularly enriched in Class I elements and one of several biotrophs that show expansion of genome size associated with high repeat content [44], [45].
In contrast to the retroelements, Class II DNA transposons comprise only a small percentage (4.7%) of the overall repetitive elements in C. fulvum, but 46.2% of the repeats in D. septosporum, although they make up only a small portion of the genome overall. Interestingly, helitron-like DNA transposons comprise 40% of all repeats in D. septosporum and are 25.8-fold higher in terms of sequence coverage than in C. fulvum, whereas the DDE-1, hAT, and MuDR_A_B DNA transposons present in C. fulvum are not present in D. septosporum. Helitrons are transposons that replicate by a rolling-circle mechanism and are found in a wide range of eukaryotes, including the white rot fungus Phanerochaete chrysosporium [46], and are thought to have a role in genome evolution [47]. Helitron-like repeats are particularly abundant on D. septosporum chromosomes 3, 6 and 11 (Figure 3) and usually occur in clusters, sometimes along with other types of repetitive elements.

The organization of repeats in D. septosporum is striking in that for the majority of the chromosomes, most repeats are localized into just one or two large regions containing a mixture of repeat element types (Figure 3), although other small repeat clusters also occur. In many eukaryotes, centromeres are characterized by repetitive DNA [48], and therefore we propose that some of the larger complex repeat regions are centromeres, in line with similar suggestions made for other fungal genomes [49], although experimental confirmation is required. The absence of any repeat cluster from chromosome 14, along with the observation that it harbors only one telomere, suggests that it is a chromosome fragment.
Repeats in fungi are affected by repeat-induced point mutation, also referred to as RIP, a defense mechanism employed by fungi to suppress transposable element activity that was first described in Neurospora crassa [50]. RIP is a process by which DNA accumulates G:C to A:T transition mutations. It occurs during the sexual stage in haploid nuclei after fertilization but prior to meiotic DNA replication. Clear evidence of RIP was found in both the C. fulvum and D. septosporum genomes (Table 3) and is mainly confined to repeat-rich regions. In total 25.9-Mb were RIP'd in C. fulvum and 1.1-Mb in D. septosporum, which represent 42.4% and 3.7% of their genomes, respectively. RIP occurred mainly on large repeated sequences (≥500 nucleotides) that represent 97.2% of all repeats in C. fulvum and 98.0% in D. septosporum (Table 3). The high rate of RIP in repeat regions is in the same range as that seen in other Dothideomycetes such as S. nodorum (97.2%; Table S3) [42], [51]. Although RIP is present at high levels in C. fulvum, we propose that it has not been able to prevent transposon expansion possibly due to very rare sexual activity.

Of the RIP'd loci, C. fulvum has almost none (0.5%) and D. septosporum little (16.9%) outside the main classified repeat regions. This is different from N. crassa (Table S3), where 35.2% of all RIP'd loci are predicted to be non-repeat-associated. For N. crassa it has been shown that even single gene duplication events are prey to the RIP machinery, thereby exemplifying its efficiency and sensitivity [50]. Clearly such sensitivity is not applicable to C. fulvum and D. septosporum, nor for three other studied Dothideomycetes (Table S3). In the Dothideomycete phytopathogenic fungus Leptosphaeria maculans, RIP slippage is found in regions adjacent to repetitive elements. In that species RIP has occurred in genes encoding small secreted proteins, such as the effector genes AvrLm6 [52] and AvrLm1 [53] that are located in repeat-rich regions of the genome [54]; mutations in these genes caused by the RIP process enabled the fungus to overcome Lm6 and Lm1-mediated resistance, respectively. However, we found no evidence of RIP slippage into the known effector genes of C. fulvum and related effector genes in D. septosporum.
The C. fulvum and D. septosporum genomes show extensive intrachromosomal rearrangements
One way to assist the assembly of a fragmented genome is to use synteny with a well-assembled genome of a closely related species to order the scaffolds [55]. We attempted to use the D. septosporum genome to improve the C. fulvum assembly in this way. However, although it was possible to map C. fulvum scaffolds onto the assembled D. septosporum genome (Figure S2A), individual C. fulvum scaffolds are not collinear along their length, but have only short blocks of synteny to different parts of the D. septosporum chromosomes. The syntenic regions of the C. fulvum and D. septosporum genomes are associated with just 461 of the C. fulvum scaffolds (Table 4). In contrast, the remaining >4,000 C. fulvum scaffolds are non-syntenic. A more detailed analysis with the ten largest C. fulvum scaffolds (two are shown in Figure S2B) revealed that they each match primarily to only one D. septosporum chromosome, suggesting predominantly intrachromosomal rearrangements (mesosynteny), as described for other Dothideomycete fungi [42], [56] (Condon B et al., unpublished data). As found in other fungi [43], [57] non-syntenic regions are repeat-rich; for C. fulvum 79.7% of the repeat sequences are present in non-syntenic regions (Table 4).

Non-syntenic, repeat-rich regions are enriched in genes encoding secreted proteins
Secreted proteins are important for communication of plant-pathogenic fungi with their hosts. They comprise not only enzymes required for penetration and growth on plant cell walls, but also proteins needed to compromize the basal defence system of plants by either suppressing or attacking it, as has been reported for several fungal effector proteins [58]. The percentage of proteins predicted to be secreted is similar for both C. fulvum (8.5%) and D. septosporum (7.2%), and in the same range as that predicted for other Dothideomycete fungi such as M. graminicola (9.1%) and S. nodorum (10.8%) [40], [42], [51], [59].
Genes encoding secreted proteins including effectors are subject to evolutionary selection pressure imposed by environmental and host plant factors [58], and they often show a high level of diversification. Repeat-rich, gene-poor regions have been proposed to contain genes involved in adaptation to new host plants. For example, in some Phytophthora species and in L. maculans significantly higher proportions of in planta-induced species-specific effector genes encoding secreted proteins are found in repeat-rich compared to repeat-poor regions [50], [59] and in pathogenic strains of Pyrenophora tritici-repentis transposable elements are associated with effector diversification (Manning V, Ciuffetti L, unpublished data). We hypothesized that we would find more genes encoding secreted proteins in repeat-rich regions that are less syntenic between the C. fulvum and D. septosporum genomes than in repeat-poor syntenic regions. We therefore compared the number of genes and their similarity at the nucleotide and protein levels in syntenic and non-syntenic regions of these two genomes (Table 5) using C. fulvum as the reference sequence due to its higher overall content of repeat elements in non-syntenic regions. The regions syntenic between C. fulvum and D. septosporum, representing 22.3-Mb of the C. fulvum genome, contain 70% of all predicted genes whereas 30% of the genes are located in the non-syntenic repeat-rich regions representing 38.8-Mb of the C. fulvum genome (Table 5). The syntenic regions contain most of the homologous genes that encode proteins with the highest level of conservation between the two genomes, whereas the proteins encoded by genes located in the non-syntenic, repeat-rich regions are less conserved. In syntenic regions, 89.9% of gene models have a bi-directional best BLAST hit (BDBH) to a D. septosporum gene model, with a mean predicted amino acid similarity of 85.2%, compared to non-syntenic with only 51.7% of gene models with BDBH and 65.1% amino acid similarity (Table 5). As expected, we found the repeat-rich, non-syntenic regions to have higher proportions of gene models encoding secreted proteins (10.4%, with a mean predicted amino acid similarity of 60.7%) and small secreted cysteine-rich proteins (2.8%) than in syntenic regions (7.6%, with a mean amino acid similarity of 81.1%, and 1.5% respectively) (Table 5), as has been reported for L. maculans [54].

C. fulvum and D. septosporum share functional effectors
Some C. fulvum effector homologs have previously been reported to occur in other Dothideomycete species including M. fijiensis, M. graminicola, and several Cercospora species [33], but in the D. septosporum genome we found the highest number of C. fulvum effector homologs discovered to date, including Avr4, Ecp2-1, Ecp2-2, Ecp2-3, Ecp4, Ecp5 and Ecp6. Of those, Avr4, Ecp2-1 and Ecp6 are core effectors [33] and show the highest identity (51.7%, 59.8% and 68.6% amino acid identity, respectively) with those present in C. fulvum, whilst Ecp4 and Ecp5 are pseudogenized. We were interested to know whether the D. septosporum effectors would be functional in triggering a Cf-mediated hypersensitive response (HR). Therefore, we inoculated plants of tomato cultivar Moneymaker (MM) carrying the Cf-Ecp2 resistance trait with Agrobacterium tumefaciens expressing potato virus X (PVX) containing D. septosporum Ecp2-1 and used PVX-containing C. fulvum Ecp2-1 as a positive control. D. septosporum Ecp2-1 triggered a Cf-Ecp2-1-mediated HR (Figure 4A), whilst MM tomato plants lacking Cf-Ecp2 did not show any HR when inoculated with PVX containing Ds-Ecp2-1 (results not shown). We also showed that the D. septosporum homolog of C. fulvum Avr4 is functional in triggering a Cf-4-mediated HR in Nicotiana benthamiana, as determined with an Agrobacterium transient transformation assay (Figure 4B). This is remarkable because D. septosporum infects a gymnosperm which is only distantly related to tomato, but apparently produces effectors that can be recognized by tomato Cf resistance proteins. It would be interesting to examine whether gymnosperms carry functional homologs of the well-studied Cf tomato resistance gene homologs [33], [60] or other major R genes that could confer resistance to D. septosporum. Major R genes have been shown to be involved in resistance of some pine species to Cronartium spp. rust pathogens [61], [62] and are thought to function in a gene-for-gene manner [63].

In C. fulvum, adaptation to resistant tomato cultivars is sometimes associated with deletion of effector genes [64]. Presence of repeats or location near a telomere can cause repeat-associated gene deletion [65]. We analyzed the location of all cloned C. fulvum effector genes in its genome. Many scaffolds containing an effector gene are very small (Figure 5), suggesting that they are surrounded by large repeats hampering assembly into larger scaffolds. The location of the C. fulvum effectors is shown in Figure 5 and the types of flanking repeats are detailed in Table S4. The well-characterized effector gene Avr9 is located on a very small (20-kb) scaffold (Figure 5) and is likely flanked on both sides by repeats; on one side there are 11-kb of repeats on the scaffold and on the other side probably also repeats just outside the region shown that prevented further scaffold assembly. This suggests that the absolute correlation found between deletion of the Avr9 gene in C. fulvum and overcoming Cf-9-mediated resistance [64] is most likely due to the close proximity of Avr9 to large, unstable repeat regions. As well as causing deletions, transposons can contribute to genome plasticity by mutation due to transposition into coding sequences. During co-evolution, transposons have inserted into effector genes causing their inactivation and overcoming Cf-mediated resistance in C. fulvum, as has been reported for inactivation of both Avr2 [64] and Avr4E [66]. The C. fulvum homologous effector genes present in D. septosporum are also often in close proximity to repeat-rich areas that may represent centromeres (Figure 3), but the biological significance of this is not yet clear.
