
Secondary Metabolism and Development Is Mediated by LlmF Control of VeA Subcellular Localization in
Secondary metabolism and development are linked in Aspergillus through the conserved regulatory velvet complex composed of VeA, VelB, and LaeA. The founding member of the velvet complex, VeA, shuttles between the cytoplasm and nucleus in response to alterations in light. Here we describe a new interaction partner of VeA identified through a reverse genetics screen looking for LaeA-like methyltransferases in Aspergillus nidulans. One of the putative LaeA-like methyltransferases identified, LlmF, is a negative regulator of sterigmatocystin production and sexual development. LlmF interacts directly with VeA and the repressive function of LlmF is mediated by influencing the localization of VeA, as over-expression of llmF decreases the nuclear to cytoplasmic ratio of VeA while deletion of llmF results in an increased nuclear accumulation of VeA. We show that the methyltransferase domain of LlmF is required for function; however, LlmF does not directly methylate VeA in vitro. This study identifies a new interaction partner for VeA and highlights the importance of cellular compartmentalization of VeA for regulation of development and secondary metabolism.
Published in the journal:
. PLoS Genet 9(1): e32767. doi:10.1371/journal.pgen.1003193
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003193
Summary
Secondary metabolism and development are linked in Aspergillus through the conserved regulatory velvet complex composed of VeA, VelB, and LaeA. The founding member of the velvet complex, VeA, shuttles between the cytoplasm and nucleus in response to alterations in light. Here we describe a new interaction partner of VeA identified through a reverse genetics screen looking for LaeA-like methyltransferases in Aspergillus nidulans. One of the putative LaeA-like methyltransferases identified, LlmF, is a negative regulator of sterigmatocystin production and sexual development. LlmF interacts directly with VeA and the repressive function of LlmF is mediated by influencing the localization of VeA, as over-expression of llmF decreases the nuclear to cytoplasmic ratio of VeA while deletion of llmF results in an increased nuclear accumulation of VeA. We show that the methyltransferase domain of LlmF is required for function; however, LlmF does not directly methylate VeA in vitro. This study identifies a new interaction partner for VeA and highlights the importance of cellular compartmentalization of VeA for regulation of development and secondary metabolism.
Introduction
Small bioactive secondary metabolites are molecules that have great importance to humankind and can be broadly characterized by their impact on human health. Members of the filamentous fungal genus Aspergillus are prolific producers of secondary metabolites, both useful (penicillin, lovastatin) and detrimental (aflatoxin, sterigmatocystin, gliotoxin), therefore they are excellent organisms to study the genetic regulation of secondary metabolism.
Genes responsible for the production of secondary metabolites are often clustered on chromosomes, vaguely reminiscent of bacterial operons, and these gene clusters are co-regulated [1]. Relatively recently a global regulator of secondary metabolism, LaeA, was described in the Aspergilli [2]. LaeA has since been shown to be part of the velvet complex, composed of LaeA, VeA, VelB, that couples secondary metabolism with developmental processes including asexual and sexual development [3]. Moreover, LaeA has been found to direct formation of a second velvet-like complex composed of VelB and VosA [4]. Both complexes function to orchestrate secondary metabolism with developmental differentiation in a light dependent fashion [3]–[5].
The core components of the velvet complex are found in all filamentous fungi studied to date and function to regulate important pathways, including pathogenicity in both plants and humans. For example, null mutants of A. fumigatus laeA produce fewer secondary metabolites and are hypovirulent in the mouse model of invasive aspergillosis [6], [7] while both VeA and LaeA homologs in the plant pathogens A. flavus, Fusarium fujikuroi, F. verticillioides, and Cochliobolus heterostrophus are involved in regulation of virulence and toxin production [8]–[12]. Recent work in F. graminearum has identified that both the veA and velB homologs are virulence factors [13]–[16], however these data indicate the heterotrimeric LaeA-VeA-VelB complex might be slightly different in this organism, as FgVeA did not interact with FgLaeA or FgVelB in a yeast-two-hybrid assay [13], [15]. Taken together, the velvet complex control of secondary metabolism and development appears to be conserved in filamentous fungi [5], however its cellular operational mechanism remains an enigma.
Current knowledge of the velvet complex functionality in Aspergillus centers on the importance of VeA as a bridging factor between VelB and the putative methyltransferase LaeA. Together, this heterotrimeric complex regulates developmental differentiation in response to light. LaeA is a constitutively nuclear protein that harbors a methyltransferase domain required for function [2], [17] and VelB is a velvet–like protein that forms a heterodimer with VeA [3]. In A. nidulans, VeA-VelB is required for sexual development, while VeA-LaeA is required for production of the mycotoxin sterigmatocystin [3].
An important aspect of VeA is its subcellular localization in response to illumination. Stinnett et al. [18] first reported that VeA is found in the cytoplasm under light conditions and is mainly nuclear under dark conditions, while the truncated VeA1 is blind to light and fails to accumulate in the nucleus in the dark. It has also been demonstrated that transcription of veA is relatively constant over all developmental stages [19] and this correlates with protein levels [4]. Therefore, the subcellular location of VeA has the potential to control protein interaction partners as well as to direct developmental and chemical responses to environmental cues.
While VelB-VeA-LaeA has been shown to be a stable complex that interacts in the nucleus, VeA interacts with several other proteins, including the red light sensing protein FphA, and thus indirectly with the blue light sensing proteins LreA and LreB [20], [21]. The VeA-family of proteins (VeA, VelB, VelC, VosA) all contain a characteristic velvet domain [5], while VosA also has a characterized C terminal transcriptional activation domain [5], [22]. Bayram and Braus [5] have speculated that the velvet domain may function as a protein-protein interaction domain. Calvo [23] and others (i.e. [5]) hypothesize that VeA could function as a scaffold protein directing transcriptional response to changes in environmental cues, such as illumination. Working under this hypothesis, the velvet complex may contain additional accessory proteins that could influence development and secondary metabolism.
We used a reverse genetics approach to identify several putative methyltransferases in A. nidulans that have sequence homology to LaeA, and then set out to determine if any of these putative methyltransferases were linked to the velvet complex. Here we describe the LaeA-like methyltransferase, LlmF, that is a negative regulator of sexual development and secondary metabolism in A. nidulans. We report that LlmF interacts with VeA and functions to mediate subcellular localization of VeA. This study identifies a new interaction partner of VeA while illustrating the importance of cellular compartmentalization as a form of regulating secondary metabolism and development.
