
Insulin/IGF-1 and Hypoxia Signaling Act in Concert to Regulate Iron Homeostasis in
Iron plays an essential role in many biological processes, but also catalyzes the formation of reactive oxygen species (ROS), which can cause molecular damage. Iron homeostasis is therefore a critical determinant of fitness. In Caenorhabditis elegans, insulin/IGF-1 signaling (IIS) promotes growth and reproduction but limits stress resistance and lifespan through inactivation of the DAF-16/FoxO transcription factor (TF). We report that long-lived daf-2 insulin/IGF-1 receptor mutants show a daf-16–dependent increase in expression of ftn-1, which encodes the iron storage protein H-ferritin. To better understand the regulation of iron homeostasis, we performed a TF–limited genetic screen for factors influencing ftn-1 gene expression. The screen identified the heat-shock TF hsf-1, the MAD bHLH TF mdl-1, and the putative histone acetyl transferase ada-2 as activators of ftn-1 expression. It also revealed that the HIFα homolog hif-1 and its binding partner aha-1 (HIFβ) are potent repressors of ftn-1 expression. ftn-1 expression is induced by exposure to iron, and we found that hif-1 was required for this induction. In addition, we found that the prolyl hydroxylase EGL-9, which represses HIF-1 via the von Hippel-Lindau tumor suppressor VHL-1, can also act antagonistically to VHL-1 in regulating ftn-1. This suggests a novel mechanism for HIF target gene regulation by these evolutionarily conserved and clinically important hydroxylases. Our findings imply that the IIS and HIF pathways act together to regulate iron homeostasis in C. elegans. We suggest that IIS/DAF-16 regulation of ftn-1 modulates a trade-off between growth and stress resistance, as elevated iron availability supports growth but also increases ROS production.
Published in the journal:
. PLoS Genet 8(3): e32767. doi:10.1371/journal.pgen.1002498
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002498
Summary
Iron plays an essential role in many biological processes, but also catalyzes the formation of reactive oxygen species (ROS), which can cause molecular damage. Iron homeostasis is therefore a critical determinant of fitness. In Caenorhabditis elegans, insulin/IGF-1 signaling (IIS) promotes growth and reproduction but limits stress resistance and lifespan through inactivation of the DAF-16/FoxO transcription factor (TF). We report that long-lived daf-2 insulin/IGF-1 receptor mutants show a daf-16–dependent increase in expression of ftn-1, which encodes the iron storage protein H-ferritin. To better understand the regulation of iron homeostasis, we performed a TF–limited genetic screen for factors influencing ftn-1 gene expression. The screen identified the heat-shock TF hsf-1, the MAD bHLH TF mdl-1, and the putative histone acetyl transferase ada-2 as activators of ftn-1 expression. It also revealed that the HIFα homolog hif-1 and its binding partner aha-1 (HIFβ) are potent repressors of ftn-1 expression. ftn-1 expression is induced by exposure to iron, and we found that hif-1 was required for this induction. In addition, we found that the prolyl hydroxylase EGL-9, which represses HIF-1 via the von Hippel-Lindau tumor suppressor VHL-1, can also act antagonistically to VHL-1 in regulating ftn-1. This suggests a novel mechanism for HIF target gene regulation by these evolutionarily conserved and clinically important hydroxylases. Our findings imply that the IIS and HIF pathways act together to regulate iron homeostasis in C. elegans. We suggest that IIS/DAF-16 regulation of ftn-1 modulates a trade-off between growth and stress resistance, as elevated iron availability supports growth but also increases ROS production.
Introduction
In order to survive in a changing environment, organisms have evolved abilities to sense their surroundings and adaptively adjust their physiology. For example, the nematode Caenorhabditis elegans is capable of postponing reproduction if conditions are unsuitable for growth and reproduction by forming dauer larvae [1], [2], [3]. This developmentally arrested third larval stage is resistant to starvation and other stressors, allowing the animal to survive until conditions improve. Should this occur, dauer larvae can re-enter the normal reproductive life cycle.
The decision between reproductive growth and survival with enhanced stress resistance is controlled by a complex sensory/signaling network that includes the insulin/IGF-1 signaling (IIS) pathway [2]. Mutants with reduced IIS exhibit constitutive dauer larva formation, but can also form adults that are resistant to a range of stressors, including reactive oxygen species (ROS), UV irradiation, heat stress and ER stress [4], [5], [6]. IIS controls this response through the DAF-16/FoxO transcription factor, which enters the nucleus under adverse conditions and affects gene regulation [7], [8]. DAF-16 promotes increased expression of many genes encoding proteins that protect against stress, including superoxide dismutases, drug metabolizing enzymes and molecular chaperones 9,10. DAF-16 is also required for the longevity of IIS mutants, for example those with defects in the DAF-2 insulin/IGF-1 receptor [11]. Both of these roles of DAF-16, the promotion of stress resistance and longevity, will improve the chances of living through periods of adversity. Whether the same downstream mechanisms cause increased stress protection and longevity remains unclear [12].
One factor contributing to growth and stress resistance is cellular iron availability. Free intracellular iron is toxic to the cell due to its role in catalyzing the Fenton reaction, which generates hydroxyl radicals from hydrogen peroxide:However, while free intracellular iron is harmful to the cell, iron is also an important element for a large number of cellular processes, including electron transport, deoxyribonucleotide synthesis, cellular detoxification, the cell cycle, oxygen transport and many others [13], [14]. Lack of iron is thought to affect the health of up to a billion people worldwide [15].
As well as nutritional iron deficiency, disruption of mechanisms that regulate iron homeostasis can also lead to a number of serious diseases in humans, such as anemias and iron overload disorders [16], [17]. The maintenance of appropriate iron levels is therefore important to viability and is tightly regulated by a number of proteins. These include ferritins, which form 24-subunit spherical nanocages that are each able to safely store up to 4500 atoms of iron. Heavy chain ferritins (H-ferritins) contain a ferroxidase centre, which has the capacity to convert Fe(II) to Fe(III) when the iron atom enters the complex [18].
The C. elegans genome contains two H-ferritin genes, ftn-1 and ftn-2 [19]. ftn-1 is predominantly expressed in the intestine, while ftn-2 is expressed in many cell types [19], [20]. In vertebrates, regulation of ferritin gene expression in response to iron levels is achieved both transcriptionally [21], and post-transcriptionally by the actions of iron regulatory proteins (IRPs) which bind to iron responsive elements (IREs) in the 5′ UTR of ferritin mRNAs [22]. Expression of C. elegans ferritin genes is also sensitive to iron levels: iron supplementation increases ftn-1 expression, while iron chelation has the opposite effect. However, ftn-1 and ftn-2 lack IRE sequences in their 5′ UTRs and iron-dependent regulation seems to be achieved solely through transcriptional regulation [23]. The mechanism by which this occurs remains unknown, but iron-dependent regulation of ftn-1 requires a 63 bp iron-dependent element (IDE) in its promoter [20].
Research on the regulation of ftn-1 in C. elegans has contributed to our understanding of ‘restless leg syndrome’, a human disease linked to iron deficiency in the brain. A haplotype of the gene MEIS1 has been associated with inheritance of the syndrome [24] but the gene's function was unknown. The involvement of the C. elegans ortholog unc-62 in regulating iron homeostasis was tested and a repressive role for this gene in ftn-1 regulation was identified. This regulation may be conserved in humans, since reduced MEIS1 expression seems to cause increased expression of human ferritin as well as of an iron transporter [25]. Thus, ftn-1 regulation in C. elegans can serve as a model for understanding the mechanisms of iron homeostasis in humans, and of human disease.
In this study, we explore the biology of iron homeostasis in C. elegans by investigating further the regulation of ftn-1. We show that ftn-1 is transcriptionally regulated by IIS/DAF-16, and then perform a genetic screen using RNA mediated interference (RNAi) to identify factors influencing expression of a Pftn-1::gfp reporter. We identify several transcription factors known to act with IIS to regulate lifespan as factors that also regulate ftn-1 expression. We also reveal a major role for the hypoxia signaling pathway in ftn-1 regulation and iron homeostasis.