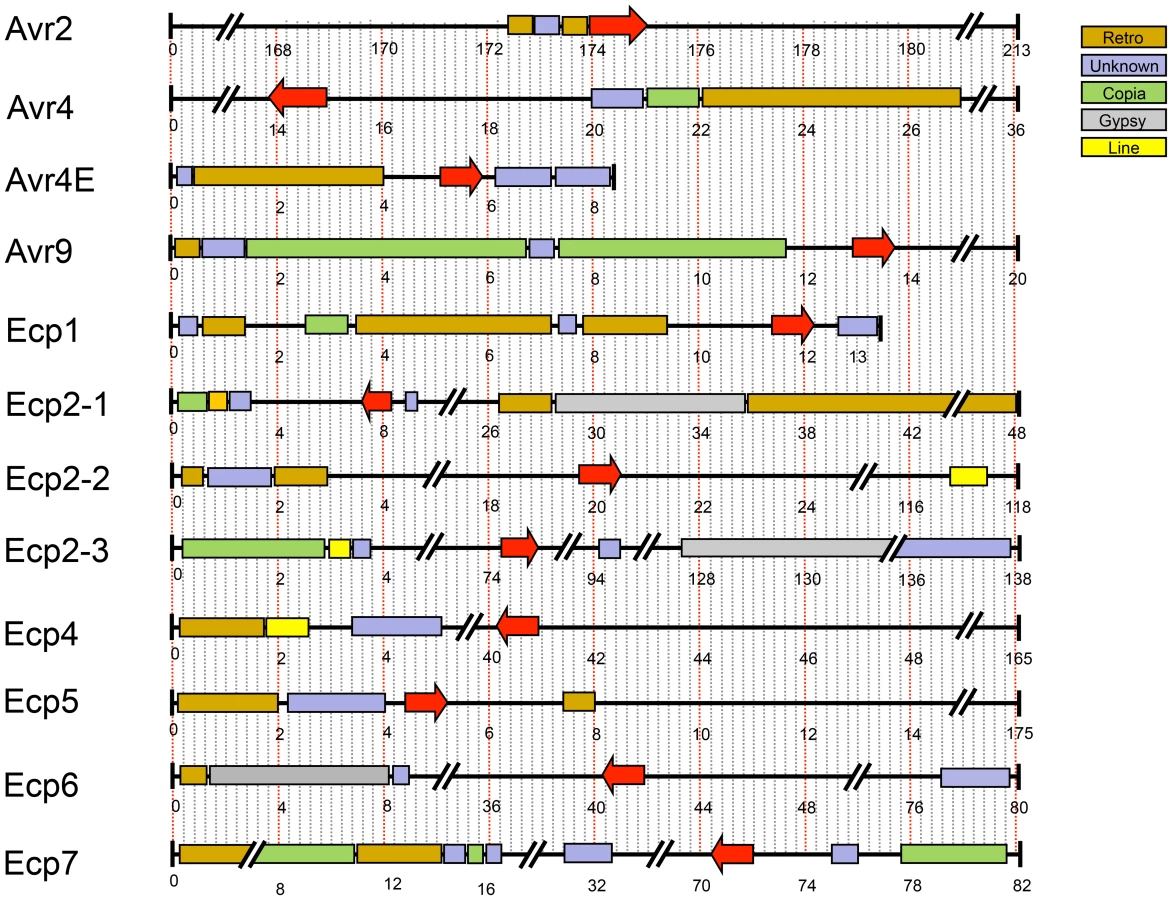
Pseudogenization of two D. septosporum effector genes, Ecp4 and Ecp5, homologous to those reported for C. fulvum [67], could point to host adaptation in the DNB fungus at the pine genus, species or cultivar level. Future population analysis of both fungal strains and host genotypes will reveal the mechanism behind this phenomenon.
New hydrophobin genes in C. fulvum
Another class of well-studied C. fulvum small cysteine-rich secreted proteins is the hydrophobins. These amphipathic proteins are implicated in developmental processes in filamentous fungi and are localized on the outer surface of fungal cell walls [68]. They are divided into class I and class II hydrophobins based on sequence differences that also correlate with their different solubility [68].
Six hydrophobin genes (Hcf-1 to Hcf-6) had previously been identified from C. fulvum [66], [67]. We identified five additional hydrophobin genes in the C. fulvum genome [two class I (Cf187601 and Cf189770) and three class II (Cf197052, Cf188363 and Cf183780)] (Figure S3), which makes C. fulvum the Ascomycete species with the largest number of hydrophobin genes reported so far. In the D. septosporum genome only four hydrophobin genes were found, one of which (Ds75009) is predicted to encode a class II hydrophobin and was highly expressed both in culture and in planta. Based on EST data the 11 C. fulvum hydrophobin genes show a range of different expression patterns. Of the six class I C. fulvum hydrophobins, two were only expressed in culture [Cf184635 (Hcf-2) and Cf189850 (Hcf-4)], three were expressed both in culture and in planta [Cf189770, Cf187601 and Cf193176 (Hcf-1)], and one was not expressed in culture or in planta [Cf184193 (Hcf-3)]. Three of the class II C. fulvum hydrophobins were only expressed in culture [Cf197052, Cf188363 and Cf193013 (Hcf-5)], whilst Cf193331 (Hcf-6) and Cf183780 were expressed neither in culture nor in planta. None of the C. fulvum hydrophobin genes were expressed in planta only. It has been proposed that hydrophobins may act as ‘stealth’ factors, preventing the invading fungus from detection by its host plant [69] or protecting it against deleterious effects of plant chitinases and β-1,3 glucanases as reported for C. fulvum [70]. Early functional studies focused on the hydrophobin genes Hcf-1 (Cf193176) and Hcf-2 (Cf184635). Knocking down expression of Hcf-1, Hcf-2, or both genes by homology-dependent gene silencing did not compromise virulence [71],[72]; a similar result was reported for knock-down mutants of class I Hcf-3 and Hcf-4 and class II Hcf-6 genes [73]. A phylogenetic tree (Figure S3) shows that the four class I genes (Hcf-1 to Hcf-4) are paralogs, suggesting functional redundancy that might explain the lack of a phenotype; functional redundancy may also exist between different classes. It would be interesting to examine the role in virulence of the two most similar hydrophobin class I and class II genes of C. fulvum and D. septosporum (Cf 189770/Ds67650 and Cf197052/Ds75009, respectively) either by knock-out or knock-down strategies.
Carbohydrate active enzyme gene and expression profiles reflect adaptation to different host plants
Because C. fulvum and D. septosporum have very different plant hosts and pathogenic lifestyles, we expected that their capacity to degrade carbohydrates would also differ and that this might be reflected in their gene complements and expression profiles. We compared numbers of genes predicted to encode carbohydrate-active enzymes (CAZymes) [74] in these two fungi to those in other fungi representative of different lifestyles. As seen for grouped families of CAZyme genes in Table 6 (e.g., GH family of glycoside hydrolases), both C. fulvum and D. septosporum have gene numbers in the same range as hemibiotrophic and necrotrophic fungi, and many more than the obligate biotroph B. graminis f. sp. hordei. Despite this, both C. fulvum and D. septosporum have fewer predicted cellulolytic enzyme genes (e.g., GH6, GH7) as well as fewer genes classified in carbohydrate binding module gene families (e.g., CBM1) than most of the other fungi shown except for M. graminicola (Table 6, Table S5). The reduced number of predicted genes for cell wall-degrading enzymes in M. graminicola was hypothesized to represent an adaptation to avoid host defenses during stealth pathogenicity [59], which also may apply to C. fulvum and D. septosporum. However, it is known that even a small number of genes can enable high levels of enzymatic activity, as has been shown for the strongly cellulolytic fungus Trichoderma reesei [75].

Next we focused on CAZyme gene families that appear to differ in gene number between C. fulvum and D. septosporum. Because small differences in gene number could be due to mis-annotation, only families that differed by two or more genes were considered and examples of these are shown in Table 6 (full data in Table S5). Potentially interesting is the expansion of genes associated with pectin degradation in C. fulvum. For example, in the GH28 family that includes many pectinolytic enzymes, C. fulvum has 15 genes whilst D. septosporum has only four. A higher pectinolytic activity in C. fulvum is concordant with the higher pectin content of its host, tomato, compared to the pine host of D. septosporum [76], [77], but larger numbers of genes encoding pectin-degrading enzymes have generally been associated with a necrotrophic rather than a biotrophic lifestyle in fungi [78]. High pectinolytic activity is observed in fungi such as Botrytis cinerea [79], [80] that invades soft, pectin-rich plant tissues causing a water-soaked appearance of the infected tissues [43]. However, during colonization of tomato leaves by C. fulvum this type of symptom is never observed [79], [80]. Instead of contributing to the destruction of host cell walls, the C. fulvum pectinolytic enzymes may facilitate local modification of primary cell walls of mesophyll cells allowing the fungus to thrive in the apoplast of tomato leaves, as suggested for the ectomycorrhizal fungus Laccaria bicolor that thrives on plant roots [81].
Although C. fulvum has a large arsenal of pectinolytic genes compared to D. septosporum, not all of them appear to be functional. For example, two of the six GH78 and one of the two GH88 pectinolytic genes are pseudogenized in C. fulvum, whilst the corresponding D. septosporum families do not contain pseudogenes. Another constraint to function is that gene expression appears to be tightly regulated. As shown in Table 6, none of the 15 C. fulvum GH28 genes appear to be expressed in planta, whilst all four D. septosporum GH28 genes are expressed. Indeed in all gene families with predicted pectinolytic function shown in Table 6 (GH28, GH78, GH88, GH95, PL1, PL3), expression in planta was only detected for 2 of the 31 C. fulvum genes, whilst all 6 genes in these pectinolytic gene families were expressed in D. septosporum. It is possible that C. fulvum pectinases are only expressed very locally to modulate complex primary cell wall structures. The location and accessibility of pectin structures embedded in the cell wall is an important consideration for its enzymatic degradation. For instance, the Basidiomycete Schizophyllum commune grows predominantly on beech and birch wood which is poor in pectin [82]. However, the pectin in these cell walls is concentrated around the bored pits that are used by S. commune to enter the wood, explaining why this fungus contains a higher number of pectinase genes than would be expected based on the overall host pectin content. Differences in pectinolytic gene content and expression between C. fulvum and D. septosporum may therefore be related to their different strategies of host invasion and subsequent colonization.