Results
In silico identification of putative LaeA-like methyltransferases
Since LaeA has been shown to regulate many but not all secondary metabolite clusters and VeA has several interaction partners, including LaeA, VelB, and FphA [23], we hypothesized that other LaeA-like methyltransferases could play a role in velvet complex developmental regulation. The primary amino acid sequence of LaeA (AN0807) indicates that it contains an S-adenosyl methionine (SAM) binding domain [2] that is required for function [17]. To determine if there are other putative LaeA-like methyltransferases in A. nidulans, predicted amino acid sequences were obtained for all SAM binding domain proteins from the Aspergillus nidulans genome database at the Broad Institute and aligned with known methyltransferases from S. cerevisiae, Sc. pombe, and A. flavus. A bootstrapped phylogenic tree was inferred from this analysis (Figure 1). This analysis identified nine putative ORFs that have sequence homology to LaeA and no homologies to previously characterized yeast methyltransferases (LlmA – AN2165, LlmB – AN8945, LlmC – AN7933, LlmD – AN5416, LlmE – AN5091, LlmF – AN6749, LlmG – AN5874, LlmI – AN8833, and LlmJ – AN9193). One of these putative ORFs, llmE (AN5091), has previously been characterized and resides in a location that is transcriptionally repressed by telomere position effect [24].

Preliminary characterization of LaeA-like methyltransferases
In order to ascertain functions of the LaeA-like methyltransferase (Llm) proteins, transcriptional profiling from a wild type A. nidulans strain (RDIT9.32) was conducted at several distinct developmental growth time points (Figure 2). Northern analysis of the llm genes show that some are transcribed at nearly all developmental stages (llmA, llmB, and llmI), while others are transcribed at very low levels (llmD, llmF, llmG, and llmJ), and llmC is mainly transcribed during late asexual development (Figure 2). To assess the roles of each Llm on secondary metabolism, null mutants were created for all of the putative Llm's and confirmed by Southern blot (Figure S1 and data not shown). Preliminary characterization of secondary metabolite production indicated that ΔllmF produced increased levels of sterigmatocystin (Figure 3), whereas the other eight Llm's had minor impacts on sterigmatocystin (Figure S2, [24]). Therefore we chose to focus our efforts on characterization of LlmF (AN6749). While llmF appears to be expressed at low levels throughout development, there is an increase in llmF transcription during later stages of both vegetative and asexual growth conditions, while there is no increase of llmF during sexual development (Figure 2).

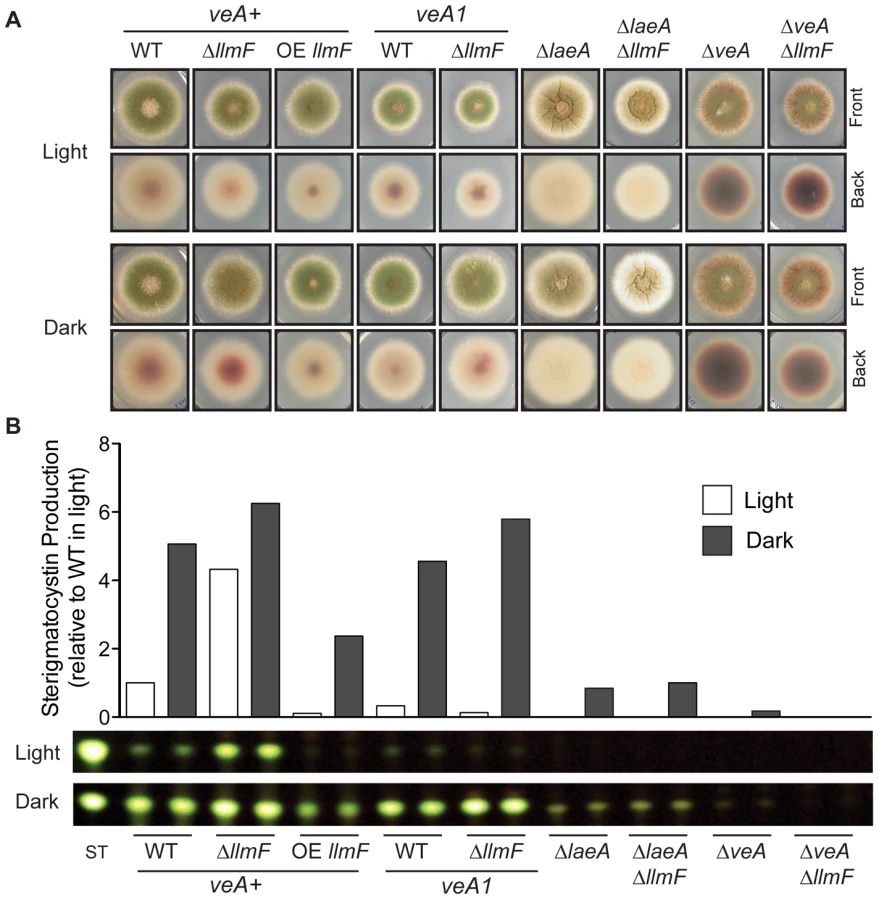
LlmF is a negative regulator of sterigmatocystin
To gain further insight into llmF regulation of secondary metabolism, llmF was over-expressed with the constitutive gpdA promoter (data not shown). We assessed sterigmatocystin production in the llmF mutants, and oppositely to ΔlaeA mutants, deletion of llmF results in increased sterigmatocystin levels. Conversely, over-expression (OE) of llmF reduced sterigmatocystin production (Figure 3). Interestingly, we did not detect an increase in sterigmatocystin production in a ΔllmF mutant harboring the veA1 allele (Figure 3B). To test the requirement of LaeA and VeA in LlmF regulation of sterigmatocystin; double mutants were created and these analyses suggest that both LaeA and VeA are required for negative regulation of sterigmatocystin by LlmF (Figure 3).
LlmF interacts directly with VeA
Since LlmF mediated regulation of sterigmatocystin was dependent on the velvet complex, we hypothesized that LlmF could interact directly with LaeA or VeA. A cDNA was cloned for LlmF (Figure S3) and a directed yeast-two-hybrid approach was taken to ascertain protein-protein interactions. These results indicated that LlmF interacts with VeA but not the truncated VeA1 or LaeA (Figure 4A). However, the yeast-two-hybrid assay is historically prone to false-positives and in addition we have noticed inconsistencies with VeA1 in this assay, for example Bait-LaeA does not interact with Prey-VeA1 (Figure 4A). Therefore, we also assessed the LlmF-VeA interaction with an in vitro GST pull-down assay with recombinantly expressed GST-LlmF and His6-VeA-S-tag (Figure 4B). As an assay control, GST-VelB and GST-LaeA were also used to pull-down His6-VeA-S-tag (Figure 4B). Finally, we performed an in vivo pull-down experiment using tandem affinity purification (TAP) tagged LlmF combined with S-tagged VeA and VeA1. After purification of TAP-LlmF, both VeA-S-tag and VeA1-S-tag were detected via immunoblotting (Figure 4C). Unlike the yeast-two-hybrid results, the in vivo pull-down data suggest that LlmF interacts with both VeA and the truncated VeA1 under these conditions.