Results
ftn-1 expression is regulated by insulin/IGF-1 signaling
To ascertain whether ftn-1 expression might be regulated by insulin/IGF-1 signaling (IIS) and daf-16, we examined published microarray-derived mRNA profiles comparing daf-2 and daf-16; daf-2 mutants [26], [27]. These implied that ftn-1 mRNA levels are greatly elevated (47-fold increase) in daf-2 compared to daf-16; daf-2 animals. This we were able to confirm using qRT-PCR (Figure 1A). The increase in ftn-1 mRNA levels in daf-2 mutants was fully daf-16 dependent. Loss of daf-16 also decreased ftn-1 mRNA levels in daf-2(+) animals.

We then created a transgenic C. elegans line bearing a Pftn-1::gfp transcriptional reporter containing 3.8 kb of sequence upstream of the ftn-1 start codon. This was generated by microinjection of transgene DNA, and the resulting extrachromosomal transgene arrays were then chromosomally integrated. The Pftn-1::gfp transgene showed strong expression throughout the intestine, consistent with previous reports [20]. Effects of daf-2 and daf-16 upon Pftn-1::gfp expression paralleled those seen in ftn-1 mRNA levels (Figure 1B, 1C). This confirms that ftn-1 is regulated by IIS, and shows that this regulation occurs principally in the intestine.
RNAi screen identifies more regulators of ftn-1 expression
We then used the Pftn-1::gfp reporter as the basis of an RNAi screen to investigate the mechanisms by which ftn-1 is regulated (Figure 1D). The initial aim of this screen was to identify pathways that work coordinately with IIS, and regulatory factors that act downstream of DAF-16. Expression of the integrated GFP (green fluorescent protein) reporter was intensified by mutation of daf-2 and sensitivity to RNAi was increased by introducing the rrf-3(pk1426) mutation. The resulting strain, of genotype rrf-3(pk1426); daf-2(m577ts); wuIs177 [Pftn-1::gfp], was raised at 15°C until the L4 stage, then transferred to RNAi plates and incubated at 25°C (non-permissive temperature for daf-2(m577)). GFP fluorescence levels were measured in a plate-reader two days later.
Given our interest in mechanisms of gene regulation, the RNAi screen was restricted to 812 genes encoding predicted transcription factors or other proteins associated with gene regulation [28]. RNAi of a number of these genes led to altered Pftn-1::gfp expression. In an initial screen, RNAi of 30 genes reduced Pftn-1::gfp expression by ≥20% (Table S1) and we investigated these more thoroughly in several genetic backgrounds. For 10 of these genes, not including daf-16, RNAi consistently and robustly reduced Pftn-1::gfp expression in multiple trials (data not shown). We then verified the effect of RNAi on levels of mRNA from the endogenous ftn-1 gene. This identified four genes where RNAi robustly reduced ftn-1 mRNA levels: hsf-1, mdl-1, ada-2 and elt-2 (Figure 2A, Table S1).

The heat-shock factor hsf-1 was previously shown to mediate effects of IIS on gene expression [29]. The screen also confirmed that the GATA transcription factor elt-2 plays a role in ftn-1 regulation. This is consistent with the role of elt-2 as an activator of intestinal gene expression [30]; moreover, elt-2 is the only previously described transcriptional activator of ftn-1 expression [20]. Thus, identification of hsf-1 and elt-2 in this unbiased screen is evidence of the efficacy of the screen. ada-2 encodes a homolog of the Ada2 subunit of various histone acetyl transferase (HAT) complexes that activate gene expression by modifying chromatin via histone acetylation [31]. It is possible that, ada-2 influences ftn-1 expression via effects on chromatin status.
More notable is the identification of the MAD-like transcription factor mdl-1 as an activator of ftn-1 expression. mdl-1 plays a role in the protective effects of reduced IIS against a tumorous germline phenotype [32] and is upregulated in IIS mutants [10], [26], [32]. We confirmed that the null mutation mdl-1(tm311) reduces ftn-1 mRNA levels in daf-2 mutants (Figure 2B).
To explore whether these four factors might be acting downstream of DAF-16, we tested whether RNAi reduces ftn-1 expression in a daf-16; daf-2 double mutant. The results imply that only MDL-1 does not require DAF-16 to activate ftn-1 expression. This suggests that mdl-1 acts downstream of daf-16, or possibly in parallel to IIS, to regulate ftn-1 expression (Figure 2C). Given that mdl-1 is a direct transcriptional target of DAF-16 [33], the former seems more likely.
Unexpectedly, RNAi of hsf-1 markedly increased ftn-1 expression in a daf-16; daf-2 background (Figure 2C). The effects of hsf-1 RNAi (Figure 2A) imply that HSF-1 and DAF-16 act together to activate ftn-1 expression, as previously shown for other genes [29]. That loss of hsf-1 in daf-16; daf-2 mutants increases expression of ftn-1 could imply a repressive role of HSF-1 in the absence of DAF-16. Alternatively, this increase might merely reflect a stressed state in the worms, caused by loss of both hsf-1 and daf-16 at 25°C (see Discussion).
Since ftn-1 is known to be responsive to iron levels, we also tested whether DAF-16, HSF-1 or MDL-1 are required for iron-dependent regulation of ftn-1. daf-16, hsf-1 and mdl-1 mutants were treated with iron (25 mM ferric ammonium citrate, FAC) and ftn-1 transcript levels measured by qRT-PCR. Iron-induced up-regulation of ftn-1 was unchanged in each case (Figure S1A), i.e. these three factors do not mediate effects of iron on ftn-1 expression.
hif-1 and daf-2 act additively to repress ftn-1 expression
RNAi of 28 genes further increased expression of the Pftn-1:gfp reporter (Table S2), already induced by daf-2(m577). Of note was the large increase in expression upon RNAi of unc-62, a transcription factor with a conserved role in ferritin regulation and, for its human ortholog, a possible role in the iron-related disorder ‘restless leg syndrome’ [25].
Also among the repressors of Pftn-1::gfp expression identified were hif-1, encoding the hypoxia-inducible factor, and aha-1, its binding partner (HIFβ, also called ARNT). RNAi of either gene strongly increased Pftn-1::gfp expression, in the original daf-2(m577); Pftn-1::gfp strain (Table S2) but also in two separate integrants of the Pftn-1::gfp reporter in a daf-2(+) background (Figure 3A, 3B and data not shown).

We verified this activity of hif-1 by using the loss of function mutation hif-1(ia4), which proved to greatly increase ftn-1 mRNA levels (Figure 3C) and Pftn-1::gfp expression (Figure 3D). In a hif-1(ia4) mutant background, RNAi of aha-1 did not further increase Pftn-1::gfp expression (Figure 3D), indicating that hif-1 and aha-1 act together to repress ftn-1 expression.
The finding that hif-1 RNAi increases Pftn-1::gfp expression in a daf-2 mutant background suggests that hif-1 influences ftn-1 expression independently of IIS. Consistent with this, hif-1 or aha-1 RNAi increased Pftn-1::gfp expression in the absence of daf-16 (Figure 3E). Results were similar at both 25°C and 20°C and at L4 and adult stages (Figure 3E and data not shown). In addition, RNAi of hif-1 increased ftn-1 transcript levels in daf-16 mutants, and also in hsf-1 and mdl-1 mutants (Figure S1B), indicating that none of these factors act downstream of HIF-1 to regulate ftn-1 expression.