In addition to increased numbers of pectinolytic genes compared to D. septosporum, C. fulvum has more genes for enzymes that degrade hemicelluloses (e.g., families GH31, GH35 and GH39) [83] and hemicellulose-pectin complexes (GH43) (Table 6). It also contains 11 genes (compared to 4 in D. septosporum) encoding CE5 enzymes; these include cutinases that are required for early recognition and colonization of the host by fungal pathogens [84], [85]. The presence of so many genes encoding enzymes for plant cell wall and cuticle degradation in a biotrophic fungus like C. fulvum that enters its host via stomata is unexpected. However, the number of cutinase genes, and other secreted lipase genes is particularly low in the D. septosporum genome compared to other Dothideomycetes, a feature shared with the other tree pathogens Mycopshaerella populorum and M. populicola [42].
Overall our comparison shows a similar complement of CAZy genes between C. fulvum and D. septosporum, but an increased number of particular CAZyme families in C. fulvum including genes encoding pectin - and hemicellulose-degrading enzymes. However, a large proportion of genes in the C. fulvum CAZyme families lack expression in planta and some genes are pseudogenized.
C. fulvum and D. septosporum share a broad range of carbohydrate substrates
A second aspect of carbohydrate metabolism that we considered was a comparison of growth on defined and complex carbon substrates (Figure 6 and Figure S4; www.fung-growth.org). It was anticipated that growth profiles could illuminate differences between pathogens with dicot and gymnosperm hosts and show correlations with their respective gene complements. In a study of polysaccharide hydrolysis activities of many fungal pathogens, King et al. [86] showed preferential substrate utilization based on host specificity (dicot or monocot). In general D. septosporum grows more slowly on minimal control medium [87] than C. fulvum, but surprisingly overall the growth profiles of the two fungi are similar on most substrates (Figure 6 and Figure S4; Table S6) and both appear to utilize a broader range of substrates than M. graminicola (Figure 6). This is not only the case for the oligomeric and polymeric carbon substrates, requiring CAZymes for degradation, but also for monomeric carbon substrates, suggesting a diverse and efficient carbon catabolism in C. fulvum and D. septosporum. The good growth of D. septosporum on sucrose is particularly striking, suggesting that it can utilize sucrose available in apoplastic fluid during its early biotrophic colonization phase.

In terms of complex carbon sources, D. septosporum shows a slightly better capacity than C. fulvum to utlise apple and citrus pectin (Figure 6 and Figure S4). This seems to contradict the higher pectinolytic gene numbers in C. fulvum compared to D. septosporum, but is supported by the expression of fewer C. fulvum pectinolytic genes during infection of tomato when compared to the D. septosporum pectinolytic genes during infection of pine needle (Table 6). Interestingly, good growth on pectin is also observed for M. graminicola, despite an even lower number of putative pectinases than D. septosporum. This suggests that regulation of expression is a more dominant factor in pectin degradation by these plant pathogens than the number of pectinase-encoding genes in their genomes. In contrast, pectinase gene numbers correlate well with growth profiles of Aspergillus nidulans, A. oryzae and A. niger [88]. Compared to growth on controls lacking a carbon source, D. septosporum also showed slightly better growth than C. fulvum on lignin. This would be consistent with the higher proportion of lignin in pine needles, estimated to be 25–30% of dry weight [89], compared to less than 10% in dicots [90]. However, due to the very slow growth of both fungi and the non-uniform growth habit of D. septosporum on these media, firm conclusions about their abilities to utilize lignin cannot be made.
Adaptations for coping with chemical and structural defences
Tomato plants produce the antimicrobial saponin, tomatine. The tomato pathogen Fusarium oxysporum produces α-tomatinase, which functions as a virulence factor as it degrades tomatine into the non-toxic compounds tomatidine and lycotetraose [91]. A gene predicted to encode α-tomatinase, classified as a GH10 enzyme, was found in the C. fulvum genome (JGI ID 188986) but is absent from the D. septosporum genome. Another gene found only in C. fulvum shows predicted similarity to the GH5 family enzyme hesperidin 6-O-α-L-rhamnosyl-β-glucosidase that can degrade hesperidin [77]. Hesperidin occurs most abundantly in citrus fruits [92] and is a member of the flavonoid group of compounds that is well known for its antimicrobial activity. Flavonoid-degrading enzymes such as hesperidin 6-O-α-L-rhamnosyl-β-glucosidase might enable C. fulvum to detoxify hesperidin or related compounds present in tomato.
Chemical defence molecules in pine needles include antimicrobial monoterpenes. Thus it is expected that D. septosporum is adapted to tolerate or degrade these compounds whilst C. fulvum is not. Recent work on the pine pathogen Grosmannia clavigera revealed several classes of genes that are upregulated in response to terpene treatment [93]. After 36 h, major classes of upregulated genes included those involved in β-oxidation as well as mono-oxygenases and alcohol/aldehyde dehydrogenases that may be involved in activating terpenes for β-oxidation. A drug transporter, GLEAN_8030, was functionally analyzed and found to be required for tolerance of the fungus against terpenes, enabling G. clavigera to grow on media containing these compounds. A search for three of these genes, including GLEAN_8030, showed that both C. fulvum and D. septosporum genomes contain putative homologs and share a similar gene complement to each other (Table S7). However, since these genes have not all been functionally characterized in G. clavigera and all are predicted to encode proteins involved in general metabolic processes, further work is required to determine the roles of the homologs found in both C. fulvum and D. septosporum.
As well as chemical mechanisms, plants employ basal structural defence mechanims including lignification of cell walls [94], [95]. Due to the abundance of lignin in pine needles that block access to usable cellulose, fungal pathogens and saprophytes living on pines have a particularly challenging environment [96]. For D. septosporum to complete its lifecycle, degradation of pine needle tissue must occur so that conidiophores bearing conidia can erupt through the epidermis (Figure 1H), which contains lignin [97]. This is in contrast to C. fulvum whose conidiophores emerge from tomato leaves through stomatal pores (Figure 1D). Thus, we investigated genes that may be involved in lignin degradation.
Some saprophytic fungi utilize oxidoreductases, particularly class-II peroxidases such as lignin peroxidases, manganese peroxidases and laccases, and a number of H2O2-producing enzymes, to achieve lignin breakdown [98], [99]. However, the number of genes encoding oxidoreductases in D. septosporum is no higher than those of other Dothideomycetes (C. fulvum, M. graminicola and S. nodorum) that infect plants with lower levels of lignin (Table S8). D. septosporum appears to have a similar complement of laccase genes as C. fulvum and only one distant relative of a class-II peroxidase, missing in C. fulvum, but also present in M. graminicola and S. nodorum. Interestingly, the classical Ascomycete laccases found in C. fulvum, D. septosporum and M. graminicola bear a carbohydrate-binding domain (CBM20, putative starch binding domain). This type of laccase is only found in Dothideomycetes but the significance of this novel modular structure is unclear. Brown-rot saprophytes such as Serpula lacrymans have a reduced complement of ligninolytic genes compared to lignin-degrading white-rot fungi and are proposed to initially weaken lignocellulose complexes by non-enzymatic use of hydroxyl radicals prior to enzymatic assimilation of accessible carbohydrates [81]. It is likely that D. septosporum uses a similar strategy to breach the lignin-rich components of pine needles, as complete degradation of this polymer is not required to complete its life cycle.
The secondary metabolite gene complement of C. fulvum and D. septosporum
Secondary metabolites (SMs) are important compounds for the colonization of specific ecological niches by fungi. In particular, plant-pathogenic fungi can produce non-specific and host-specific toxic SMs [100]. SMs also include mycotoxins that contaminate food and feed and are harmful to mammals [100]. The only currently known SMs produced by C. fulvum and D. septosporum are cladofulvin and dothistromin, respectively [101], [102]; both compounds are anthraquinone pigments. In fungi, SM biosynthetic pathways often involve enzymes encoded in gene clusters [103] and always require the activity of at least one of four key enzymes: polyketide synthase (PKS), non-ribosomal peptide synthetase (NRPS), terpene cyclase (TC) or dimethylallyl tryptophan synthase (DMATS) [104]. It has been suggested that loss of SM biosynthetic pathways is associated with biotrophy [44], thus we searched for SM gene pathways in both genomes. Surprisingly, the biotroph C. fulvum has twice the number of key SM genes (23 in total) compared to the hemibiotroph D. septosporum (11 in total) (Table 7), of which 14 and 9, respectively, are organized into gene clusters along with other SM-related genes. The numbers of key SM enzyme-encoding genes are comparable to those of M. graminicola, but are lower than those in most other sequenced Dothideomycetes [42]. Like all Ascomycetes [105], the majority of key SM enzymes in C. fulvum and D. septosporum are PKSs, NRPSs and hybrid PKS-NRPSs. Annotation of all key SM genes was manually checked and two truncated (Pks4 and Nps1) and five pseudogenized (Pks9, Hps2, Nps5, Nps7 and Nps10) genes were found in the C. fulvum genome, while all D. septosporum genes except Pks4 (truncated) are predicted to encode functional enzymes. Overall, the number of predicted functional pathways suggests that C. fulvum and D. septosporum can produce at least 14 and 10 different SMs, respectively.

Surprisingly, only three of the key SM genes are predicted to belong to biosynthetic pathways shared between the two species (Table S9) suggesting a diverse SM repertoire. This is much lower than expected given the overall level of similarity in gene content between the two genomes, and suggests that this SM repertoire is under strong selection. The three common genes are predicted to be involved in production of a pigment related to melanin (Pks1), a siderophore (Nps2) and dothistromin (PksA) based on similarities to other characterized genes. In C. fulvum, the three other functional non-reducing PKS enzymes are candidates for production of cladofulvin.
The genomic locations of the 11 biosynthetic SM genes in D. septosporum do not show any enrichment at sub-telomeric positions, as reported for Aspergillus spp. and Fusarium graminearum [106], [107], or near putative centromeres (Figure 3). However, 8 out of the 11 genes are located on chromosomes smaller than 2-Mb (chromosomes 8 to 12; Figure 3). The genomic regions immediately surrounding all 11 D. septosporum SM genes are conserved in the C. fulvum genome, although 8 of them lack the key SM gene itself and, sometimes, putative accessory genes also (Figure S5). Reciprocally, only 9 C. fulvum SM genomic regions out of 23 are conserved in D. septosporum with 6 of these lacking the key SM gene, suggesting either gain or loss of SM genes has occurred. For two of the regions where flanking genes are conserved but SM gene(s) are missing in C. fulvum (regions corresponding to those surrounding Pks3 and Nps3 in D. septosporum), the presence of repeats suggests that SM gene loss may have occurred in C. fulvum (Figure S5). The C. fulvum-specific SM loci Pks5, Pks6, Nps5/Dma1 and Nps9 include many transposable elements and genes that have similarity to genes scattered in the D. septosporum genome, often on the same chromosome, leading to the hypothesis that these SM loci were assembled by gene relocation as recently proposed for the fumonisin gene cluster in F. verticillioides [108].