LaeA and LlmF can bind S-adenosyl methionine
Previous studies on LaeA had indicated that it was a likely methyltransferase based on site directed mutagenesis of the S-adenosyl methionine (SAM) binding domain, which resulted in a non-functional protein [17]. A multiple sequence alignment [25] of A. nidulans LlmF, A. nidulans LaeA, A. flavus LaeA, A. fumigatus LaeA, Cochliobolus heterostrophus LaeA, and Fusarium fujikuroi LaeA confirm that all of these proteins contain a SAM binding domain which can be further classified into conserved motifs as previously described [26], [27] (Figure 5A). We tested the functionality of the SAM binding domain by using an ultraviolet (UV) light cross-linking experiment of 3H-SAM (Figure 5B). As a control, GST and GST-LaeA were included in the experiment and these results indicate that both LaeA and LlmF have the ability to bind SAM (Figure 5B). Competitive inhibition of the binding site was achieved by co-incubation of 3H-SAM with S-adenosyl homocysteine (SAH) and under these conditions a reduction in 3H-SAM binding was observed in both GST-LlmF and LlmF. GST-LlmF but not GST-LaeA showed a reduction in 3H-SAM binding under these conditions, which could suggest that GST-LaeA binds SAM more tightly than LlmF.

The SAM binding domain of LlmF is required for negative regulation of sexual development
In order to assess the role of the SAM binding site of LlmF, we constructed a SAM binding site mutant in vivo by site directed mutagenesis of motif I (Figure 5A). Mutation of two conserved glycine residues to alanine residues in motif I rendered LaeA non-functional [17], therefore we mutated the same residues in LlmF (G91A and G93A). To analyze the effect of this mutation in vivo, llmFG91A,G93A (llmFSAM) was driven by the gpdA promoter in a ΔllmF background. As abnormalities in sexual development - ranging from cleistothecial morphological aberrancies to alterations in sexual to asexual spore ratios - are clearly and consistently observed in veA, velB and laeA mutants [4], [28], we investigated the impacts of llmF on sexual development.
Macroscopic observations during sexual developmental induction of ΔllmF and OE llmF mutants showed that ΔllmF favored sexual development and OE llmF asexual development. Additionally, the OE llmFSAM mutant displayed a phenotype more similar to the ΔllmF mutant (Figure 6A). To more accurately measure llmF effect on sexual development, ascospores and conidia were quantified from sexually induced culture conditions and represented as a ratio of ascospores to conidia (Figure 6B). The ΔllmF strain produced an increased ratio of ascospores to conidia and consistent with the negative regulation of sexual development the OE llmF strain produces decreased ratio of ascospores to conidia. Mirroring the macroscopic images, the OE llmFSAM mutant produced a similar ratio to the ΔllmF strain (Figure 6B). Moreover, we measured production of sterigmatocystin from these four strains. This further confirms that LlmFSAM is not functional as the OE llmFSAM mutant produced levels of sterigmatocystin intermediate to wild type and ΔllmF, in contrast to the reduced levels of the OE llmF strain (Figure 6C). Finally, a northern blot from total RNA isolated from the same conditions confirmed the strains were correct and the OE llmFSAM construct was properly expressed (Figure 6D). Taken together these data indicate that LlmF is a negative regulator of sexual development and secondary metabolism and the SAM binding site of LlmF is required for function.

LlmF has limited effect on velvet complex transcription
Since the velvet complex has been implicated in transcriptional control of secondary metabolism and development, we hypothesized that the observed phenotypes could be explained by differential transcription of velvet complex members in the ΔllmF and OE llmF backgrounds. Specifically, we hypothesized that LlmF could negatively regulate transcription of veA, laeA, or velB which could explain the increased secondary metabolism and sexual development in the ΔllmF background. To test this hypothesis, a northern blot was done on RNA isolated from asexual and sexual developmental induced cultures. These data refute this hypothesis, as LlmF has minimal effect on transcription of veA, laeA, or velB (Figure 7). This is in contrast to the impact of loss or over-expression of velvet complex members on each other in which deletions of veA, velB, and laeA have previously been shown to substantially increase transcription of the other members of the complex and, where examined, over-expression reduced expression of other gene members [3], [11]. However, consistent with the conidial phenotype of llmF mutants, the key asexual developmental activator brlA shows decreased expression in the ΔllmF strain and increased expression in the OE llmF strain (Figure 7).

Cytoplasmic LlmF alters VeA localization in response to light
VeA has previously been shown to be a light regulated protein; in the light VeA is localized mainly in the cytoplasm, however in the dark VeA is almost exclusively nuclear [18]. On the other hand, LaeA is located in the nucleus independent of light but requires nuclear VeA for control of secondary metabolism and some aspects of sexuality [2], [3]. Since VeA is required for LaeA function, it seemed plausible that LlmF and LaeA could be competing for VeA binding, either directly or indirectly. Additionally, llmF mutants do not show phenotypes in a mutant veA1 background suggesting that nucleo-cytoplasmic localization of VeA could be altered compared to wild type. To test this hypothesis, we first tagged LlmF with GFP at both the N and C terminus independently. By phenotypic analysis, C terminal LlmF-GFP constructs were nonfunctional; therefore we used the functional N terminal GFP-LlmF strains and determined LlmF was located primarily in the cytoplasm independent of light (Figure 8A). Additionally, the localization of GFP-LlmF in a veA1 background was tested and displayed similar localization (Figure 8A).

While LaeA is constitutively nuclear and LlmF cytoplasmic, we reasoned that competition for VeA binding could be assessed by the subcellular localization of VeA. To test this hypothesis, VeA-GFP was visualized in ΔllmF and OE llmF genetic backgrounds. Interestingly, VeA-GFP was mis-localized in response to light conditions in both the ΔllmF and OE llmF backgrounds. Specifically, slightly more VeA accumulated in the nucleus under light conditions in a ΔllmF background compared to wild type whereas VeA failed to accumulate in the nucleus under dark conditions in an OE llmF background, phenocopying the VeA1-GFP localization pattern (Figure 8B and 8C). These results indicate that increased nuclear VeA could be responsible for the increased production of sterigmatocystin in ΔllmF strains and the cytoplasmic location of VeA in the OE llmF background the reason for decreased sterigmatocystin and sexual development in this strain.
LlmF does not interact with VelB or the nuclear importin α
Nuclear import of VeA in response to light is mediated by importin α (KapA) [18], [29]. Additionally, VelB is hypothesized to be a cytoplasmic protein that enters the nucleus through its interaction with VeA [3]. Hence, the model for VeA nuclear import is as follows: KapA recognizes and binds the nuclear localization signal of VeA and along with VelB, the KapA-VeA-VelB transient complex is transferred to the nuclear pore. To determine if LlmF could be altering VeA localization through disruption of VeA-VelB interaction or KapA-VeA interaction, we tested for the ability for LlmF to interact with either KapA or VelB. Figure 9A illustrates that neither KapA nor VelB are interaction partners for LlmF in the yeast-two-hybrid assay. These data also indicate that KapA is capable of recognizing VelB in the absence of VeA, however previous data suggests that VelB requires VeA for nuclear import [3].