Stabilization of HIF-1 reduces Pftn-1::gfp expression
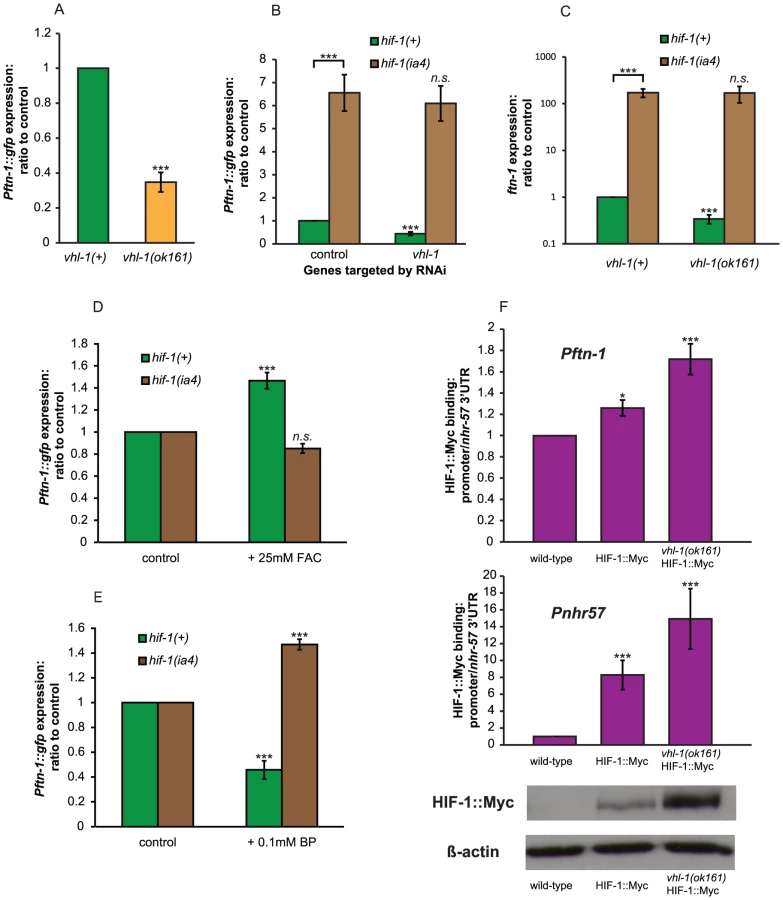
If HIF-1 is a repressor of ftn-1 expression, then elevation of HIF-1 levels should decrease expression of Pftn-1::gfp. Loss of vhl-1 (von Hippel-Lindau factor) leads to increased HIF-1 protein levels in C. elegans [34]. As expected, the deletion mutation vhl-1(ok161) markedly decreased expression of Pftn-1::gfp (Figure 4A). Moreover, RNAi of vhl-1 reduced Pftn-1::gfp expression in hif-1(+) but not hif-1(ia4) animals (Figure 4B), and genetic deletion of vhl-1 led to a reduction in ftn-1 transcript levels that is also completely dependent on hif-1 (Figure 4C). These results imply that HIF-1 acts downstream of VHL-1 as a repressor of ftn-1 expression.
The induction of Pftn-1::gfp expression by iron requires hif-1
The prolyl hydroxylase EGL-9 hydroxylates the P621 residue of HIF-1, which causes VHL-1 to bind to HIF-1, leading to proteasomal degradation [34]. This hydroxylation reaction requires iron as a cofactor, suggesting that regulation of ftn-1 expression by iron might involve the HIF-1 pathway. We therefore tested whether the effects of iron on ftn-1 expression are hif-1 dependent.
As expected given previous findings [23], both expression of Pftn-1::gfp and ftn-1 mRNA levels were increased upon supplementation with iron (ferric ammonium citrate, FAC) and decreased upon treatment with the iron chelator 2′-2 bipyridil (BP) (Figure 4D, 4E and Figure S2A and S2B). This is consistent with the previous observation that BP treatment greatly increases HIF-1 protein levels in C. elegans [34], since increased HIF levels would be expected to further repress ftn-1 expression. By contrast, in hif-1(ia4) mutants, addition of iron did not increase either Pftn-1::gfp expression or ftn-1 mRNA levels. This implies that hif-1 mediates the induction of ftn-1 expression by iron. Iron chelation did not decrease ftn-1::gfp and ftn-1 expression in hif-1(ia4), but instead increased it. The cause of this induction remains unexplained. One possibility is that BP treatment leads to cellular stress and induction of other stress response regulators (e.g. DAF-16), which can activate ftn-1 expression in the absence of the repressive effects of HIF-1 (see Discussion).
HIF-1 binds to the ftn-1 promoter
In order to investigate whether HIF-1 represses ftn-1 expression by directly binding to the ftn-1 promoter, we carried out a chromatin immunoprecipitation (ChIP) assay using C. elegans expressing Myc-tagged HIF-1 [35] and an anti-Myc antibody. We used three lines: wild type (N2), ZG429 [hif-1::myc] and GA654 [hif-1::myc vhl-1(ok161)]. Given that vhl-1 mutants have elevated HIF-1 levels and reduced ftn-1 mRNA levels (Figure 4C), greater levels of HIF-1::Myc binding to the ftn-1 promoter should be detectable in vhl-1 mutants, if the interaction is in fact direct.
We first checked that our ChIP protocol allowed us to measure binding by HIF-1::Myc by testing binding to the promoter of a known HIF-1 target gene, nhr-57. We designed one set of primers to amplify the region of the promoter containing two putative hypoxia response elements (HREs) and another set of primers targeting an area within the 3′ UTR of this gene. Quantity of qRT-PCR amplified promoter DNA was then compared to the 3′ UTR quantity as a test of enrichment of the promoter in our ChIP DNA pools. This amplification from the 3′ UTR (to which HIF-1 is not expected to bind) allowed us to control for input quantity. We saw a large (8.3-fold) enrichment of the nhr-57 promoter sequence in the HIF-1::Myc lines and an even greater (14.9-fold) enrichment when HIF-1::Myc was stabilized by deletion of vhl-1 (Figure 4F). Relative amounts of HIF-1::Myc were monitored by Western blotting of the same ChIP samples using the same aliquot of anti-Myc antibody used for ChIP, and we were able to confirm that vhl-1(ok161) increases HIF-1::Myc protein levels (Figure 4F).
We then measured binding to the ftn-1 promoter through qRT-PCR against the promoter sequence of ftn-1. For this, we used a primer pair specific to the IDE sequence. These results were again normalized against the same nhr-57 3′UTR in order to correct for differences in input quantity. While weaker than binding to Pnhr-57, enrichment of the Pftn-1 sequence in HIF-1::Myc and stabilized HIF-1::Myc lines was statistically significantly different to that seen in wild-type controls (Figure 4F). This is evidence that HIF-1 represses ftn-1 expression through direct binding to its promoter.
Iron-dependent regulation of ftn-1 is partially vhl-1–dependent
The repression of ftn-1 expression by HIF-1 and the requirement for iron in the degradation of HIF-1 by the proteasome suggests a possible mechanism for the iron-dependent regulation of ftn-1 in which changes in iron levels alter the level of HIF-1 protein, which in turn alter ftn-1 expression. Since the iron-dependent degradation of HIF-1 occurs via the action of VHL-1, HIF-1 protein levels in C. elegans are not sensitive to iron levels when VHL-1 is absent [36].
We found that loss of vhl-1 largely abrogated the induction of Pftn-1::gfp expression by iron supplementation, though there was still a significant induction of lesser magnitude (Figure 5A). Reduction of Pftn-1::gfp expression by iron chelation was not affected by loss of vhl-1 (Figure 5B). Taken together, this suggests that regulation of ftn-1 by iron may occur partially, but not exclusively, through changes in HIF-1 protein levels regulated by iron-dependent degradation.

Evidence that hif-1 mediates iron-dependent regulation via the iron-dependent element
The induction of ftn-1 levels by iron requires a 63 bp element (the iron-dependent element, or IDE) in the gene's promoter [20]. We wondered whether the hif-1 pathway might mediate the effects of iron on IDE-mediated gene expression. A reporter strain carrying a ftn-1 promoter lacking the IDE is insensitive to changes in iron levels [20]. Using these same reporters we found that absence of the IDE abolished hif-1 RNAi-induced induction of expression (Figure 5C).
Another reporter construct with just the IDE sequence fused to a minimal promoter and driving GFP expression was previously shown to be responsive to iron [20]. We found that loss of hif-1 increased ide::gfp expression, demonstrating that hif-1 does promote gene expression from the IDE (Figure 5D). Moreover, addition of iron did not induce ide::gfp expression in hif-1 mutants (Figure 5D). However, in hif-1 mutants treatment with the iron chelator BP still reduced ide::gfp expression (Figure 5E). This possibly reflects an effect of BP on ftn-1 that is independent of its effects on iron levels, or the existence of a second iron-dependent factor. These results show that the IDE is subject to regulation by HIF-1 and suggest that HIF-1 mediates the effects of iron on IDE-mediated gene expression.