Dothistromin toxin genes are present in both genomes
Analysis of the D. septosporum 1.3-Mb chromosome 12 revealed that the three previously identified mini-clusters of dothistromin genes [38] are widely dispersed, confirming fragmentation of this gene cluster (Figure 3 and Figure 7). Candidates for additional dothistromin genes, previously predicted based on aflatoxin pathway genes [39], are also present. Although three of these genes (OrdB, AvnA, HexB) are located in the published VbsA mini-cluster, the others are dispersed over different regions of chromosome 12 as shown in Figure 7. The end of the Nor1 gene cluster (Nor1, AdhA, VerB) is less than 10-kb from one predicted telomere, whilst Ver1 (previously called dotA [35]) is only 81-kb from the other telomere. As expected, a gene similar to the aflatoxin AflR regulatory gene is present and, like in aflatoxin-producing species of Aspergillus, is divergently transcribed with an adjacent AflJ regulatory gene candidate. Functional analysis of these genes is in progress.

Although C. fulvum is not known to produce dothistromin, the complete set of predicted dothistromin genes is present in its genome, encoding proteins with amino acid identities ranging from 49% (AflJ) to 98% (Ver1) when compared with those of D. septosporum (Table S10). The arrangement of predicted dothistromin genes in C. fulvum reveals a high level of synteny with some rearrangements. With the exception of the Ver1 gene cluster, the mini-clusters contain the same genes in the same orientations in the two species (Figure 7A). The three mini-clusters on C. fulvum scaffold 130775 are much closer together than in D. septosporum, but are still separated from each other by considerable distances (approximately 24-kb between Est1 and the VbsA gene cluster, and 40-kb between the VbsA and Nor1 gene clusters). A comparison of the relative locations of the mini-clusters in the two species suggests inversions (AflR/J and VbsA gene clusters) as well as rearrangements over relatively small (VbsA-Nor1) and large (Ver1-AflR/J) distances. This is consistent with the overall pattern of intrachromosomal rearrangements observed between these two genomes.
Given the presence of the dothistromin biosynthetic pathway genes, we tested whether dothistromin is produced by C. fulvum. However, no dothistromin was detected by HPLC analysis of extracts from C. fulvum PDB cultures, which is a condition favorable to dothistromin production by D. septosporum. Despite the lack of dothistromin production under these conditions, a strong evolutionary constraint on dothistromin biosynthetic genes was seen by analyzing the ratio of non-synonymous to synonymous mutations (Ka/Ks) between C. fulvum and D. septosporum. The low Ka/Ks ratios seen for dothistromin genes (range 0.018–0.169) are indicative of purifying selection [109] and did not differ from the distribution observed for four housekeeping genes (Tub1, Eif3b, Pap1, Rps9; range 0.003–0.073) (P = 0.561). Evidence for purifying selection was also shown for aflatoxin pathway genes in Aspergillus flavus and A. nomius [110]. On the basis of this we propose that C. fulvum might produce dothistromin, or a metabolite related to dothistromin, under certain environmental conditions when it is required.
Regulation of secondary metabolite biosynthetic pathways suggests lifestyle adaptation at the transcriptome level
Many fungal SM biosynthetic pathways are cryptic, meaning that they are not expressed in wild-type strains under laboratory conditions. However, manipulation of genetic regulatory pathways or environmental conditions has shown that some of these cryptic pathways are functional [111], [112].
As seen for other gene families such as CAZyme genes, C. fulvum appears to be more economical in its expression of SM genes than D. septosporum, particularly in planta. In C. fulvum, EST support was obtained from in vitro conditions for all key SM genes except Hps2, Nps7 and Nps10, which are pseudogenized. The two truncated genes (Pks4 and Nps1) and the pseudogenized Nps5 genes also have EST support but the resulting proteins are unlikely to be functional. However, no evidence for in planta expression could be obtained for any of the C. fulvum key SM genes from this EST library. In contrast, all D. septosporum key SM genes have EST support from both in vitro and in planta libraries, with the unique DsPKS2 being one of the most highly expressed genes during pine needle infection.
Differences in dothistromin pathway regulation were confirmed by quantitative PCR. In D. septosporum, Ver1, PksA, AflR and VbsA show higher expression during pine infection than in controlled culture conditions used to induce dothistromin production (Figure 7B). In contrast, the same genes show a low expression level in C. fulvum during infection and in vitro (Figure 7C). Because no dothistromin could be detected in vitro, this low expression likely represents background transcription with no biological relevance. Such an expression pattern is significantly different from the upregulation of Avr4 and Avr9 genes during tomato infection (Figure S6).
SM production is associated with development in fungi and involves common regulators [113]. We searched the genomes of C. fulvum and D. septosporum for conserved regulators of development and SM production and, based on predicted protein sequences, found clear homologs for most of these genes in both fungi (Table S11). The two species appear to lack a PpoB oxygenase, but PpoA and PpoC are sufficient to produce all psi factors (oxylipins) identified in Aspergillus species [114]. In addition, C. fulvum lacks clear homologs of the G-protein regulators FlbA and RgsA, while possible homologs are found in D. septosporum. In Aspergillus species both proteins are negative regulators of G-protein signaling pathways. Neither C. fulvum nor D. septosporum have a homolog of BrlA, an essential regulator of conidiation in Aspergillus species [115], suggesting that they use another regulator for this role. Future studies analyzing expression of the SM genes, and the roles of regulatory genes, will help to determine fundamental differences in how C. fulvum and D. septosporum differentially regulate their SM gene expression.
Conclusion
We embarked upon a comparative genomics analysis of C. fulvum and D. septosporum to test for differences that might explain their host specificity and lifestyles. The comparison revealed surprising similarities, such as the presence of dothistromin toxin genes in C. fulvum and functional Avr4 and Ecp2 effector genes in D. septosporum. However, the genome sizes of the two fungi are remarkably different, mainly due to a vast expansion of transposable elements in C. fulvum, and show several key differences in gene content. Adaptation of C. fulvum to its host plant tomato is exemplified by the specific presence of a gene encoding α-tomatinase, likely involved in degradation of tomatine. In contrast, the dothistromin gene cluster is present in both fungi, but while it is strongly expressed in D. septosporum at late stages of pine needle infection, it is lowly or not expressed in C. fulvum during infection of tomato leaves. Both fungi contain additional key SM genes, but the majority of these are not in common, contrasting with the high degree of homology between the two genomes. We suggest that this lack of conservation of key SM genes in the C. fulvum and D. septosporum genomes is a consequence of different evolutionary pressures that result from their different lifestyles, either as a pathogen inside their host or possibly as a saprophyte outside their host.
Another key difference between the two fungi during pathogenesis concerns their differential gene regulation. Gene expression in C. fulvum is strictly regulated in planta, with many SM, hydrophobin and CAZy genes not expressed, while expression in D. septosporum is more constitutive. This differential regulation of expression may be crucial in determining differentiation between these fungi despite very similar gene profiles. Furthermore, this expression pattern is consistent with a biotrophic lifestyle without gene loss. Finally we suggest that the higher repeat content of the C. fulvum genome, along with evidence for gene pseudogenization (van der Burgt A et al., unpublished data) has facilitated the evolution of different lifestyles between C. fulvum and its sister species D. septosporum. Overall, our comparison of the two genomes suggests that even closely related plant pathogens could adapt to very different hosts and lifestyles by differentiating gene content and regulation, whilst retaining genetic signatures of a common ancestral way of life.
Materials and Methods
Fungal strains and growth conditions
The fungal strains of C. fulvum (race 0WU; CBS131901) and D. septosporum (strain NZE10; CBS128990) were isolated from tomato growing in an allotment garden in Wageningen, The Netherlands, in 1997, and from a needle from an eight-year-old Pinus radiata tree on the West Coast of the South Island of New Zealand in 2005, respectively. Monospore cultures, whose identities were confirmed by ribosomal ITS sequencing, were used throughout. Unless specified otherwise, cultures of these fungi were maintained on potato dextrose agar (PDA) or potato dextrose broth (PDB) media (C. fulvum) or Dothistroma Medium (DM; 5% w/v malt extract, 2.8% w/v nutrient agar or nutrient broth) at 22°C prior to use. Growth conditions used for generation of EST libraries (Protocol S1) are shown in Tables S12 and S13. Cultures were maintained for long-term storage in closed vials at −80°C stocks in 20% glycerol.
Tomato infections
Conidia of C. fulvum were harvested from two-week-old PDA plates with distilled water. The conidial suspension was filtered through Calbiochem Miracloth (EMD Millipore Chemicals, Philadelphia, PA) and washed once with water prior to calibration to 5×105 conidia/mL. Five-week-old Heinz tomato plants were sprayed on the lower side of the leaves with the conidial suspension (10 mL per plant). The plants were kept at 100% relative humidity for 48 h. The plastic-covered cages were then opened to grow the plants under regular greenhouse conditions (70% relative humidity, 23–25°C during daytime and 19–21°C at night, light/dark regime of 16/8 h, and 100 W/m2 supplemental light when the sunlight influx intensity was less than 150 W/m2). The 4th composite leaves of infected tomato plants were harvested at 2, 4, 8, 12 and 16 dpi, and immediately frozen in liquid nitrogen.
Phylogenetic comparison of fungal species
To highlight the phylogenetic relationships of C. fulvum and D. septosporum with Dothideomycetes and other fungi relevant to this study, conserved protein families were predicted by use of the MCL Markov clustering program [116] with pairwise blastp protein similarities and an inflation factor of 4. From this multi-gene family set, 51 orthologous groups of genes were identified. Predicted protein sequences were concatenated, aligned using MAFFT 6.717b [117] and a species tree calculated using RAxML 7.2.8 [118]. We also determined protein homology data based on bidirectional best hits when comparing the proteomes of eleven Dothideomycete species (Alternaria brassicicola, C. fulvum, Cochliobolus heterostrophus, D. septosporum, Hysterium pulicare, Mycosphaerella fijiensis, Mycosphaerella graminicola, Pyrenophora tritici-repentis, Rhytidhysteron rufulum, Septoria musiva and Stagonospora nodorum), together with four out-group species (Aspergillus nidulans, Fusarium graminearum, Neurospora crassa and Magnaporthe grisea).