Full-length LlmF is required for interaction with VeA
Since LlmF interacted with only VeA and not VelB or KapA, we hypothesized that VeA-LlmF interaction could be interfering with KapA recognition and subsequent nuclear import. As previous data have indicated that KapA recognizes the VeA-VelB dimer for nuclear import [3], [29], to see if LlmF and VelB interacted with VeA in the same domains, we mapped the protein interaction site of VeA-LlmF using the directed yeast-two-hybrid assay. Testing the interaction of full length VeA with 5 different truncations of LlmF showed that only full length LlmF is capable of interacting with VeA in this assay (Figure 9B). Similarly, when 5 different truncations of VeA were tested as interaction partners with LlmF, similar results were obtained in which full length VeA is required for interaction with full length LlmF (Figure 9C).
Simultaneously, we were able to refine the interaction domains of VeA-LaeA and VeA-VelB. Previously Bayram et al. [3] showed that VelB interacts with the N terminus of VeA (1–300 aa) while LaeA interacts with the C terminus of VeA (276–573 aa). Here our data suggest that VelB interacts with the N terminus of VeA (1–235 aa), however the first 28 aa are necessary for this interaction as VelB did not interact with just the velvet domain of VeA (29–235) (Figure 9C). We were also able to show that LaeA can interact with the C terminal 115 amino acids of VeA (Figure 9C). Since our in vivo LlmF SAM binding site mutant was not functional (Figure 6), we tested for interaction between VeA and LlmFSAM, and found LlmFSAM interacted with VeA (Figure 9D). Taken together, these data suggest that LlmF-VeA interaction is relatively weak in comparison to the heterotrimeric complex of VelB-VeA-LaeA.
LlmF does not methylate VeA, VelB, or KapA in vitro
Since a mutation in the SAM binding site of LlmF did not disrupt its ability to interact with VeA (Figure 9D) but did render the protein inactive in vivo (Figure 6), we hypothesized that LlmF could be directly methylating VeA or other components involved in nuclear import of VeA, such as VelB and KapA. To test this hypothesis, recombinant LlmF was incubated with tritiated SAM (3H SAM) in the presence of GST, His6-VeA-GST-S-tag, GST-VelB, and GST-KapA and subsequently, methylation was measured by fluorography. As a positive control, GST-RmtA was incubated with its substrate of histone H4 as previously described [30]. These data indicated that in vitro LlmF does not methylate histone H4, VeA, VelB, or KapA (Figure 10A and 10B).

Discussion
The multifaceted regulatory network that controls developmental differentiation and secondary metabolism in fungi is dependent on the velvet complex. Here we describe LlmF, a LaeA-like methyltransferase that constitutes a new interaction partner for VeA. LlmF is a negative regulator of sexual development and secondary metabolism through its control of VeA subcellular localization. These results provide strong evidence for a complex system regulating VeA subcellular compartmentalization as an important layer of control for development and secondary metabolism.
Using an in silico approach we identified several loci from the A. nidulans genome annotation that have sequence homology to LaeA (Figure 1), one of which (LlmF) was able to interact with VeA in a yeast-two-hybrid assay, an in vitro GST pull-down assay, and an in vivo pull-down experiment (Figure 4), thus identifying LlmF as a potential LaeA mimic or competitor. Deletion and overexpression (OE) LlmF mutants established that LlmF exhibited properties opposite of those of LaeA, specifically that LlmF repressed both sterigmatocystin and sexual development (Figure 3 and Figure 6). Interestingly, we also observed that in strains where llmF was overexpressed, sexual development was decreased in the dark, a phenotype reminiscent of the veA1 allele. Using double mutants of ΔllmF with veA1, ΔlaeA, and ΔveA it was apparent that LlmF control of secondary metabolism and development required LaeA and the full length VeA protein. Jiang et al. [13] also reported finding putative methyltransferases other than LaeA that were capable of interacting with VeA in F. graminearum, suggesting that LlmF may be conserved in other filamentous fungi.
Compartmentalization of proteins in eukaryotic cells is vital for proper growth and development. For example, several DNA binding transcription factors have been shown to shuttle between the cytoplasm and nucleus thereby directing gene transcription through subcellular localization [31]–[36] and proper localization of metabolic enzymes to organelles such as the peroxisome or mitochondria are critical for cellular function [37]–[39]. Here we observe the consequences of mis-localization of VeA mediated by LlmF for fungal development. Previous studies showed that VeA shuttles between the cytoplasm and nucleus in response to light [18]. VeA is hypothesized to act as a scaffold protein and mediate developmental pathways in response to illumination through regulation of its subcellular location [5], [23]. When the fungus is exposed to conditions favoring asexual development, e.g. light, VeA is located mainly in the cytoplasm. However, when there is no light, there is increased nuclear transport of the VeA-VelB heterodimer via the importin α (KapA), where they interact with the nuclear LaeA to induce sexual development and secondary metabolism (Figure 11). Our data implicate LlmF as a VeA cytoplasmic retention molecule contributing to proper location of VeA at the right developmental time point.

Deletion of the red light sensing phytochrome photoreceptor FphA results in increased VeA accumulation in the nucleus under light conditions, reminiscent of the ΔllmF background [20]. Blumenstein et al. [40] report that FphA is a cytoplasmic protein and functions to repress sexual development under red light. This would be consistent with the phenotypes observed with ΔllmF, however subsequent bimolecular fluorescence complementation (BiFC) assays suggest that FphA interacts with VeA in the nucleus [21]. It is also important to note that In vivo tandem affinity tag purification (TAP) of VeA resulted in co-purification of VelB, LaeA, and KapA [3], however did not pull-down LlmF or FphA. A plausible explanation for this phenomenon is that different culture conditions were used, most notably Bayram et al. [3] used sexual developmental culture conditions in the TAP purification of VeA. We suggest that these data indicate that LlmF (and potentially other proteins) may form a transient interaction with VeA in the cytoplasm thereby controlling its subcellular localization, while the stable LaeA-VeA-VelB complex forms in the nucleus.