Loss of egl-9 increases ftn-1 expression
As previously described, loss of vhl-1 decreases expression of Pftn-1::gfp (Figure 4A). This is expected given that HIF-1 represses ftn-1 expression and that loss of vhl-1 increases HIF-1 levels [34]. The prolyl hydroxylase EGL-9 targets HIF-1 for proteasomal degradation, and loss of egl-9 causes a similarly large increase in HIF-1 protein levels as loss of vhl-1 [36]. We therefore expected that loss of egl-9, like that of vhl-1, would reduce Pftn-1::gfp expression. In fact, deletion of egl-9 caused an 11-fold increase in Pftn-1::gfp expression (Figure 6A) and a ∼950-fold increase in ftn-1 mRNA levels (Figure 6B). Animals with a different allele, egl-9(n586), also showed increased ftn-1 mRNA levels (Figure S3A). Visible Pftn-1::gfp expression remained restricted to the intestine in wild type, vhl-1 mutants and egl-9 mutants.

vhl-1-independent effects of EGL-9 on HIF-1 target gene expression have been observed previously [36]. Our findings suggest that in the case of ftn-1 regulation, egl-9 can act independently of, and antagonistically to, vhl-1. As expected, loss of egl-9 induced ftn-1 expression even in the absence of vhl-1 (Figure S3B and S3C). However, egl-9 RNAi did not increase ftn-1 transcript or Pftn1::gfp expression in the absence of hif-1 (Figure 6B and Figure S3D). This implies that the inhibition of ftn-1 expression by EGL-9 also requires hif-1.
Thus, egl-9 and vhl-1 inhibit and activate expression of ftn-1, respectively, and both activities require hif-1. One possibility is that EGL-9 inhibits ftn-1 expression by stimulating HIF-1 activity via an as yet unidentified pathway.
Discussion
In this study, we have investigated the regulation of the inducible C. elegans ferritin gene ftn-1, a key determinant of iron homeostasis. We reveal that expression of this gene is coordinately regulated by insulin/IGF-1 and HIF signaling, pathways previously known to interact in the regulation of stress resistance and lifespan. Our findings imply that the HIF pathway is required for gene regulation in response to iron levels in C. elegans, and that IIS controls iron homeostasis, potentially increasing free iron availability to support growth.
Insulin/IGF-1 signaling (IIS) regulates growth and iron homeostasis
IIS and DAF-16 play a pivotal role in the organismal decision between growth and diapause. Under growth-promoting conditions, DAF-16 is inactivated through cytoplasmic retention, which facilitates reproductive growth [8], [37]. In the absence of sufficient food or given exposure to certain forms of stress, DAF-16 enters the nucleus and transcriptionally specifies a survival program. This entails delayed reproduction, enhanced stress resistance and increased lifespan. Modulation of DAF-16 activity is therefore crucial for ensuring an optimal response to the worm's environment; with growth and reproduction under conditions that are propitious to growth, and developmental arrest, stress protection and increased longevity under conditions that are not.
Regulation of ftn-1 by DAF-16 suggests the existence of a trade-off between growth and stress resistance involving iron homeostasis. A role for ferritin in regulating growth via its effects on iron homeostasis has been described previously in mammalian cells [38]. This study found that Myc, a bHLH transcription factor with a major role in promoting cellular proliferation, can repress H-ferritin expression. Overexpression of ferritin in cells carrying activated Myc led to a decrease in in vitro clonogenicity, and this effect could be rescued by addition of iron, suggesting that Myc–mediated repression of ferritin expression favors growth by increasing iron availability. The study identified DNA synthesis as a possible mechanism for iron-dependent control of cellular proliferation by c-Myc, as DNA synthesis is increased by c-Myc in a manner dependent on ferritin repression and the associated increases in iron availability. This finding is consistent with the requirement for iron in the activity of ribonucleotide reductase, the rate-limiting enzyme in DNA synthesis. Similar mechanisms may be at play in the regulation of ferritin expression by IIS. When conditions favor growth, and IIS is increased, reduced ftn-1 expression is expected to increase iron availability, thus fulfilling a key requirement for growth.
While free iron is required for growth, it can also cause harm by catalyzing the Fenton reaction, which increases levels of ROS and molecular damage. When conditions are not suitable for growth, IIS is reduced, and increased ftn-1 expression is expected to lower levels of free iron and of ROS, thereby protecting against stress. Consistent with this, induced over-expression of ftn-1 causes resistance to oxidative stress (S. Valentini and D. Gems, unpublished results). Thus, upregulation of ftn-1 likely contributes to the broader increase in cytoprotection seen when IIS is reduced.
Reduced IIS also increases levels of autophagy in C. elegans [39], [40] and autophagy releases iron from ferruginous materials, such as mitochondrial metalloproteins [41]. This predicts that reduced IIS will increase free iron levels, and concomitant elevation of ftn-1 expression could ensure that iron released by autophagy does not cause molecular damage.
Transcriptional activators of ftn-1 expression
Using an RNAi screen we identified new regulators of ftn-1, including hsf-1 and mdl-1. It was previously shown that in daf-2 mutants the heat shock factor HSF-1 acts in concert with DAF-16 to promote expression of small heat shock proteins and other molecular chaperones, which contribute to longevity [29]. We find that hsf-1 is also involved in the induction of ftn-1 in daf-2 mutants, since loss of hsf-1 reduced ftn-1 expression in daf-2 but not daf-16; daf-2 mutants.
The MAD-like transcription factor mdl-1 is also regulated by IIS. Microarray and qRT-PCR studies showed it to be up-regulated in daf-2 mutants [10], [26], [32]. mdl-1 also contributes to the resistance of daf-2 mutants to germline tumor formation in the gld-1 tumor model, and to daf-2 mutant longevity [32]. That MDL-1 activates ftn-1 expression is consistent with the role of mammalian MAD as an inhibitor of Myc, which represses ferritin expression (see above); however, C. elegans does not possess any clear ortholog of Myc [42], [43].
A study of DAF-16 binding sites did not provide evidence that ftn-1 is a direct regulatory target of DAF-16 [33], but suggested that mdl-1 might be. Given that ftn-1 may be a direct target of MDL-1 [44], [45], one possibility is that activation of mdl-1 expression by DAF-16 leads to increased ftn-1 expression. This hypothesis predicts that abrogation of mdl-1 expression should decrease ftn-1 expression more in daf-2 than in daf-16; daf-2 animals, but this is not the case (Figure 2A, 2C). This could imply that mdl-1 regulates ftn-1 independently of daf-16, at least in part.
ftn-1 is negatively regulated by hif-1 and aha-1
We discovered that loss of hif-1 or its binding partner aha-1 greatly increased ftn-1 expression in daf-2 mutants. This implicated hypoxia signaling in the regulation of ftn-1.
The HIF transcription factor is composed of an α and a β subunit, encoded by the genes hif-1 and aha-1 in C. elegans. HIF regulates the transcriptional response to hypoxia in both worms and vertebrates and, as expected, worms lacking hif-1 are hypersensitive to hypoxia [46]. Levels of HIFβ protein are relatively stable, whereas HIFα is constantly being degraded by the proteasome under normal, non-hypoxic conditions. In both worms and higher organisms, this occurs because the HIFα/HIF-1 protein is hydroxylated at conserved proline residues by prolyl hydroxylase (PHD), encoded by the egl-9 gene in worms. After hydroxylation by PHD/EGL-9, the von Hippel-Lindau protein VHL-1 binds to HIFα, which targets it for degradation [34], [47].
PHDs require oxygen, iron and 2-oxoglutarate for the hydroxylation reaction. When cells are kept under hypoxic conditions or when an iron chelator is added, the proline residue in HIFα is not hydroxylated and the HIFα protein accumulates [48]. That loss of hif-1 has such dramatic effects on gene expression under normoxic conditions demonstrates that HIF-1 affects gene regulation even at the very low levels of HIF-1 found when it is being hydroxylated and degraded. Similarly, it was previously observed that loss of hif-1 can increase C. elegans lifespan under normoxic conditions [49]. Consistent with this, we find statistically significant levels of binding of the non-stabilized HIF-1::Myc protein to both ftn-1 and nhr-57 promoters (Figure 4F).