Repetitive sequences and transposable elements
Repeat sequences in both genomes were identified using RECON [119]. To group repetitive elements together into different families the default RECON output was parsed to include families with 10 or more elements. The parsed RECON repeat library was used to determine the extent of the repetitive fraction in the D. septosporum and C. fulvum genomes using RepeatMasker [120] and to annotate repetitive families and identify structural features, such as Long Terminal Repeats (LTRs) and Terminal Inverted Repeats (TIRs), using BLAST.
Repeat-induced point mutation (RIP)
Sequences that had undergone Repeat-Induced Point mutation (RIP) were identified according to the composite RIP index (CRI) method [121]. The CRI was calculated for each 500-nt sequence window, which was shifted at each 25-nt step. Sequences were identified as having been subjected to RIP when the RIP product, RIP substrate and composite RIP indices were at least 1.2, at most 0.8 and at least 1.0 respectively. As a final constraint, a series of overlapping sequence windows had to exceed 750 nt in length and the CRI value of any of the windows peaked to 1.5 in order to be scored as a RIP'd locus.
Syntenic and non-syntenic regions
Syntenic regions shared between C. fulvum and D. septosporum were detected ab initio on their repeat-masked genome sequences using promer [122], blastp and a suite of custom made python scripts. A script called blastpmer obtained all translated ORFs above a threshold nucleotide length from both query and subject genomes, performed a blastp on these ORFs, and subsequently filtered on expected value and high-scoring segment pair (HSP) length. Protein matches (using C. fulvum as query and D. septosporum as subject) were obtained with promer (–maxmatch) and blastpmer (–ORF 500 nt –HSP 250 nt –expect 1e-9). Both genomes were masked for these protein matches before being subjected to a second round of searching for weaker and shorter protein similarities, again using promer (–b 50 –c 15 –l 5 –maxmatch) and blastpmer (–ORF 300 nt –HSP 110 nt –e 1e-7). These four searches yielded 57,270, 44,865, 1,864 and 2,367 matches, respectively, many of which were redundant and overlapping. This large set was reduced to 24,480 unique matches by removing all except the best alignment for each unique genomic locus. This step removed overlapping alignments with different phases or orientations, and excluded suboptimal alignments caused by paralogs and common protein domains. The product of amino acid similarity and match length was employed as a final alignment quality score. Matches were ordered by query scaffold position and joined into linked syntenic regions according to the following criteria: (i) adjacent matches were identical on the query and subject scaffolds; (ii) matches had the same strand orientation; and (iii) maximum and average nucleotide distance between adjacent matches on the query and subject scaffolds were <10-kb and <5-kb, respectively. This step resulted in a reduction to 1,875 collinear match regions, of which 1,277 were >5-kb. For comparison of protein-coding genes in syntenic versus non-syntenic areas, gene models were classified as syntenic if they overlapped with any of the 1,875 collinear syntenic areas. Thus, subsets of 9,890 syntenic and 4,237 non-syntenic genes were inferred for C. fulvum.
To investigate mesosynteny on a whole-genome scale, a refined synteny dataset was created with correction for inversions and rearrangements, and removal of spurious, small alignments. Match regions were compared and merged further if (i) adjacent groups had opposite orientations; or (ii) groups with identical query and subject scaffolds were separated by at least one (group of) matches on a conflicting subject scaffold, but maximum and average nucleotide distances between match regions were at most 20-kb or on average <10-kb apart; and finally (iii) match regions <5-kb were rejected. The final refined dataset contained 1,103 syntenic regions between 5 and 226-kb (average 22,194-bp), representing 22,700 matches from the original 24,480 unique matches.
C. fulvum and D. septosporum-specific proteins
To identify potential C. fulvum and D. septosporum-specific proteins, the total protein sets from both fungi were used in comparative blastMatrix [123] searches against sequences from the nine additional members of the Dothideomycetes listed in the phylogenetics section.
C. fulvum and D. septosporum secretome analysis
Initially, subcellular localizations for all C. fulvum and D. septosporum proteins were predicted using WoLF PSORT (wolfpsort.org; [124]). Only proteins containing a signal peptide and a signal peptide cleavage site, but lacking transmembrane (TM) domains or proteins containing a single TM that overlaps with the secretion signal, were selected. Signal peptides and cleavage sites were predicted using SignalP version 3.0 [125], where a final D-Score cut-off of 0.5 was used to increase specificity while retaining sensitivity. Subsequently, all proteins with signal peptides (1,886 and 1,591 for C. fulvum and D. septosporum, respectively) were analyzed for the presence of TM domains using the web servers Phobius [126] and TMHMM (version 2.0; [127]). The servers identified different, partially overlapping, sets of proteins with putative TM domains. On average Phobius detected 22% more TM domain proteins than did TMHMM, and about 75% of the predictions were shared between the servers. For further analyses, all proteins with putative TM domains as predicted by either of the two servers were removed from the dataset. Then, the proteins that contain a putative mitochondrial targeting signal as predicted by TargetP version 1.1 [128] were removed. Finally, proteins containing a potential GPI-anchor signal as predicted by the PredGPI web service were discarded [129].
Functional analysis of D. septosporum Avr4 by A. tumefaciens-mediated transient gene expression in N. benthamiana
A C. fulvum Avr4 (Cf-Avr4) gene homolog was identified in the genome of D. septosporum (Ds-Avr4) by blastp, with an E-value of 1×10−4. To determine Cf-4-mediated HR-inducing ability of Ds-Avr4 of D. septosporum, the Agrobacterium tumefaciens-mediated transient gene expression assay (ATTA) was performed in N. benthamiana as described by Van der Hoorn et al. [130]. The Cf-Avr4 and Ds-Avr4 genes were each fused to a PR-1A signal peptide sequence [131] for secretion into the apoplast. Subsequently a Gateway cloning strategy was performed to clone them into a pK2GW7 binary expression vector [132] containing the CaMV 35S promoter. A. tumefaciens (strain GV3101) was finally transformed with pK2GW7 binary vectors containing Cf-Avr4 or Ds-Avr4 genes by electroporation. Agroinfiltration of Cf-4 transgenic N. benthamiana leaves with Cf-Avr4- and Ds-Avr4-containing A. tumefaciens clones was performed as described by van der Hoorn et al. [130]. Photographs were taken at six days post inoculation.
Heterologous expression of the D. septosporum Ecp2 (Ds-Ecp2-1) gene in MM-Cf-Ecp2 tomato plants
Three D. septosporum homologs of C. fulvum Ecp2 genes (Ds-Ecp2-1, Ds-ecp2-2 and Ds-Ecp2-3) were identified as described for Avr4. A binary Potato Virus X (PVX)–based vector, pSfinx, was used for transient expression of the Cf-Ecp2-1 ortholog, Ds-Ecp2-1, in MM-Cf-Ecp2 tomato lines based on methodology described by Hammond-Kosack et al. [131]. The recombinant viruses were obtained by cloning Ds-Ecp2-1 (an intron-less gene), encoding the mature protein, downstream of the PR-1A signal sequence for secretion into the apoplast and under the control of the CaMV 35S promoter. Recombinant pSfinx::Ecp2-1, corresponding to the C. fulvum Ecp2 (Cf-Ecp2-1), and pSfinx::Empty viruses were as published [33]. A. tumefaciens (GV3101) was transformed with the pSfinx::Ds-Ecp2-1 construct by electroporation. A. tumefaciens strains containing the pSfinx constructs for the expression of Cf-Ecp2-1 and Ds-Ecp2-1 proteins were inoculated on MM-Cf-Ecp2 tomato lines containing the cognate R gene, and MM-Cf-0 tomato lines that contain no R genes, mediating recognition of the Ecp2-1 effector. Photographs were taken four weeks post inoculation.
Analyses of hydrophobin-encoding genes
All six previously reported hydrophobin genes from C. fulvum [133] were found in the automated gene predictions performed on the genome sequence. Five of the hydrophobins (Hcf-1 to Hcf-5) are predicted to contain an interpro motif common in fungal hydrophobins (IPR001338), while Hcf-6 has an interpro motif, which is restricted to Ascomycetes only (IPR010636). To identify putative hydrophobin-encoding genes in other genomes, all secreted gene models of C. fulvum, D. septosporum and M. graminicola were computationally annotated using Interpro scan and Gene Ontology terms. Then, gene models with IPR001338 and IPR010636 Interpro scan terms were identified as putative hydrophobin candidates. Also, a HMM profile search (which was built based on the conserved cysteine motifs in class I hydrophobins) was performed to identify hydrophobins missed by standard similarity searches. In this way five additional hydrophobin genes were identified in the C. fulvum genome. Hydrophobin sequences were aligned with ClustalW and edited in GeneDoc software. Then a consensus phylogenetic tree of predicted hydrophobin amino acid sequences was constructed using MEGA5 software [134] performing the minimum-evolution algorithm with default parameters and 1000 bootstrap replications.
Analyses of carbohydrate-active (CAZy) enzymes
The carbohydrate-active enzyme catalogs of C. fulvum and D. septosporum were compared with the corresponding catalogs from other Dothideomycete fungi [42]. The boundaries of the carbohydrate-active modules and associated carbohydrate-binding modules of the proteins encoded by each fungus in the comparison were determined using the BLAST and HMM-based routines of the Carbohydrate-Active-EnZymes database ([74]; www.cazy.org). For determining the growth profiles on different carbohydrate substrates Aspergillus minimal medium [87] adjusted to pH 6.0 and containing 1.5% agar (Invitrogen, 30391–049) was used. Carbon sources were added at concentrations as indicated in the text and using standard methods as described at www.fung-growth.org. Duplicate plates were inoculated with 2 µL of a suspension containing 500 conidia/µL. Cultures were grown at 22–25°C for two weeks for C. fulvum and four weeks for D. septosporum, and representative plates were photographed.
Secondary metabolite gene analysis
Genes encoding polyketide synthases (PKSs), non-ribosomal peptide synthases (NRPSs), hybrids of PKS and NRPS, terpene cyclases (TCs) and dimethylallyl tryptophan synthases (DMATSs) were sought in the two genomes using tblastn/blastp and several Ascomycete protein sequences as queries (Ace1 for PKS and hybrids; MGG_00022.7 protein for NRPS; tri5, cps/ks, all TCs from B. cinerea for TCs; Dma1 from Claviceps purpurea for DMATSs). For each tblastn/blastp hit, search for conserved domains (CDS at NCBI, InterproScan) and blastp analysis at NCBI and InterproScan confirmed the functional annotation. The locus of each key gene was analyzed for genes that could potentially be involved in a biosynthetic pathway. Functional annotation of downstream and upstream genes was confirmed using blastp at NCBI. In addition, homologs to genes that were shown to be involved in the regulation of fungal development and secondary metabolism were sought using tblastn/blastp with the sequences of the characterized proteins as queries.