Our data present evidence that cytoplasmic proteins interacting with VeA (LlmF and possibly FphA) control VeA localization, leading to downstream impacts on development and secondary metabolism. Here we present a model (Figure 11) where VeA retention by LlmF likely involves post-translational modifications precluding nuclear localization of VeA with a possible secondary effect of occlusion of KapA or VelB from cytoplasmic VeA. Post-translational modifications are known to both inhibit and enhance nuclear uptake depending on the cargo protein. For example, phosphorylation enhances the nuclear localization of the large tumor antigen of simian-virus 40 (SV40); conversely phosphorylation inhibits nuclear transport of Msn2p, a yeast transcriptional regulator of stress responses [31] and similarly inhibits nuclear transport of AflR [41]. Recently MpkB (Fus3) has been shown to be the kinase responsible for phosphorylating VeA, however VeA localization was not altered in the ΔmpkB background. [42]. More relevant to the putative methyltransferase ability of LlmF, methylation has also been linked to nuclear transport, as methylation of arginine residues in the NLS of RNA helicase A by the PRMT1 is required for transport into the nucleus [43].
To address if the putative methyltransferase ability of LlmF contributed to VeA localization, we constructed a SAM binding domain mutant of LlmF. Using the yeast-two-hybrid assay we determined that LlmFSAM was still capable of binding to VeA, however was not functional in vivo. Although this does not preclude a role for LlmF in blocking KapA/VelB access to VeA, this result does suggest that the main role of LlmF in directing VeA localization involved a methylation activity. Thus we tested the ability of LlmF to methylate the known proteins involved in VeA nuclear import, which are VeA, VelB, and KapA. LlmF was unable to methylate any of these proteins in an in vitro methylation assay, which suggests another methylation substrate of LlmF is involved in VeA import.
Understanding the dynamics of the velvet complex cellular compartmentalization is crucial towards identifying regulatory networks of the fungal cell. Since VeA protein levels are constant over all developmental stages of the fungus, the subcellular localization of VeA plays a large role in proper response to environmental cues, i.e. development and secondary metabolism. It is not surprising then, that several mechanisms could exist to ensure proper localization of VeA such as mediation by LlmF. Future studies on proteins or small molecules that alter the cellular location of VeA could provide a novel form of controlling production of secondary metabolites, which has implications for reduction of contaminating toxins in food supplies as well as increasing production of beneficial metabolites for industrial applications.
Materials and Methods
Fungal strains used in this study are listed in Table S1, plasmids used in this study are listed in Table S2, and primers used in this study are listed in Table S3. All strains were maintained on glucose minimal medium (GMM) [44] at 37°C and when appropriate were supplemented with 5 mM uracil, 5 mM uridine, 0.5 µM pyridoxine-HCl, 1 g L-tryptophan per liter, 0.125 g L-methionine per liter, and 1 µM p-aminobenzoate. Standard techniques were used for nucleic acid manipulations according to [45].
In silico identification of putative methyltransferases in A. nidulans
Bioinformatic analysis of the A. nidulans genome was done using a combination of the Broad Institute Aspergillus Comparative database (http://www.broadinstitute.org/annotation/genome/aspergillus_group/) and the Aspergillus Genome Database [46]. SAM binding domain containing proteins were identified by retrieving all predicted proteins in the genome that contained an annotated methyltransferase domain from the Aspergillus Comparative database at the Broad Institute. The resulting 80 protein sequences were then manually culled and those that corresponded to polyketide synthases (15) or resided inside secondary metabolite gene clusters (9) were removed from the analysis. Clr4p from Schizosaccharomyces pombe, StcP from A. flavus, and 36 previously described methyltransferase from Saccharomyces cerevisiae were manually added from PubMed to the analysis. A ClustalW alignment and inferred phylogenetic tree was completed by the use of MegAlign Software (DNASTAR). Bootstrapping was performed using 1000 trials to determine tree branch lengths using MegAlign.
Construction of mutant fungal strains
To assess the function of 8 LaeA-like methyltransferases (AN2165, AN8945, AN7933, AN5416, AN6749, AN5874, AN8833, and AN9193) gene disruption constructs were created to independently replace the putative ORFs with the A. fumigatus pyrG gene. Gene replacement constructs were created using fusion PCR [47]–[49]. Briefly, PCR products corresponding to approximately 1 kb upstream and downstream of the ORF were amplified from genomic DNA, subsequently gel purified, and then PCR-fused to flank either side of the A. fumigatus pyrG gene. The fusion PCR products were then used to transform RJMP1.49 [50] according to previously described procedures [51] with a minor modification of embedding protoplasts in molten 0.75% agar. Single gene replacement mutants were identified by PCR and subsequent Southern analysis (Figure S1 and data not shown). Prototrophic gene replacement strains as well as double mutants were constructed by sexual recombination (Table S1). Creation of VeA and VeA1 S-tagged strains were achieved via insertion of the C terminal S-tag construct from pAO81 [48] into RJMP1.27 and RJMP1.1. An llmF over-expression vector (pJMP22) was constructed by PCR fusion of the gpdA promoter to the ORF of llmF and subsequently an EcoRI-NotI fragment was cloned into the pyroA targeting vector pJW53 [52]. A SAM binding domain mutant (LlmFG91A, G93A) was constructed by quick-change mutagenesis (Stratagene) of pJMP22 to construct pJMP105. C-terminal and N-terminal llmF GFP fusions were constructed by insertion of GFP (amplified from pFNO3 and pSK505 respectively) into pJMP22 via PCR mediated insertion [53] to construct pJMP23 (OE llmF-GFP) and pJMP102 (OE GFP-GA5-llmF). The N terminal TAP tag was PCR amplified from pME2968 [54] and used to replace the GFP fragment in pJMP102 via PCR mediated insertion, yielding pJMP106 (OE TAP-GA5-llmF). Over-expression (OE) of llmF, GFP, llmFSAM, and TAP fusions was accomplished by transformation of pJMP22, pJMP23, pJMP102, pJMP105, and pJMP106 into RJMP1.59. Strains confirmed by Southern blot (data not shown) were subsequently crossed to prototrophy (Table S1).
Fungal physiology experiments
Cultures for analysis of sporulation and production of secondary metabolites were set up by overlay inoculation of 1×106 spores in molten GMM top agar containing 0.75% agar and subsequent incubation at 37°C. Three 10 mm cores were taken from each plate and homogenized in 3 mL of sterile water. Analysis of sterigmatocystin was done from cultures grown in constant light and constant dark for 4 days. Secondary metabolites were extracted with an equal volume of ethyl acetate and the organic layer was dried down. The dried extract was resuspended in 100 µL of ethyl acetate, 10 µL was spotted on silica backed thin layer chromatography plates (Whatman #4410-221), and separated using toluene: ethyl acetate: acetic acid (8∶1∶1) as a solvent. To visualize sterigmatocystin, the TLC plates were dried and sprayed with 15% aluminum chloride in ethanol and imaged under UV light (254 nm).
Sporulation was measured as previously described [24] by counting ascospores and conidia with a hemocytometer from sexual induced overlay-inoculated cultures that were grown for 5 days on GMM.