Since iron is a required cofactor for hydroxylation of HIF by PHD, levels of iron affect those of HIF. For example, in C. elegans, depletion of iron using the iron chelator 2-2′ bipyridyl stabilizes HIF-1 [34], and feeding mice a low-iron diet leads to increased HIFα levels [50]. This increase in HIF-1 levels is not without consequence: chelation of iron has also been shown to increase expression of the C. elegans HIF-1 target gene nhr-57, indicating that the stabilization of HIF upon loss of iron leads to HIF-1-dependent changes in gene expression [51]. In vertebrates, HIF activates expression of genes involved in regulating iron homeostasis, including heme oxygenase [52], the transferrin receptor [53], [54], ceruloplasmin [55], DMT1 [56] and possibly ferroportin [57]. Loss of HIF-2α in mice causes decreased iron levels in the plasma and livers of mice [56]. It has therefore been suggested that HIF can act as an iron sensor: low iron levels lead to HIF stabilization, which leads to changes in gene expression that increase iron levels [57]. The results presented here support this hypothesis.
Iron-dependent regulation of ftn-1 via hif-1
The repression of ferritin expression by hif-1/aha-1 is consistent with a role of HIF in increasing iron availability. By this view, lower ferritin expression upon HIF activation would reduce iron storage capacity, thereby increasing iron availability. We therefore investigated whether HIF mediates iron-dependent regulation of ftn-1, and this proved to be the case: ftn-1 regulation by iron is blocked in hif-1 mutants. In wild-type animals iron supplementation increases ftn-1 expression while iron depletion decreases it. By contrast, in hif-1 mutants iron supplementation does not increase ftn-1 expression.
Treatment of hif-1 mutants with the iron chelator 2-2′ bipyridyl (BP) caused a large increase, rather than decrease, of ftn-1 expression. This was unexpected, but we noticed that BP treated worms were somewhat sickly in appearance. One possibility is that toxicity of BP in hif-1 mutants triggers other stress response mediators (e.g. DAF-16) that activate ftn-1 expression. This is consistent with our observation that stressful conditions tend to induce expression of this reporter. Similar to treatment with BP, RNAi of hsf-1 in daf-16(mgDf50); daf-2(m577) animals raised at 25°C also caused the worms to have a sickly appearance and induced Pftn-1::gfp expression (Figure 2C). Moreover, we observed that starved animals also show elevated Pftn-1::gfp expression (data not shown).
The requirement for hif-1 in the iron-dependent regulation of ftn-1 suggests that this regulation may occur via iron-dependent degradation of HIF-1. However, our data implies that this is not the whole story. Mutants of vhl-1 have constitutively stabilized HIF-1 and its levels cannot therefore respond to changes in iron (or oxygen) levels [34], [36]. While the increase in Pftn-1::gfp expression upon treatment with iron was greatly reduced in vhl-1 mutants, Pftn-1::gfp expression was still elevated compared to the control treatment. This implies that iron-dependent degradation of HIF-1 is not the sole mechanism by which ftn-1 is regulated in response to iron levels.
The control of ftn-1 expression by iron was previously shown to be mediated by the 63 bp iron-dependent element (IDE) in the ftn-1 gene promoter [20]. This implied the presence of an unknown iron-responsive transcriptional activator exerting effects upon the IDE. Our findings strongly suggest that this factor is HIF. We found that loss of hif-1 increases ide::gfp expression. Moreover, in the absence of hif-1, iron supplementation failed to induce ide::gfp expression. Furthermore, Romney et al. (2008) identified three conserved elements (called DR elements), with the consensus sequence: CACGTA(C/G)(C/A/G) in the IDE to which they attribute the responsiveness of ftn-1 expression to iron levels. This DR sequence has homology to the E-box motif, which led Romney et al. to suggest that the iron-sensory pathway includes a basic helix-loop-helix (bHLH) transcription factor. Both HIF-1 and AHA-1 belong to this family of proteins. In fact, the conserved DR sequence described by Romney et al. contains the putative C. elegans hypoxia response element (HRE) [58] (in reverse orientation). Moreover, using ChIP, we found that epitope-tagged HIF-1 bound to the region of the promoter containing the IDE. Taken together, these results support the view that HIF-1 acts as an iron sensor in C. elegans, suppressing ftn-1 expression by binding to the IDE, although the mechanism by which iron levels are detected has not yet been identified.
Our discovery of the role of HIF in iron homeostasis in C. elegans has notable implications in terms of the evolution of HIF as an iron sensor. The effects of iron on HIF levels in higher organisms has been viewed in the context of HIF's role in stimulating erythropoiesis. Since erythropoiesis requires large quantities of iron, it was proposed that the purpose of the HIF-mediated induction of genes involved in increasing iron availability is to supply iron for erythropoiesis [57]. That HIF regulates iron homeostasis in nematodes implies that the evolution of this function predates the emergence of a circulatory system. The sensitivity of hypoxia signaling to oxygen, iron and ROS, which interact and produce oxidative damage to the cell, further suggests that HIF may have an ancestral role in fine-tuning the response to different levels of these potentially toxic substances.
Antagonistic regulation of ftn-1 by vhl-1 and egl-9
Against expectation, loss of egl-9 increased expression of Pftn-1::gfp, rather than decreasing it. This does not merely indicate that EGL-9 regulates targets other than HIF-1, since the induction is hif-1 dependent. Given that loss of egl-9 or vhl-1 cause similar increases in HIF-1 protein levels [36], this finding suggests that increased HIF protein levels can be associated with both increased and decreased ftn-1 expression.
That both decreased expression upon vhl-1 deletion and increased expression upon egl-9 deletion require hif-1 is difficult to reconcile. However, vhl-1 independent effects of EGL-9 on HIF-1 target gene expression have been observed previously [36]. HIF-1 target genes are often more highly induced by loss of egl-9 than of vhl-1, despite identical levels of HIF-1 stabilization in each case [36]. This implies that regulation of HIF-1 target gene expression by EGL-9 occurs via both VHL-1-dependent and independent mechanisms. Additionally, mutations in egl-9 protect worms against infection by Pseudomonas aeruginosa and this effect is dependent on HIF-1. However, stabilization of HIF-1 by other means is insufficient to achieve this effect, again showing that EGL-9 can act via mechanisms other than HIF-1 stabilization [59].
We find that the effects of loss of egl-9 on ftn-1 do not require vhl-1 either. But in contrast to the other examples cited above, regulation of ftn-1 by the vhl-1-dependent and independent pathways downstream of EGL-9 act antagonistically, the former repressing ftn-1 expression and the latter activating it. Thus, our findings imply that EGL-9 not only represses HIF-1 activity by the well-characterized VHL-1-dependent pathway, but also modulates HIF-1 activity by an unknown mechanism (Figure 7). Prolyl hydroxylases are sensitive proteins capable of responding not only to iron and oxygen levels, but also to cues from metabolism [60] and to ROS [61], [62]. One or more of these may trigger the VHL-independent activity of EGL-9.

Coordinate regulation of ftn-1 by IIS and hypoxia signaling
Previous studies have suggested that hypoxia and IIS might act in concert to regulate gene expression. daf-2 mutants are highly resistant to hypoxia [63] and microarray studies found an over-representation of genes containing hypoxia response elements (HRE) among IIS/DAF-16 regulated genes [26]. A study using murine embryonic fibroblasts found that the FOXO3a transcription factor inhibits HIF-1 mediated gene regulation [64]. We were therefore interested in investigating whether hypoxia signaling and IIS interact to regulate ftn-1.