Ka/Ks calculations were carried out to estimate evolutionary constraints on putative dothistromin genes (PksA, VbsA, Ver1, HexA, AvfA, CypA and MoxA) in comparison to four housekeeping genes (Tub1 JGI PIDs Cf-186859 Ds-68998, Eif3b Cf-190521 Ds-75033, Pap1 Cf-190301 Ds-180959 and Rps9 Cf-196996 Ds-92035). DNA sequences from D. septosporum and C. fulvum were aligned with the codon-aware multiple sequence alignment software, RevTrans [135]. Sequence alignments were trimmed in codon units to remove missing data across both species with the sequence editor, Jalview [136]. The non-synonymous/synonymous amino acid ratio (Ka/Ks or ω) was obtained using the Ka/Ks Calculator [137] with the algorithm of Nei and Gojobori [138]. Statistical differences between Ka/Ks values for dothistromin and housekeeping genes were determined using Student's two-sided t test [139]. For determination of dothistromin production, previously published extraction and hplc methods were followed [140].
Quantitative PCR
For quantification of dothistromin gene expression in D. septosporum, RNA was extracted from sporulating lesions on Pinus radiata needles collected from a forest in New Zealand (in planta sample) or grown in PDB or B5 [141] broths for 6 days as described previously [140]. cDNA synthesis and relative quantitative RT-PCR were carried out using primers and methods described earlier [140], with three biological replicates and two technical replicates. For C. fulvum, similar protocols were followed except that tomato infections, RNA extraction and cDNA synthesis followed the protocols of van Esse et al. [142] and four biological replicates were used. Oligonucleotides were designed with Primer3Plus [143] and are shown in Table S14. Their efficiency and specificity were tested on a genomic DNA dilution series. For both species, quantitative PCR was performed with the Applied Biosystems 7300 Real-Time PCR system (Applied Biosystems, USA) using the default parameters. Raw data were analyzed using the 2−ΔCt method [144].
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. van KanJAL, van den AckervekenGFJM, de WitPJGM (1991) Cloning and characterization of cDNA of avirulence gene Avr9 of the fungal pathogen Cladosporium fulvum, causal agent of tomato leaf mold. Mol Plant-Microbe Interact 4 : 52–59.
2. ThommaBPHJ, Van EsseHP, CrousPW, De WitPJGM (2005) Cladosporium fulvum (syn. Passalora fulva), a highly specialized plant pathogen as a model for functional studies on plant pathogenic Mycosphaerellaceae. Mol Plant Pathol 6 : 379–393.
3. GoodwinSB, DunkleLD, ZismannVL (2001) Phylogenetic analysis of Cercospora and Mycosphaerella based on the internal transcribed spacer region of ribosomal DNA. Phytopathology 91 : 648–658.
4. BradshawRE, ZhangSG (2006) Biosynthesis of dothistromin. Mycopathologia 162 : 201–213.
5. BarnesI, CrousPW, WingfieldBD, WingfieldMJ (2004) Multigene phylogenies reveal that red band needle blight of Pinus is caused by two distinct species of Dothistroma, D. septosporum and D. pini. Stud Mycol 50 : 551–565.
6. JenkinsJA (1948) The origin of the cultivated tomato. Econ Bot 2 : 379–392.
7. CookeMC (1883) New American fungi. Grevillea XII: 32.
8. De WitPJGM (1992) Molecular characterization of gene-for-gene systems in plant-fungus interactions and the application of avirulence genes in control of plant-pathogens. Annu Rev Phytopathol 30 : 391–418.
9. EnyaJ, IkedaK, TakeuchiT, HorikoshiN, HigashiT, et al. (2009) The first occurrence of leaf mold of tomato caused by races 4.9 and 4.9.11 of Passalora fulva (syn. Fulvia fulva) in Japan. J Gen Plant Pathol 75 : 76–79.
10. IidaY, IwadateY, KubotaM, TeramiF (2010) Occurrence of a new race 2.9 of leaf mold of tomato in Japan. J Gen Plant Pathol 76 : 84–86.
11. BednarovaM, PalovcikovaD, JankovskyL (2006) The host spectrum of Dothistroma needle blight Mycosphaerella pini E. Rostrup - new hosts of Dothistroma needle bight observed in the Czech Republic. J For Sci 52 : 30–36.
12. BradshawRE (2004) Dothistroma (red-band) needle blight of pines and the dothistromin toxin: a review. For Pathol 34 : 163–185.
13. WoodsAJ, CoatesKD, HamannA (2005) Is an unprecedented Dothistroma needle blight epidemic related to climate change? Bioscience 55 : 761–769.
14. Brown A, Webber JF (2008) Red band needle blight of conifers in Britain. Edinburgh, UK: Forestry Commission. 1–8 p.
15. WoodsA (2011) Is the health of British Columbia's forests being influenced by climate change? If so, was this predictable? Can J Plant Pathol 33 : 117–126.
16. LazarovitsG, HigginsVJ (1976) Ultrastucture of susceptible, resistant and immune reactions of tomato to races of Cladosporium fulvum. Can J Bot 54 : 235–249.
17. LazarovitsG, HigginsVJ (1976) Histological comparison of Cladosporium fulvum race 1 on immune, resistant and susceptible tomato varieties. Can J Bot 54 : 224–234.
18. De WitPJGM (1977) A light and scanning-electron microscopic study of the infection of tomato plants by virulent and avirulent races of Cladosporium fulvum. Neth J Plant Pathol 83 : 109–122.
19. MuirJA, CobbJFW (2005) Infection of radiata and bishop pine by Mycosphaerella pini in California. Can J For Res 35 : 2529–2538.
20. GadgilPD (1967) Infection of Pinus radiata needles by Dothistroma pini. N Z J Bot 5 : 498–503.
21. StergiopoulosI, GroenewaldM, StaatsM, LindhoutP, CrousPW, et al. (2007) Mating-type genes and the genetic structure of a world-wide collection of the tomato pathogen Cladosporium fulvum. Fungal Genet Biol 44 : 415–429.
22. GroenewaldM, BarnesI, BradshawRE, BrownAV, DaleA, et al. (2007) Characterization and distribution of mating type genes in the Dothistroma needle blight pathogens. Phytopathology 97 : 825–834.
23. TomšovskýM, TomešováV, PalovčíkováD, KostovčíkM, RohrerM, et al. (2012) The gene flow and mode of reproduction of Dothistroma septosporum in the Czech Republic. Plant Pathol (In Press) DOI: 10.1111/j.1365-3059.2012.02625.x.
24. DaleAL, LewisKJ, MurrayBW (2011) Sexual Reproduction and Gene Flow in the Pine Pathogen Dothistroma septosporum in British Columbia. Phytopathology 101 : 68–76.
25. HirstP, RichardsonTE, CarsonSD, BradshawRE (1999) Dothistroma pini genetic diversity is low in New Zealand. N Z J For Sci 29 : 459–472.
26. JoostenMHAJ, De WitPJGM (1999) The tomato-Cladosporium fulvum interaction: A versatile experimental system to study plant-pathogen interactions. Annu Rev Phytopathol 37 : 335–367.
27. De WitPJGM, JoostenMHAJ, ThommaBPHJ, StergiopoulosI (2009) Gene-for-gene models and beyond: the Cladosporium fulvum-tomato pathosystem. The Mycota V: 135–156.
28. De JongeR, Van EsseHP, KombrinkA, ShinyaT, DesakiY, et al. (2010) Conserved fungal LysM effector Ecp6 prevents chitin-triggered immunity in plants. Science 329 : 953–955.
29. De KockMJD, BrandwagtBF, BonnemaG, De WitPJGM, LindhoutP (2005) The tomato Orion locus comprises a unique class of Hcr9 genes. Mol Breed 15 : 409–422.
30. van EsseHP, BoltonMD, StergiopoulosI, de WitPJGM, ThommaBPHJ (2007) The chitin-binding Cladosporium fulvum effector protein Avr4 is a virulence factor. Mol Plant-Microbe Interact 20 : 1092–1101.
31. van den BurgHA, HarrisonSJ, JoostenMHAJ, VervoortJ, de WitPJGM (2006) Cladosporium fulvum Avr4 protects fungal cell walls against hydrolysis by plant chitinases accumulating during infection. Mol Plant-Microbe Interact 19 : 1420–1430.
32. StergiopoulosI, KourmpetisYAI, SlotJC, BakkerFT, De WitPJGM, et al. (2012) In silico characterization and molecular evolutionary analysis of a novel superfamily of fungal effector proteins. Mol Biol Evol 29 : 3371–3384.
33. StergiopoulosI, Van den BurgHA, ÖkmenB, BeenenH, KemaGHJ, et al. (2010) Tomato Cf resistance proteins mediate recognition of cognate homologous effectors from fungi pathogenic on dicots and monocots. Proc Natl Acad Sci, USA 107 : 7610–7615.
34. ShawGJ, ChickM, HodgesR (1978) A 13C-NMR study of the biosynthesis of the anthraquinone dothistromin by Dothistroma pini. Phytochemistry 17 : 1743–1745.
35. SchwelmA, BarronNJ, BakerJ, DickM, LongPG, et al. (2009) Dothistromin toxin is not required for dothistroma needle blight in Pinus radiata. Plant Pathol 58 : 293–304.
36. BradshawRE, JinHP, MorganBS, SchwelmA, TeddyOR, et al. (2006) A polyketide synthase gene required for biosynthesis of the aflatoxin-like toxin, dothistromin. Mycopathologia 161 : 283–294.
37. SchwelmA, BarronNJ, ZhangS, BradshawRE (2008) Early expression of aflatoxin-like dothistromin genes in the forest pathogen Dothistroma septosporum. Mycol Res 112 : 138–146.
38. ZhangS, SchwelmA, JinHP, CollinsLJ, BradshawRE (2007) A fragmented aflatoxin-like gene cluster in the forest pathogen Dothistroma septosporum. Fungal Genet Biol 44 : 1342–1354.
39. SchwelmA, BradshawRE (2010) Genetics of dothistromin biosynthesis of Dothistroma septosporum: an update. Toxins 2 : 2680–2698.
40. GrigorievIV, NordbergH, ShabalovI, AertsA, CantorM, et al. (2012) The genome portal of the Department of Energy Joint Genome Institute. Nucleic Acids Res 40: D26–32.