Assaying protein–protein interactions: Yeast-two-hybrid
To determine if LlmF could physically interact with VeA, a directed yeast-two-hybrid system was used based on the LexA DNA binding domain [55], which has previously been used to map interaction sites of the VelB-VeA-LaeA complex [3]. Total RNA was extracted from WIM126 grown in both asexual and sexual developmental conditions, the RNA was pooled and cDNA was constructed. A PCR product was obtained using LlmF gene specific primers (Table S3) and subsequently cloned into pACT2 (Clonetech) and confirmed by sequencing. This cDNA was subsequently moved into the ‘bait’ vector (pTLexA) and the ‘prey’ vector (pGAD424). The appropriate plasmids were then transformed into the Saccharomyces cerevisiae L40 host [56] according to previously described protocols [57], and protein-protein interactions were measured using the histidine growth reporter as well as the lacZ X-gal colorimetric reporter.
Recombinant protein purification and GST pull-downs
Heterologous expression and subsequent purification of proteins was conducted in E. coli BL21 (λDE3) using a combination of GST fusion pGEX (GE Healthcare) and N terminal 6X histidine - C terminal S-tag fusion vector pRSF-1b (EMD Biosciences). Briefly, GST fusions were constructed for LaeA, LlmF, VelB, and KapA which were purified from cell lysates using glutathione sepharose 6B (GE healthcare) following manufacturers recommendations. LlmF was cleaved from GST-LlmF using the Thrombin Cleavage Capture Kit (EMD Biosciences). An E. coli codon-optimized version of VeA was obtained from Reinhard Fischer, which was subsequently PCR amplified and cloned into pBluescript II SK - (pJMP70). An EcoRI-XhoI fragment from pJMP70 was sub-cloned into pRSF-1b (EMD Biosciences) that yielded a His6-VeA-S-tag fusion protein, which was purified using Ni-NTA resin (Qiagen) according to manufacturer's recommendations. A C terminal GST tag from pJMP89 was inserted into the XhoI site of pJMP126 to construct pJMP134 (His6-VeA-GST-S-tag), which was subsequently tandem purified with Ni-NTA resin followed by glutathione sepharose 6B (GE healthcare).
GST pull-down experiments were conducted essentially as previously described [45]. Briefly, 10 µg of purified GST tagged proteins (GST-LaeA, GST-VelB, GST-LlmF), 25 µL of glutathione sepharose 4B (GE healthcare), and 50 µL of purified His6-VeA-S-tag were co-incubated for 3 hours at 4°C in PBS buffer (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, 1 mM DTT, pH 7.3). The resin was subsequently washed 4 times with PBS buffer, boiled for 10 minutes in 2X NuPage LDS sample buffer (Invitrogen), electrophoresed on a 9% Bis-Tris polyacrylamide gel, transferred to nitrocellulose membrane, and stained with 0.1% Ponceau S solution (Sigma). Immunodetection of His6-VeA-S-tag was performed using a Rabbit α-S-tag antibody at 1∶2,000 dilution (Immunology Consultants Laboratory, Inc) followed by Goat anti-Rabbit-HRP 1∶15,000 dilution (Pierce) and chemiluminescence detection with Pierce West Pico Substrate (Pierce).
In vivo TAP pull-down experiments
One-liter liquid shaking cultures of GMM were inoculated with 5×105 spores/mL and grown for 30°C at 230 RPM for 36 hours. Mycelia was harvested by filtration through miracloth (EMD Biosciences), frozen in liquid nitrogen, and ground via cryo-compaction using a mixer mill (Retsch MM 400). Tandem affinity purification was conducted similarly as previously described [54] with a few minor modifications. Ground mycelia were extracted in buffer B250 (100 mM Tris-HCl, 250 mM NaCl, 10% glycerol, 1 mM EDTA, 1 mM DTT, 1 mM PMSF, 0.05% Tergitol-NP40, pH 7.5, and Roche Complete Protease inhibitors) and crude lysate was obtained by centrifugation at 47,810 x g for 30 minutes. The clarified crude lysate was incubated with 350 µL of IgG Sepharose 6 Fast Flow (GE Healthcare) for 3 hours on a rocking platform at 4°C and subsequently loaded into a 10 mL chromatography column (BioRad Poly-Prep). The IgG Sepharose was washed twice with 10 mL of buffer W250 (40 mM Tris-HCl, 250 mM NaCl, 1 mM PMSF, 1 mM DTT, 0.1% Tergitol-NP40, pH 8.0), once with 10 mL of buffer W150 (40 mM Tris-HCl, 150 mM NaCl, 1 mM PMSF, 1 mM DTT, 0.1% Tergitol-NP40, pH 8.0), and once with 10 mL of buffer TCB (40 mM Tris-HCl, 150 mM NaCl, 0.5 mM EDTA, 1 mM DTT, 0.1% Tergitol-NP40, pH 8.0). TEV cleavage was done by incubating the washed IgG Sepharose in 1 mL of TCB with 80 µL of rTEV (kind gift from Ivan Rayment, UW-Madison) overnight at 4°C on a rocking platform. The eluate was mixed with 500 µL of equilibrated calmodulin sepharose 4B (GE Healthcare) in 6 mL of CBB (40 mM Tris-HCl, 150 mM NaCl, 1 mM magnesium acetate, 1 mM imidazole, 2 mM CaCl2, 10 mM 2-mercaptoethanol, 0.1% Tergitol-NP40, pH 8.0) and incubated for 1 hour at 4°C on a rocking platform. The calmodulin sepharose was subsequently washed three times with 1 mL of CBB with 0.02% Tergitol-NP40 and finally eluted twice with 500 µL of CEB (40 mM Tris-HCl, 150 mM NaCl, 1 mM magnesium acetate, 1 mM imidazole, 20 mM EGTA, 10 mM 2-mercaptoethanol, pH 8.0). The eluate was TCA precipitated, resuspended in NuPAGE LDS sample buffer (Invitrogen), electrophoresed in a 10% Bis-Tris polyacrylamide gel, and transferred to Immobilon-P PVDF membrane (Millipore). Immunodetection of VeA and VeA1-S-tag was accomplished using Rabbit-α-S-tag at a 1∶2,000 dilution followed by Goat-α-Rabbit-Cy5 (GE Healthcare ECL Plex) at a 1∶2,000 dilution and imaged using a Typhoon FLA9000 fluorescent imager. The blot was stripped in 62.5 mM Tris-HCl, pH 6.5, 2% SDS, 100 mM 2-mercaptoethanol at 50°C for 30 minutes and washed twice in TBS (50 mM Tris-HCl, 150 mM NaCl, pH 7.4). The blot was then re-probed for detection of TAP-LlmF with Rabbit-α-calmodulin (Millipore) at a 1∶1,000 dilution and detected with the ECL Plex system as described above.