One model for the joint regulation of ftn-1 by hif-1 and IIS/DAF-16 that we initially considered is that loss of hif-1 activates DAF-16 which in turn activates ftn-1. DAF-16 is a stress-responsive transcription factor, so it seemed possible that stress caused by loss of HIF triggers a DAF-16-mediated cytoprotective response. There is evidence that loss of hif-1 does indeed have this effect on DAF-16: hif-1 mutants are long-lived and this lifespan extension has been shown to require daf-16 [49]. However, several observations argue against the idea that DAF-16 mediates HIF-1 effects. Firstly, HIF-1 binds directly to the ftn-1 promoter (Figure 4F). Secondly, the effects of loss of hif-1 on ftn-1 expression do not require daf-16 (Figure 3E). Finally, loss of hif-1 can further induce the expression of Pftn-1::gfp in daf-2 mutants.
The fact that daf-16 and hif-1 have opposite effects on ftn-1 expression bears consideration. Recent reports have found that HIF-1 overexpression extends lifespan [49], suggesting that HIF-1 activity has a similar effect to increased DAF-16 activity. Whether this occurs through the activation of a similar set of genes is unknown. Our finding that HIF-1 and DAF-16 can have opposing effects on gene expression suggests that the relationship between the two gene-sets is complex. Regulation of ftn-1 by HIF-1 and DAF-16 could be a special case in which DAF-16 regulation occurs as part of a broad response to lower oxidative stress whereas HIF-1 acts as an iron sensor. Further work is required in order to establish whether antagonistic regulation by DAF-16 and HIF-1 is specific to ftn-1 or whether it represents a more general pattern of gene regulation by the two pathways.
In summary, this study maps out a complex gene-regulatory network controlling expression of ftn-1 and, by extension, iron homeostasis in the nematode C. elegans. This reveals the acute sensitivity of iron homeostasis to environmental conditions, allowing fine tuning of iron availability in the face of variability of factors that increase free iron (increased environmental iron, growth arrest, increased autophagy) and decrease it (reduced environmental iron, increased growth). Our results also underscore the value of C. elegans as a model system for understanding mammalian iron homeostasis, and the pathologies that can result from its breakdown.
Materials and Methods
Nematode culture and strains
Maintenance and culture of C. elegans was carried out as published [65], [66], [67]. The following strains were used: CB5602 vhl-1(ok161), DR1563 daf-2(e1370), DR1567 daf-2(m577), GA300 daf-16(mgDf50); daf-2(m577), GA633 daf-2(m577); wuIs177 [Pftn-1::gfp lin-15(+)], GA636 rrf-3(pk1426); daf-2(m577); wuIs177 [Pftn-1::gfp lin-15(+)], GA639 daf-16(mgDf50); wuIs177 [Pftn-1::gfp lin-15(+)], GA640 wuIs176 [Pftn-1::gfp lin-15(+)], GA641 wuIs177 [Pftn-1::gfp lin-15(+)], GA642 hif-1(ia4); wuIs177 [Pftn-1::gfp lin-15(+)], GA643 daf-16(mgDf50); daf-2(m577); wuIs177 [Pftn-1::gfp lin-15(+)], GA654 unc-119(ed3) vhl-1(ok161) iaIs128[Phif-1::hif-1a::myc unc-119(+)], GA675 xtEx79 [Δpes-10(+63)::GFP-his, pha-1(+)], GA676 hif-1(ia4) xtEx79 [Δpes-10(+63)::GFP-his, pha-1(+)], GA688 pha-1(e2123ts) xtEx79 [Δpes-10(+63)::GFP-his, pha-1(+)], GA688 pha-1(e2123ts); hif-1(ia4) xtEx79 [Δpes-10(+63)::GFP-his, pha-1(+)], GA694 wuIs176 [Pftn-1::gfp lin-15(+)] egl-9(sa307)], GA1200 mdl-1(tm311), GA1203 daf-2(e1370); mdl-1(tm311), GA1204 daf-2(m577); mdl-1(tm311), GR1307 daf-16(mgDf50), JT307 egl-9(sa307), N2, PS3551 hsf-1(sy441), UZ96 pha-1(e2123ts) xtEx79 [Δpes-10(+63)::GFP-his, pha-1(+)], XA6900 pha-1(e2123ts) qaEx6902 [Pftn-1(Δ63)::[Δpes-10::GFP-his, pha-1(+)], XA6902 pha-1(e2123ts) qaEx6902 [Pftn-1::[Δpes-10::GFP-his, pha-1(+)] and ZG31 hif-1(ia4). ZG429 unc-119(ed3) iaIs128[Phif-1::hif-1a::myc unc-119(+)] Worms were maintained at 20°C unless otherwise indicated.
Strain constructions
Multiple mutants were created using standard methodologies and the presence of genomic deletions was tested via PCR. Genotyping was carried out by lysis of parent animals using proteinase K (Sigma) and subsequent PCR using the following primers. For daf-16(mgDf50): daf-16F1, gccactttattggaatttgagc; and daf-16R1, atcctcccatagaaggaccatt. For hif-1(ia4): hif-1_ex_fwd1, gctcctcctactccacctttg, hif-1_ex_rev1, gtgacgagttgtgaatgcacc, hif-1_int_rev1.2, tcggcgatggtgtcttcagtc. For rrf-3(pk1426): rrf-3_ex_fwd1, gagttcgcatcaagtttcac, rrf-3_ex_rev1, tgccttcgtacatttcaacc and rrf-3_int_rev2, ggtatttattgcttcctgccac. For vhl-1(ok161): DA75, gctgtcaatcggagcactgtc, DA76, ttgctgaggtctctggggtc, and DA77, gttagctctgccacgaatacgatg. For egl-9(sa307): DA117, acaaagacaggtgttgcgaatgag, DA118, ttgtagtgatccgagcccag, and DA119, gatgcttctgatgttcttggagg.
The promoter::gfp transgene of ftn-1 was created using methods as previously described [68] and the transgenic strain was created by microinjection. The primers used for creation of the construct were: ftn-1.5'ex, tgcttactggttctgccgag, ftn-1.5'in, tgtagggtttgattgtggtttg, ftn-1.3'fus, agtcgacctgcaggcatgcaagctttgacgagctagagacatgac. Extrachromosomal arrays were integrated by X-ray irradiation.
Fluorescence measurements
The method used to quantify GFP expression was adapted from one used in an earlier study [69]. Using a worm pick, samples of forty adult worms were transferred into the wells (V-shaped) of microtitre plates (Greiner). Fluorescence was then measured in a GeniosPlus plate reader (Tecan) at wavelengths appropriate for GFP (excitation: 495 nm; emission: 535 nm) using a fixed gain of 75. Quantification of GFP expression from transgenes with low level expression was carried out using a Leica DMRXA2 microscope using a GFP filter cube (excitation: 470/40 nm; emission: 525/50 nm), an Orca C10600 digital camera (Hamamatsu) and Volocity image analysis software (Improvision).
RNAi library
The transcription factor RNAi library used for this project was generously provided by Dr. Weiqing Li (University of Washington). Similar libraries are now available commercially (geneservice.co.uk). Where RNAi robustly affected ftn-1 expression levels, RNAi plasmid inserts were sequenced to confirm their identity using the primers JJM130 (gggaagggcgatcggtgcgggcc) and JJM131 (gcgcagcgagtcagtgagcgagg).
qRT–PCR
RNA was isolated from 2-day old adults after three washes, which removed E. coli and L1 progeny from the sample. After RNA isolation cDNA was synthesized using SuperScript II reverse transcriptase (Invitrogen) using oligo dT (Invitrogen). qRT-PCR was carried out using Fast SYBR Green Master Mix (Applied Biosystems) and the 7900 HT Fast PCR system (Applied Biosystems). Normalization of transcript quantity was carried out using the geometric mean of three stably expressed reference genes Y45F10D.4, pmp-3, and cdc-42 in order to control for cDNA input, as previously described [70]. The following primers were used for this assay. Y45F10D.4: DA90, gtcgcttcaaatcagttcagc, and DA91, gttcttgtcaagtgatccgaca. pmp-3: DA88, gttcccgtgttcatcactcat, and DA89, acaccgtcgagaagctgtaga. cdc-42: DA86, ctgctggacaggaagattacg, and DA87: ctcggacattctcgaatgaag. ftn-1: ftn-1_fwd_RT2, cggccgtcaataaacagattaacg, and ftn-1_rev_RT2 cacgctcctcatccgattgc.
qRT-PCR of ChIP DNA pools was carried out for the nhr-57 promoter using DA130: cctcccgcgtctccacattcaatc and DA131: cagcgaggtctgggttttccg, the nhr-57 3′UTR using DA135: tggcacaagatatgacgaaagctg and DA136: ggcgagaaatttgttgtaggttgcc, and the ftn-1 promoter using DA139: aacagctcacgtagccaatgataag and DA140: gcatcacatgagctgcccta.