41. van der BurgtA, SeveringE, de WitP, CollemareJ (2012) Birth of new spliceosomal introns in fungi by multiplication of introner-like elements. Curr Biol 22 : 1260–1265.
42. OhmRA, FeauN, HenrissatB, SchochCL, HorwitzBA, et al. (2012) Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes fungi. PLoS Pathogens (In Press)..
43. AmselemJ, CuomoCA, van KanJAL, ViaudM, BenitoEP, et al. (2011) Genomic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet 7: e1002230 doi:10.1371/journal.pgen.1002230.
44. SpanuPD, AbbottJC, AmselemJ, BurgisTA, SoanesDM, et al. (2010) Genome expansion and gene loss in powdery mildew fungi reveal tradeoffs in extreme parasitism. Science 330 : 1543–1546.
45. SchmidtSM, PanstrugaR (2011) Pathogenomics of fungal plant parasites: what have we learnt about pathogenesis? Curr Opin Plant Biol 14 : 392–399.
46. PoulterRTM, GoodwinTJD, ButlerMI (2003) Vertebrate helentrons and other novel helitrons. Gene 313 : 201–212.
47. KapitonovVV, JurkaJ (2007) Helitrons on a roll: eukaryotic rolling-circle transposons. Trends Genet 23 : 521–529.
48. LambJC, TheuriJ, BirchlerJA (2004) What's in a centromere? Genome Biology 5 : 239.
49. RoyB, SanyalK (2011) Diversity in requirement of genetic and epigenetic factors for centromere function in fungi. Eukaryot Cell 10 : 1384–1395.
50. FreitagM, WilliamsRL, KotheGO, SelkerEU (2002) A cytosine methyltransferase homologue is essential for repeat-induced point mutation in Neurospora crassa. Proc Natl Acad Sci USA 99 : 8802–8807.
51. HaneJK, LoweRGT, SolomonPS, TanKC, SchochCL, et al. (2007) Dothideomycete-plant interactions illuminated by genome sequencing and EST analysis of the wheat pathogen Stagonospora nodorum. Plant Cell 19 : 3347–3368.
52. FudalI, RossS, BrunH, BesnardA-L, ErmelM, et al. (2009) Repeat-induced point mutation (RIP) as an alternative mechanism of evolution toward virulence in Leptosphaeria maculans. Mol Plant-Microbe Interact 22 : 932–941.
53. GoutL, FudalI, KuhnML, BlaiseF, EckertM, et al. (2006) Lost in the middle of nowhere: the AvrLm1 avirulence gene of the Dothideomycete Leptosphaeria maculans. Mol Microbiol 60 : 67–80.
54. RouxelT, GrandaubertJ, HaneJK, HoedeC, van de WouwAP, et al. (2011) Effector diversification within compartments of the Leptosphaeria maculans genome affected by Repeat-Induced Point mutations. Nat Commun 2 : 202.
55. NowrousianM, StajichJE, ChuM, EnghI, EspagneE, et al. (2010) De novo assembly of a 40 Mb eukaryotic genome from short sequence reads: Sordaria macrospora, a model organism for fungal morphogenesis. PLoS Genet 6: e1000891 doi:10.1371/journal.pgen.1000891.
56. HaneJK, RouxelT, HowlettBJ, KemaGHJ, GoodwinSB, et al. (2011) A novel mode of chromosomal evolution peculiar to filamentous Ascomycete fungi. Genome Biol 12: R45.
57. ThonMR, PanHQ, DienerS, PapalasJ, TaroA, et al. (2006) The role of transposable element clusters in genome evolution and loss of synteny in the rice blast fungus Magnaporthe oryzae. Genome Biol 7: R16.
58. StergiopoulosI, De WitPJGM (2009) Fungal effector proteins. Annu Rev Phytopathol 47 : 233–263.
59. GoodwinSB, Ben M'BarekS, DhillonB, WittenbergAHJ, CraneCF, et al. (2011) Finished genome of the fungal wheat pathogen Mycosphaerella graminicola reveals dispensome structure, chromosome plasticity, and stealth pathogenesis. PLoS Genet 7: e1002070 doi:10.1371/journal.pgen.1002070.
60. RivasS, ThomasCM (2005) Molecular interactions between tomato and the leaf mold pathogen Cladosporium fulvum. Annu Rev Phytopathol 43 : 395–436.
61. WilcoxPL, AmersonHV, KuhlmanEG, LiuBH, O'MalleyDM, et al. (1996) Detection of a major gene for resistance to fusiform rust disease in loblolly pine by genomic mapping. Proc Natl Acad Sci USA 93 : 3859–3864.
62. LiuJJ, EkramoddoullahAKM (2011) Genomic organization, induced expression and promoter activity of a resistance gene analog (PmTNL1) in western white pine (Pinus monticola). Planta 233 : 1041–1053.
63. NelsonCD, KubisiakTL, AmersonHV (2010) Unravelling and managing fusiform rust disease: a model approach for coevolved forest tree pathosystems. For Pathol 40 : 64–72.
64. van den AckervekenGFJM, van KanJAL, de WitPJGM (1992) Molecular analysis of the avirulence gene Avr9 of the fungal tomato pathogen Cladosporium fulvum fully supports the gene-for-gene hypothesis. Plant J 2 : 359–366.
65. ChumaI, IsobeC, HottaY, IbaragiK, FutamataN, et al. (2011) Multiple translocation of the AVR-Pita effector gene among chromosomes of the rice blast fungus Magnaporthe oryzae and related species. PLoS Pathog 7: e1002147 doi:10.1371/journal.ppat.1002147.
66. WesterinkN, BrandwagtBF, De WitPJGM, JoostenMHAJ (2004) Cladosporium fulvum circumvents the second functional resistance gene homologue at the Cf-4 locus (Hcr9-4E) by secretion of a stable avr4E isoform. Mol Microbiol 54 : 533–545.
67. LaugéR, GoodwinPH, De WitPJGM, JoostenMHAJ (2000) Specific HR-associated recognition of secreted proteins from Cladosporium fulvum occurs in both host and non-host plants. Plant J 23 : 735–745.
68. WesselsJGH (1994) Developmental regulation of fungal cell-wall formation. Annu Rev Phytopathol 32 : 413–437.
69. TempletonMD, RikkerinkEHA, BeeverRE (1994) Small, cysteine-rich proteins and recognition in fungal-plant interactions. Mol Plant-Microbe Interact 7 : 320–325.
70. JoostenMHAJ, VerbakelHM, NettekovenME, van LeeuwenJ, van der VossenRTM, et al. (2005) The phytopathogenic fungus Cladosporium fulvum is not sensitive to the chitinase and β-1,3-glucanase defence proteins of its host, tomato. Physiol Mol Plant Pathol 46 : 45–59.
71. SpanuP (1997) HCF-1, a hydrophobin from the tomato pathogen Cladosporium fulvum. Gene 193 : 89–96.
72. WhitefordJR, SpanuPD (2001) The hydrophobin HCf-1 of Cladosporium fulvum is required for efficient water-mediated dispersal of conidia. Fungal Genet Biol 32 : 159–168.
73. LacroixH, WhitefordJR, SpanuPD (2008) Localization of Cladosporium fulvum hydrophobins reveals a role for HCf-6 in adhesion. FEMS Microbiol Lett 286 : 136–144.
74. CantarelBL, CoutinhoPM, RancurelC, BernardT, LombardV, et al. (2009) The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res 37: D233–238.
75. MartinezD, BerkaRM, HenrissatB, SaloheimoM, ArvasM, et al. (2008) Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nat Biotechnol 26 : 553–560.
76. PutoczkiTL, GerrardJA, ButterfieldBG, JacksonSL (2008) The distribution of un-esterified and methyl-esterified pectic polysaccharides in Pinus radiata. IAWA Journal 29 : 115–127.
77. RidleyBL, O'NeillMA, MohnenDA (2001) Pectins: structure, biosynthesis, and oligogalacturonide-related signaling. Phytochemistry 57 : 929–967.
78. SprockettDD, PiontkivskaH, BlackwoodCB (2011) Evolutionary analysis of glycosyl hydrolase family 28 (GH28) suggests lineage-specific expansions in necrotrophic fungal pathogens. Gene 479 : 29–36.
79. Ten HaveA, MulderW, VisserJ, van KanJAL (1998) The endopolygalacturonase gene Bcpg1 is required for full virulence of Botrytis cinerea. Mol Plant-Microbe Interact 11 : 1009–1016.
80. van KanJAL (2006) Licensed to kill: the lifestyle of a necrotrophic plant pathogen. Trends Plant Sci 11 : 247–253.
81. EastwoodDC, FloudasD, BinderM, MajcherczykA, SchneiderP, et al. (2011) The plant cell wall-decomposing machinery underlies the functional diversity of forest fungi. Science 333 : 762–765.
82. OhmRA, de JongJF, LugonesLG, AertsA, KotheE, et al. (2010) Genome sequence of the model mushroom Schizophyllum commune. Nat Biotechnol 28 : 957–U910.
83. van den BrinkJ, de VriesRP (2011) Fungal enzyme sets for plant polysaccharide degradation. Appl Microbiol Biotechnol 91 : 1477–1492.
84. LeeMH, ChiuCM, RoubtsovaT, ChouCM, BostockRM (2010) Overexpression of a redox-regulated cutinase gene, MfCUT1, increases virulence of the brown rot pathogen Monilinia fructicola on Prunus spp.. Mol Plant-Microbe Interact 23 : 176–186.
85. SkamniotiP, GurrSJ (2008) Cutinase and hydrophobin interplay: A herald for pathogenesis? Plant Signal Behav 3 : 248–250.
86. KingBC, WaxmanKD, NenniNV, WalkerLP, BergstromGC, et al. (2011) Arsenal of plant cell wall degrading enzymes reflects host preference among plant pathogenic fungi. Biotechnol Biofuels 4 : 4.
87. de VriesRP, BurgersK, van de VondervoortPJI, FrisvadJC, SamsonRA, et al. (2004) A new black Aspergillus species, A. vadensis, is a promising host for homologous and heterologous protein production. Appl Environ Microbiol 70 : 3954–3959.
88. CoutinhoPM, AndersenMR, KolenovaK, vanKuykPA, BenoitI, et al. (2009) Post-genomic insights into the plant polysaccharide degradation potential of Aspergillus nidulans and comparison to Aspergillus niger and Aspergillus oryzae. Fungal Genet Biol 46: S161–S169.
89. BergB, DeantaRC, EscuderoA, GardenasA, JohanssonMB, et al. (1995) The chemical composition of newly shed needle litter of scots pine and some other pine species in a climatic transect. X. Long-term decomposition in a scots pine forest. Can J Bot 73 : 1423–1435.