S-adenosyl methionine binding assay
The ability of recombinant LaeA and LlmF to bind the methyl group donor S-adenosyl methionine (SAM) was determined through an in vitro ultraviolet-light (UV) crosslinking assay as previously described [58]. Briefly, 5 µg of recombinantly purified GST, GST-LaeA, GST-LlmF, and LlmF were incubated with 6 µL of 1.0 µCi/mL of 3H-SAM (Perkin Elmer) at room temperature for 20 minutes followed by irradiation with UV light on ice for an additional 30 minutes. To demonstrate active site binding, the competitive inhibitor S-adenosyl-homocysteine (SAH) was used at 1 µM. The reactions were stopped by addition of SDS-PAGE sample buffer and electrophoresed on a 10% Tricine-SDS-PAGE gel [59]. Proteins were transferred to a nitrocellulose membrane (Protran) and bound 3H-SAM was measured by 48-hour exposure to a tritium phosphor storage screen (GE Healthcare) following manufacturers recommendations.
Microscopy
Localization of proteins was visualized with a C terminal GFP fusion of VeA [18] and N terminal GFP fusion of LlmF driven by the gpdA promoter. Cells were prepared as previously described with minor modifications [60]. Briefly, approximately 5×104 conidia were inoculated in liquid GMM medium over sterile cover slips in petri dishes at 30°C overnight. Cover slips containing adherent germlings were transferred to glass slides with 1 µL of Hoechst 33258 (1 mg/mL) and imaged with a Zeiss AxioImager A10 equipped with a Zeiss EC Plan-NEOFLUAR 40X/1.3 Oil DIC objective, a series 120 X-Cite light source (EXFO), Zeiss filter set 10 for GFP, Zeiss filter set 00 for mRFP, and Zeiss filter set 49 for Hoechst. Microscope settings were identical for all samples and GFP quantification was achieved using the Zeiss AxioVision 4.7 software of 50 nuclei and 50 cytoplasmic areas for each strain.
Northern analysis
Total RNA was extracted using TriZol (Invitrogen) using manufacturers recommendations. Detection of llm transcripts was achieved by analyzing mRNA from wild-type A. nidulans (RDIT9.32) grown under several different developmental conditions (Figure 2). Mycelia from sexual developmental induction cultures was scraped from the surface of solid media plate with a glass slide, lyophilized overnight, and subsequently total RNA was extracted using TriZol (Figure 6D). Vegetative growth was conducted by inoculation of 1×106 spores/mL in 50 mL of liquid GMM at 37°C and 250 rpm. Asexual and sexual development was induced by transfer of mycelia grown for 24 hours in liquid shake to solid GMM plates and further incubated in either constant light (asexual induction) or constant dark (sexual development) at 37°C. Expression of velvet complex members was assayed from mycelia grown under asexual developmental conditions for 24 hours after transfer to solid media and sexual developmental conditions for 48 hours after transfer to solid media (Figure 7). 32P-labeled probes were made using standard techniques [45] and primers are listed in Table S3.
In vitro methylation assay
Methylation assays were conducted essentially as previously described [30], with the following minor modifications. Five micrograms of purified LlmF was incubated with 5 µL of 1.0 µCi/mL of 3H-SAM (Perkin Elmer) and 5 µg of putative substrate protein in 1X methyltransferase buffer (50 mM HEPES, 100 mM NaCl, 2 mM DTT, pH 7.5) for 1 hour at 30°C. The reactions were stopped by addition of 4X sample buffer and the entire reactions were electrophoresed via Bis-Tris SDS-PAGE. Gels were subsequently stained with Coomassie Brilliant Blue R-250, impregnated with En3Hance fluorography enhancing solution (Perkin Elmer), dried, and exposed to a tritium phosphor storage screen (GE Healthcare) for 2 weeks.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. BrownDW, YuJH, KelkarHS, FernandesM, NesbittTC, et al. (1996) Twenty-five coregulated transcripts define a sterigmatocystin gene cluster in Aspergillus nidulans. Proc Natl Acad Sci U S A 93 : 1418–1422.
2. BokJW, KellerNP (2004) LaeA, a regulator of secondary metabolism in Aspergillus spp. Eukaryot Cell 3 : 527–535.
3. BayramO, KrappmannS, NiM, BokJW, HelmstaedtK, et al. (2008) VelB/VeA/LaeA complex coordinates light signal with fungal development and secondary metabolism. Science 320 : 1504–1506.
4. Sarikaya BayramO, BayramO, ValeriusO, ParkHS, IrnigerS, et al. (2010) LaeA control of velvet family regulatory proteins for light-dependent development and fungal cell-type specificity. PLoS Genet 6: e1001226 doi:10.1371/journal.pgen.1001226.
5. BayramO, BrausGH (2012) Coordination of secondary metabolism and development in fungi: the velvet family of regulatory proteins. FEMS Microbiol Rev 36 : 1–24.
6. SuguiJA, PardoJ, ChangYC, MüllbacherA, ZaremberKA, et al. (2007) Role of laeA in the regulation of alb1, gliP, conidial morphology, and virulence in Aspergillus fumigatus. Eukaryot Cell 6 : 1552–1561.
7. BokJW, BalajeeSA, MarrKA, AndesD, NielsenKF, et al. (2005) LaeA, a regulator of morphogenetic fungal virulence factors. Eukaryot Cell 4 : 1574–1582.
8. MyungK, ZitomerNC, DuvallM, GlennAE, RileyRT, et al. (2012) The conserved global regulator VeA is necessary for symptom production and mycotoxin synthesis in maize seedlings by Fusarium verticillioides. Plant Pathology 61 : 152–160.
9. WiemannP, BrownDW, KleigreweK, BokJW, KellerNP, et al. (2010) FfVel1 and FfLae1, components of a velvet-like complex in Fusarium fujikuroi, affect differentiation, secondary metabolism and virulence. Mol Microbiol 77 : 972–994.
10. WuD, OideS, ZhangN, ChoiMY, TurgeonBG (2012) ChLae1 and ChVel1 regulate T-toxin production, virulence, oxidative stress response, and development of the maize pathogen Cochliobolus heterostrophus. PLoS Pathog 8: e1002542 doi:10.1371/journal.ppat.1002542.
11. AmaikeS, KellerNP (2009) Distinct roles for VeA and LaeA in development and pathogenesis of Aspergillus flavus. Eukaryot Cell 8 : 1051–1060.
12. DuranRM, CaryJW, CalvoAM (2009) The role of veA on Aspergillus flavus infection of peanuts, corn and cotton. Open Mycology Journal 3 : 27–36.
13. JiangJ, LiuX, YinY, MaZ (2011) Involvement of a velvet protein FgVeA in the regulation of asexual development, lipid and secondary metabolisms and virulence in Fusarium graminearum. PLoS ONE 6: e28291 doi:10.1371/journal.pone.0028291.