Statistical analysis
All results shown are the mean of at least three independent biological replicates and error bars represent the s.e.m. Statistical significance was calculated by two-way or one-way ANOVA of either raw values or log-transformed quantities, depending on circumstances.
Chromatin immunoprecipitation
The protocol for chromatin immunoprecipitation was adapted from Mukhopdhyay et al. [71]. C. elegans cultures were grown for two generations in S-media with suspended OP50 at 20°C with constant shaking at 200 rpm. The worms were collected and washed four times in PBS buffer and then re-suspended in PBS containing 1% formaldehyde. Samples were then partially lysed using 8 strokes with a 1/3 turn in a 7 cm Dunce homogenizer and then incubated for 17 minutes with gentle mixing at room temperature. Crosslinking was stopped by addition of 200 µl 2.5 mol/L Glycine solution and 20 minutes further incubation at room temperature. After four washes in PBS containing protease inhibitor tablets (Complete, Roche), samples were flash frozen and stored at −80°C. After thawing, 2 mL of HLB buffer [50 mM HEPES-KOH, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.1% (wt/vol) sodium deoxycholate, 1% (vol/vol) Triton X-100, 0.1% (wt/vol) SDS and 1× Complete protease inhibitor] was added and sonication was carried out at 70% intensity for 7 bursts of 30 seconds in the Vibracell sonicator (Sonics). Protein quantity was estimated by Bradford assay (Biorad) and 2 mg were diluted into to 500 µl of in HLB buffer. Three 50 µl aliquots were removed at this point. DNA isolated from these samples was subsequently used as input controls. Samples were precleared for 1 h in prewashed salmon sperm DNA/protein-A agarose beads (Millipore) and then incubated overnight with 10 µl of anti-Myc Ab (9b11; Cell signalling). Samples were then incubated with prewashed salmon sperm DNA/protein-A agarose beads for 2 h. The beads were then washed twice in WB1 [50 mM HEPES-KOH, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% (wt/vol) sodium deoxycholate, 1% (vol/vol) Triton X-100, 0.1% (wt/vol) SDS and 1× Complete protease inhibitor], twice in WB2 [50 mM HEPES-KOH, pH 7.5, 1 M NaCl, 1 mM EDTA, 1% (wt/vol) sodium deoxycholate, 1% (vol/vol) Triton X-100, 0.1% (wt/vol) SDS and 1× Complete protease inhibitor] and once in WB3 [50 mM Tris-HCl, pH 8, 0.25 mM LiCl, 1 mM EDTA, 0.5% (vol/vol) NP-40 and 0.5% (wt/vol) sodium deoxycholate]. Crosslinking was reversed by addition of proteinase K solution [50 mM Tris-HCl, pH 8, 25 mM EDTA, 1.25% (wt/vol) SDS, 160 µg/ml proteinase K (Qiagen)] and incubation for 2 h at 45°C and overnight at 65°C. DNA was isolated by applying solution to Qiagen PCR purification columns (Qiagen).
Supporting Information
Zdroje
1. RiddleDLAlbertPS 1997 Genetic and Environmental Regulation of Dauer Larva Development
2. HuPJ 2007 Dauer WormBook 1 19
3. CassadaRCRussellRL 1975 The dauerlarva, a post-embryonic developmental variant of the nematode Caenorhabditis elegans. Developmental Biology 46 326 342
4. LithgowGJWalkerGA 2002 Stress resistance as a determinate of C. elegans lifespan. Mechanisms of Ageing and Development 123 765 771
5. Henis-KorenblitSZhangPHansenMMcCormickMLeeSJ 2010 Insulin/IGF-1 signaling mutants reprogram ER stress response regulators to promote longevity. Proc Natl Acad Sci U S A 107 9730 9735
6. MurakamiSJohnsonTE 1996 A genetic pathway conferring life extension and resistance to UV stress in Caenorhabditis elegans. Genetics 143 1207 1218
7. HendersonSTJohnsonTE 2001 daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Current Biology 11 1975 1980
8. LinKHsinHLibinaNKenyonC 2001 Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nature Genetics 28 139 145
9. McElweeJBubbKThomasJH 2003 Transcriptional outputs of the Caenorhabditis elegans forkhead protein DAF-16 (vol 2, pg 111, 2003). Aging Cell 2 341 341
10. MurphyCTMcCarrollSABargmannCIFraserAKamathRS 2003 Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424 277 284
11. KenyonCChangJGenschERudnerATabtiangR 1993 A C. elegans mutant that lives twice as long as wild-type. Nature 366 461 464
12. GemsDDoonanR 2009 Antioxidant defense and aging in C. elegans: is the oxidative damage theory of aging wrong? Cell Cycle 8 1681 1687
13. LeNTRichardsonDR 2002 The role of iron in cell cycle progression and the proliferation of neoplastic cells. Biochim Biophys Acta 1603 31 46
14. HentzeMWMuckenthalerMUAndrewsNC 2004 Balancing acts: Molecular control of mammalian iron metabolism. Cell 117 285 297
15. AndrewsNC 2000 Iron metabolism: iron deficiency and iron overload. Annu Rev Genomics Hum Genet 1 75 98
16. MackenzieELIwasakiKTsujiY 2008 Intracellular iron transport and storage: From molecular mechanisms to health implications. Antioxidants & Redox Signaling 10 997 1030
17. KaplanJWardDMDe DomenicoI 2011 The molecular basis of iron overload disorders and iron-linked anemias. Int J Hematol 93 14 20
18. LawsonDMTreffryAArtymiukPJHarrisonPMYewdallSJ 1989 Identification of the ferroxidase center in ferritin. FEBS Letters 254 207 210
19. KimYIChoJHYooOJAhnnJ 2004 Transcriptional regulation and life-span modulation of cytosolic aconitase and ferritin genes in C. elegans. Journal of Molecular Biology 342 421 433
20. RomneySJThackerCLeiboldEA 2008 An iron enhancer element in the FTN-1 gene directs iron-dependent expression in Caenorhabditis elegans intestine. Journal of Biological Chemistry 283 716 725
21. WhiteKMunroHN 1988 Induction of ferritin subunit synthesis by iron is regulated at both the transcriptional and translational Levels. Journal of Biological Chemistry 263 8938 8942
22. TortiFMTortiSV 2002 Regulation of ferritin genes and protein. Blood 99 3505 3516
23. GourleyBLParkerSBJonesBJZumbrennenKBLeiboldEA 2003 Cytosolic aconitase and ferritin are regulated by iron in Caenorhabditis elegans. Journal of Biological Chemistry 278 3227 3234
24. WinkelmannJSchormairBLichtnerPRipkeSXiongL 2007 Genome-wide association study of restless legs syndrome identifies common variants in three genomic regions. Nature Genetics 39 1000 1006
25. CatoireHDionPAXiongLAmariMGaudetR 2011 Restless legs syndrome-associated MEIS1 risk variant influences iron homeostasis. Ann Neurol
26. McElweeJJSchusterEBlancEThomasJHGemsD 2004 Shared transcriptional signature in Caenorhabditis elegans dauer larvae and long-lived daf-2 mutants implicates detoxification system in longevity assurance. Journal of Biological Chemistry 279 44533 44543
27. McElweeJJSchusterEBlancEPiperMDThomasJH 2007 Evolutionary conservation of regulated longevity assurance mechanisms. Genome Biology 8 7 R132
28. VermeirssenVDeplanckeBBarrasaMIReece-HoyesJSArdaHE 2007 Matrix and Steiner-triple-system smart pooling assays for high-performance transcription regulatory network mapping. Nature Methods 4 659 664
29. HsuALMurphyCTKenyonC 2003 Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 300 1142 1145