90. VogelJ (2008) Unique aspects of the grass cell wall. Curr Opin Plant Biol 11 : 301–307.
91. Pareja-JaimeY, RonceroMIG, Ruiz-RoldánMC (2008) Tomatinase from Fusarium oxysporum f. sp. lycopersici is required for full virulence on tomato plants. Mol Plant-Microbe Interact 21 : 728–736.
92. BartheGA, JourdanPS, McIntoshCA, MansellRL (1988) Radioimmunoassay for the quantitative determination of hesperidin and analysis of its distribution in Citrus sinensis. Phytochemistry 27 : 249–254.
93. DiGuistiniS, WangY, LiaoNY, TaylorG, TanguayP, et al. (2011) Genome and transcriptome analyses of the mountain pine beetle-fungal symbiont Grosmannia clavigera, a lodgepole pine pathogen. Proc Natl Acad Sci USA 108 : 2504–2509.
94. HammerschmidtR, BonnenAM, BergstromGC (1983) Association of lignification with non-host resistance of cucurbits. Phytopathology 73 : 829–829.
95. XuL, ZhuL, TuL, LiuL, YuanD, et al. (2011) Lignin metabolism has a central role in the resistance of cotton to the wilt fungus Verticillium dahliae as revealed by RNA-Seq-dependent transcriptional analysis and histochemistry. J Exp Bot 62 : 5607–5621.
96. BergB, WessenB, EkbohmG (1982) Nitrogen level and decomposition in scots pine needle litter. Oikos 38 : 291–296.
97. LukjanovaA, MandreM (2008) Anatomical structure and localisation of lignin in needles and shoots of Scots pine (Pinus sylvestris L.) growing in a habitat with varying environmental characteristics. For Stud 49 : 37–46.
98. KirkTK, FarrellRL (1987) Enzymatic “combustion”: the microbial degradation of lignin. Annu Rev Microbiol 41 : 465–505.
99. Ruiz-DueñasFJ, MartínezAT (2009) Microbial degradation of lignin: how a bulky recalcitrant polymer is efficiently recycled in nature and how we can take advantage of this. Microb Biotechnol 2 : 164–177.
100. Collemare J, Lebrun M-H (2012) Fungal secondary metabolites: ancient toxins and novel effectors in plant-microbe interactions. In: Martin F, Kamoun S, editors. Effectors in Plant-Microbe Interactions: Blackwell Publishing Ltd. pp. 379–402.
101. DaviesDG, HodgeP (1974) Chemistry of quinones. 5. Structure of cladofulvin, a bianthraquinone from Cladosporium fulvum Cooke. J Chemi Soc-Perkin Trans 1 : 2403–2405.
102. BradshawRE, BhatnagarD, GanleyRJ, GillmanCJ, MonahanBJ, et al. (2002) Dothistroma pini, a forest pathogen, contains homologs of aflatoxin biosynthetic pathway genes. Appl Environ Microbiol 68 : 2885–2892.
103. KellerNP, HohnTM (1997) Metabolic pathway gene clusters in filamentous fungi. Fungal Genet Biol 21 : 17–29.
104. KellerNP, TurnerG, BennettJW (2005) Fungal secondary metabolism - From biochemistry to genomics. Nat Rev Microbiol 3 : 937–947.
105. CollemareJ, BillardA, BoehnertHU, LebrunMH (2008) Biosynthesis of secondary metabolites in the rice blast fungus Magnaporthe grisea: the role of hybrid PKS-NRPS in pathogenicity. Mycol Res 112 : 207–215.
106. GalaganJE, CalvoSE, CuomoC, MaLJ, WortmanJR, et al. (2005) Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature 438 : 1105–1115.
107. CuomoCA, GueldenerU, XuJ-R, TrailF, TurgeonBG, et al. (2007) The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science 317 : 1400–1402.
108. KhaldiN, WolfeKH (2011) Evolutionary origins of the fumonisin secondary metabolite gene cluster in Fusarium verticillioides and Aspergillus niger. Int J Evol Biol 2011 : 423821.
109. YangZ, BielawskiJP (2000) Statistical methods for detecting molecular adaptation. Trends Ecol Evol 15 : 496–503.
110. EhrlichKC, YuJJ, CottyPJ (2005) Aflatoxin biosynthesis gene clusters and flanking regions. J Appl Microbiol 99 : 518–527.
111. BergmannS, FunkAN, ScherlachK, SchroeckhV, ShelestE, et al. (2010) Activation of a silent fungal polyketide biosynthesis pathway through regulatory cross talk with a cryptic nonribosomal peptide synthetase gene cluster. Appl Environ Microbiol 76 : 8143–8149.
112. BrakhageAA, SchroeckhV (2011) Fungal secondary metabolites - Strategies to activate silent gene clusters. Fungal Genet Biol 48 : 15–22.
113. BayramO, BrausGH (2011) Coordination of secondary metabolism and development in fungi: The velvet family of proteins. FEMS Microbiol Rev 35 : 1–24.
114. TsitsigiannisDI, KellerNP (2006) Oxylipins act as determinants of natural product biosynthesis and seed colonisation in Aspergillus nidulans. Mol Microbiol 59 : 882–892.
115. AdamsTH, BoylanMT, TimberlakeWE (1988) BrlA is necessary and sufficient to direct conidiophore development in Aspergillus nidulans. Cell 54 : 353–362.
116. EnrightAJ, Van DongenS, OuzounisCA (2002) An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res 30 : 1575–1584.
117. KatohK, TohH (2008) Recent developments in the MAFFT multiple sequence alignment program. Briefings Bioinf 9 : 286–298.
118. StamatakisA, HooverP, RougemontJ (2008) A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol 57 : 758–771.
119. BaoZR, EddySR (2002) Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res 12 : 1269–1276.
120. Smit AFA, Hubley R, Green P (1996–2010) RepeatMasker Open-3.0. Available: http://www.repeatmasker.org. Accessed 12 December 2011.
121. LewisZA, HondaS, KhlafallahTK, JeffressJK, FreitagM, et al. (2009) Relics of repeat-induced point mutation direct heterochromatin formation in Neurospora crassa. Genome Res 19 : 427–437.
122. DelcherAL, SalzbergSL, PhillippyAM (2003) Using MUMmer to identify similar regions in large sequence sets. Curr Protoc Bioinf 10.3.1–10.3.18.
123. ParkJ, ParkB, JungK, JangS, YuK, et al. (2008) CFGP: a web-based, comparative fungal genomics platform. Nucleic Acids Res 36: D562–D571.
124. HortonP, ParkK-J, ObayashiT, FujitaN, HaradaH, et al. (2007) WoLF PSORT: protein localization predictor. Nucleic Acids Res 35: W585–W587.
125. BendtsenJD, NielsenH, von HeijneG, BrunakS (2004) Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340 : 783–795.
126. KaellL, KroghA, SonnhammerELL (2007) Advantages of combined transmembrane topology and signal peptide prediction - the Phobius web server. Nucleic Acids Res 35: W429–W432.
127. KroghA, LarssonB, von HeijneG, SonnhammerELL (2001) Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J Mol Biol 305 : 567–580.
128. EmanuelssonO, NielsenH, BrunakS, von HeijneG (2000) Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol 300 : 1005–1016.
129. PierleoniA, MartelliPL, CasadioR (2008) PredGPI: a GPI-anchor predictor. BMC Bioinformatics 9 : 392.
130. van der HoornRAL, LaurentF, RothR, de WitPJGM (2000) Agroinfiltration is a versatile tool that facilitates comparative analyses of Avr9/Cf-9-induced and Avr4/Cf-4-induced necrosis. Mol Plant-Microbe Interact 13 : 439–446.
131. Hammond-KosackKE, StaskawiczBJ, JonesJDG, BaulcombeDC (1995) Functional expression of a fungal avirulence gene from a modified Potato-Virus-X genome. Mol Plant-Microbe Interact 8 : 181–185.
132. KarimiM, InzeD, DepickerA (2002) GATEWAY vectors for Agrobacterium-mediated plant transformation. Trends Plant Sci 7 : 193–195.
133. WhitefordJR, SpanuPD (2002) Hydrophobins and the interactions between fungi and plants. Mol Plant Pathol 3 : 391–400.
134. TamuraK, PetersonD, PetersonN, StecherG, NeiM, et al. (2011) MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28 : 2731–2739.
135. WernerssonR, PedersenAG (2003) RevTrans: Multiple alignment of coding DNA from aligned amino acid sequences. Nucl Acids Res 31 : 3537–3539.
136. WaterhouseAM, ProcterJB, MartinDMA, ClampM, BartonGJ (2009) Jalview version 2 — a multiple sequence alignment editor and analysis workbench. Bioinformatics 25 : 1189–1191.
137. ZhangZ, LiJ, ZhaoXQ, WangJ, WongGK, et al. (2006) KaKs Calculator: Calculating Ka and Ks through model selection and model averaging. Genomics Proteomics Bioinf 4 : 259–263.
138. NeiM, GojoboriT (1986) Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol 3 : 418–426.
139. Student (1908) The probable error of a mean. Biometrika 6 : 1–25.
140. ChettriP, CalvoAM, CaryJW, DhingraS, GuoYA, et al. (2012) The veA gene of the pine needle pathogen Dothistroma septosporum regulates sporulation and secondary metabolism. Fungal Genet Biol 49 : 141–151.
141. GamborgOL, MillerRA, OjimaK (1968) Nutrient requirements of suspension cultures of soybean root cell. Exp Cell Res 50 : 151–158.
142. van EsseHP, Van 't KloosterJW, BoltonMD, YadetaKA, van BaarlenP, et al. (2008) The Cladosporium fulvum virulence protein Avr2 inhibits host proteases required for basal defense. Plant Cell 20 : 1948–1963.
143. UntergasserA, NijveenH, RaoX, BisselingT, GeurtsR, et al. (2007) Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res 35: W71–75.
144. LivakKJ, SchmittgenTD (2001) Analysis of relative gene expression data using real time quantitative PCR and the 2−ΔΔCT method. Methods 25 : 402–408.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 11
Nejčtenější v tomto čísle
- Mechanisms Employed by to Prevent Ribonucleotide Incorporation into Genomic DNA by Pol V
- Inference of Population Splits and Mixtures from Genome-Wide Allele Frequency Data
- Zcchc11 Uridylates Mature miRNAs to Enhance Neonatal IGF-1 Expression, Growth, and Survival
- Histone Methyltransferases MES-4 and MET-1 Promote Meiotic Checkpoint Activation in