30. McGheeJDFukushigeTKrauseMWMinnemaSEGoszczynskiB 2009 ELT-2 is the predominant transcription factor controlling differentiation and function of the C. elegans intestine, from embryo to adult. Developmental Biology 327 551 565
31. PoulinGDongYFraserAGHopperNAAhringerJ 2005 Chromatin regulation and sumoylation in the inhibition of Ras-induced vulval development in Caenorhabditis elegans. Embo Journal 24 2613 2623
32. Pinkston-GosseJKenyonC 2007 DAF-16/FOXO targets genes that regulate tumor growth in Caenorhabditis elegans. Nature Genetics 39 1403 1409
33. SchusterEMcElweeJJTulletJMDoonanRMatthijssensF 2010 DamID in C. elegans reveals longevity-associated targets of DAF-16/FoxO. Mol Syst Biol 6 399
34. EpsteinACRGleadleJMMcNeillLAHewitsonKSO'RourkeJ 2001 C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107 43 54
35. ZhangYShaoZZhaiZShenCPowell-CoffmanJA 2009 The HIF-1 hypoxia-inducible factor modulates lifespan in C. elegans. PLoS ONE 4 e6348 doi:10.1371/journal.pone.0006348
36. ShaoZYZhangYPowell-CoffmanJA 2009 Two Distinct Roles for EGL-9 in the Regulation of HIF-1-Mediated Gene Expression in Caenorhabditis elegans. Genetics 183 821 829
37. LeeRYHenchJRuvkunG 2001 Regulation of C. elegans DAF-16 and its human ortholog FKHRL1 by the daf-2 insulin-like signaling pathway. Current Biology 11 1950 1957
38. WuKJPolackADalla-FaveraR 1999 Coordinated regulation of iron-controlling genes, H-ferritin and IRP2, by c-MYC. Science 283 676 679
39. MelendezATalloczyZSeamanMEskelinenELHallDH 2003 Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301 1387 1391
40. HansenMChandraAMiticLLOnkenBDriscollM 2008 A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet 4 e24 doi:10.1371/journal.pgen.0040024
41. KurzTTermanABrunkUT 2007 Autophagy, ageing and apoptosis: the role of oxidative stress and lysosomal iron. Arch Biochem Biophys 462 220 230
42. YuanJTirabassiRSBushABColeMD 1998 The C-elegans MDL-1 and MXL-1 proteins can functionally substitute for vertebrate MAD and MAX. Oncogene 17 1109 1118
43. PickettCLBreenKTAyerDE 2007 A C. elegans Myc-like network cooperates with semaphorin and Wnt signaling pathways to control cell migration. Developmental Biology 310 226 239
44. GersteinMBLuZJVan NostrandELChengCArshinoffBI 2010 Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science 330 1775 1787
45. modencode.org (accessed May 2011)
46. JiangHQGuoRPowell-CoffmanJA 2001 The Caenorhabditis elegans hif-1 gene encodes a bHLH-PAS protein that is required for adaptation to hypoxia. Proceedings of the National Academy of Sciences of the United States of America 98 7916 7921
47. KaelinWG 2005 Proline hydroxylation and gene expression. Annu Rev Biochem 74 115 128
48. MoleDR 2010 Iron Homeostasis and its interaction with prolyl hydroxylases. Antioxidants & Redox Signaling 12 445 458
49. LeiserSFBegunAKaeberleinM 2011 HIF-1 modulates longevity and healthspan in a temperature-dependent manner. Aging Cell 10 318 326
50. PeyssonnauxCZinkernagelASSchuepbachRARankinEVaulontS 2007 Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). Journal of Clinical Investigation 117 1926 1932
51. BishopTLauKWEpsteinACKimSKJiangM 2004 Genetic analysis of pathways regulated by the von Hippel-Lindau tumor suppressor in Caenorhabditis elegans. PLoS Biol 2 e289 doi:10.1371/journal.pbio.0020289
52. LeePJJiangBHChinBYIyerNVAlamJ 1997 Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. Journal of Biological Chemistry 272 5375 5381
53. LokCNPonkaP 1999 Identification of a hypoxia response element in the transferrin receptor gene. Journal of Biological Chemistry 274 24147 24152
54. TacchiniLBianchiLBernelli-ZazzeraACairoG 1999 Transferrin receptor induction by hypoxia - HIF-1-mediated transcriptional activation and cell-specific post-transcriptional regulation. Journal of Biological Chemistry 274 24142 24146
55. MukhopadhyayCKMazumderBFoxPL 2000 Role of hypoxia-inducible factor-1 in transcriptional activation of ceruloplasmin by iron deficiency. Journal of Biological Chemistry 275 21048 21054
56. MastrogiannakiMMatakPKeithBSimonMCVaulontS 2009 HIF-2alpha, but not HIF-1alpha, promotes iron absorption in mice. Journal of Clinical Investigation 119 1159 1166
57. PeyssonnauxCNizetVJohnsonRS 2008 Role of the hypoxia inducible factors in iron metabolism. Cell Cycle 7 28 32
58. ShenCNettletonDJiangMKimSKPowell-CoffmanJA 2005 Roles of the HIF-1 hypoxia-inducible factor during hypoxia response in Caenorhabditis elegans. J Biol Chem 280 20580 20588
59. ShaoZZhangYYeQSaldanhaJNPowell-CoffmanJA 2010 C. elegans SWAN-1 Binds to EGL-9 and regulates HIF-1-mediated resistance to the bacterial pathogen Pseudomonas aeruginosa PAO1. PLoS Pathog 6 e1001075 doi:10.1371/journal.ppat.1001075
60. KoivunenPHirsilaMRemesAMHassinenIEKivirikkoKI 2007 Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates - Possible links between cell metabolism and stabilization of HIF. Journal of Biological Chemistry 282 4524 4532
61. GeraldDBerraEFrapartYMChanDAGiacciaAJ 2004 JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell 118 781 794
62. GuzyRDSchumackerPT 2006 Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Experimental Physiology 91 807 819
63. ScottBAAvidanMSCrowderCM 2002 Regulation of hypoxic death in C. elegans by the insulin/IGF receptor homolog DAF-2. Science 296 2388 2391
64. EmerlingBMWeinbergFLiuJLMakTWChandelNS 2008 PTEN regulates p300-dependent hypoxia-inducible factor 1 transcriptional activity through Forkhead transcription factor 3a (FOXO3a). Proc Natl Acad Sci U S A 105 2622 2627
65. BrennerS 1974 The genetics of Caenorhabditis elegans. Genetics 77 71 94
66. SulstonJEHodjkinJ 1988 The nematode Caenorhabditis elegans Cold Spring Harbor Laboratory Press 587 606
67. StiernagleT 2006 Maintenance of C. elegans WormBook 1 11
68. HobertO 2002 PCR fusion-based approach to create reporter gene constructs for expression analysis in transgenic C. elegans. Biotechniques 32 728 730
69. WolffSMaHBurchDMacielGAHunterT 2006 SMK-1, an essential regulator of DAF-16-mediated longevity. Cell 124 1039 1053
70. HoogewijsDHouthoofdKMatthijssensFVandesompeleJVanfleterenJR 2008 Selection and validation of a set of reliable reference genes for quantitative sod gene expression analysis in C. elegans. Bmc Molecular Biology 9
71. MukhopadhyayADeplanckeBWalhoutAJTissenbaumHA 2008 Chromatin immunoprecipitation (ChIP) coupled to detection by quantitative real-time PCR to study transcription factor binding to DNA in Caenorhabditis elegans. Nat Protoc 3 698 709
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 3
Nejčtenější v tomto čísle
- PIF4–Mediated Activation of Expression Integrates Temperature into the Auxin Pathway in Regulating Hypocotyl Growth
- Metabolic Profiling of a Mapping Population Exposes New Insights in the Regulation of Seed Metabolism and Seed, Fruit, and Plant Relations
- A Splice Site Variant in the Bovine Gene Compromises Growth and Regulation of the Inflammatory Response
- Comprehensive Research Synopsis and Systematic Meta-Analyses in Parkinson's Disease Genetics: The PDGene Database