14. MerhejJ, UrbanM, Hammond-KosackKE, Richard-ForgetF, BarreauC (2011) The velvet gene, FgVe1, affects fungal development and positively regulates trichothecene biosynthesis and pathogenicity in Fusarium graminearum. Mol Plant Pathol doi:10.1111/j.1364-3703.2011.00755.x.
15. JiangJ, YunY, LiuY, MaZ (2012) FgVELB is associated with vegetative differentiation, secondary metabolism and virulence in Fusarium graminearum. Fungal Genet Biol 49 : 653–662.
16. LeeJ, MyongK, KimJ-E, KimH-K, YunS-H, et al. (2012) FgVelB globally regulates sexual reproduction, mycotoxin production and pathogenicity in the cereal pathogen Fusarium graminearum. Microbiology 158 : 1723–1733.
17. BokJW, NoordermeerD, KaleSP, KellerNP (2006) Secondary metabolic gene cluster silencing in Aspergillus nidulans. Mol Microbiol 61 : 1636–1645.
18. StinnettSM, EspesoEA, CobenoL, Araujo-BazanL, CalvoAM (2007) Aspergillus nidulans VeA subcellular localization is dependent on the importin alpha carrier and on light. Mol Microbiol 63 : 242–255.
19. KimH, HanK, KimK, HanD, JahngK, et al. (2002) The veA gene activates sexual development in Aspergillus nidulans. Fungal Genet Biol 37 : 72–80.
20. PurschwitzJ, MüllerS, KastnerC, SchöserM, HaasH, et al. (2008) Functional and physical interaction of blue - and red-light sensors in Aspergillus nidulans. Curr Biol 18 : 255–259.
21. PurschwitzJ, MüllerS, FischerR (2009) Mapping the interaction sites of Aspergillus nidulans phytochrome FphA with the global regulator VeA and the White Collar protein LreB. Mol Genet Genomics 281 : 35–42.
22. NiM, YuJH (2007) A novel regulator couples sporogenesis and trehalose biogenesis in Aspergillus nidulans. PLoS ONE 2: e970 doi:10.1371/journal.pone.0000970.
23. CalvoAM (2008) The VeA regulatory system and its role in morphological and chemical development in fungi. Fungal Genet Biol 45 : 1053–1061.
24. PalmerJM, MallaredyS, PerryDW, SanchezJF, TheisenJM, et al. (2010) Telomere position effect is regulated by heterochromatin-associated proteins and NkuA in Aspergillus nidulans. Microbiology 156 : 3522–3531.
25. EdgarRC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32 : 1792–1797.
26. KaganRM, ClarkeS (1994) Widespread occurrence of three sequence motifs in diverse S-adenosylmethionine-dependent methyltransferases suggests a common structure for these enzymes. Arch Biochem Biophys 310 : 417–427.
27. PetrossianTC, ClarkeSG (2009) Multiple motif scanning to identify methyltransferases from the yeast proteome. Mol Cell Proteomics 8 : 1516–1526.
28. ShaabanMI, BokJW, LauerC, KellerNP (2010) Suppressor mutagenesis identifies a velvet complex remediator of Aspergillus nidulans secondary metabolism. Eukaryot Cell 9 : 1816–1824.
29. Araújo-BazanL, DhingraS, ChuJ, Fernández-MartínezJ, CalvoAM, et al. (2009) Importin alpha is an essential nuclear import carrier adaptor required for proper sexual and asexual development and secondary metabolism in Aspergillus nidulans. Fungal Genet Biol 46 : 506–515.
30. TrojerP, DanglM, BauerI, GraessleS, LoidlP, et al. (2004) Histone methyltransferases in Aspergillus nidulans: evidence for a novel enzyme with a unique substrate specificity. Biochemistry (Mosc) 43 : 10834–10843.
31. NardozziJD, LottK, CingolaniG (2010) Phosphorylation meets nuclear import: a review. Cell Commun Signal 8 : 32.
32. WhitmarshAJ, DavisRJ (2000) Regulation of transcription factor function by phosphorylation. Cell Mol Life Sci 57 : 1172–1183.
33. BernreiterA, RamónA, Fernández-MartínezJ, BergerH, Araújo-BazanL, et al. (2007) Nuclear export of the transcription factor NirA is a regulatory checkpoint for nitrate induction in Aspergillus nidulans. Mol Cell Biol 27 : 791–802.
34. BergerH, PachlingerR, MorozovI, GollerS, NarendjaF, et al. (2006) The GATA factor AreA regulates localization and in vivo binding site occupancy of the nitrate activator NirA. Mol Microbiol 59 : 433–446.
35. BeckT, HallMN (1999) The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature 402 : 689–692.
36. PelegY, AddisonR, AramayoR, MetzenbergRL (1996) Translocation of Neurospora crassa transcription factor NUC-1 into the nucleus is induced by phosphorus limitation. Fungal Genet Biol 20 : 185–191.
37. Maggio-HallLA, KellerNP (2004) Mitochondrial beta-oxidation in Aspergillus nidulans. Mol Microbiol 54 : 1173–1185.
38. ChandaA, RozeLV, KangS, ArtymovichKA, HicksGR, et al. (2009) A key role for vesicles in fungal secondary metabolism. Proc Natl Acad Sci U S A 106 : 19533–19538.
39. HynesMJ, MurraySL, KhewGS, DavisMA (2008) Genetic analysis of the role of peroxisomes in the utilization of acetate and fatty acids in Aspergillus nidulans. Genetics 178 : 1355–1369.
40. BlumensteinA, VienkenK, TaslerR, PurschwitzJ, VeithD, et al. (2005) The Aspergillus nidulans phytochrome FphA represses sexual development in red light. Curr Biol 15 : 1833–1838.
41. ShimizuK, HicksJK, HuangTP, KellerNP (2003) Pka, Ras and RGS protein interactions regulate activity of AflR, a Zn(II)2Cys6 transcription factor in Aspergillus nidulans. Genetics 165 : 1095–1104.
42. BayramO, BayramÖS, AhmedYL, MaruyamaJ-I, ValeriusO, et al. (2012) The Aspergillus nidulans MAPK module AnSte11-Ste50-Ste7-Fus3 controls development and secondary metabolism. PLoS Genet 8: e1002816 doi:10.1371/journal.pgen.1002816.
43. SmithWA, SchurterBT, Wong-StaalF, DavidM (2004) Arginine methylation of RNA helicase a determines its subcellular localization. J Biol Chem 279 : 22795–22798.
44. ShimizuK, KellerNP (2001) Genetic involvement of a cAMP-dependent protein kinase in a G protein signaling pathway regulating morphological and chemical transitions in Aspergillus nidulans. Genetics 157 : 591–600.
45. Sambrook J, Russell DW (2001) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
46. ArnaudMB, ChibucosMC, CostanzoMC, CrabtreeJ, InglisDO, et al. (2010) The Aspergillus Genome Database, a curated comparative genomics resource for gene, protein and sequence information for the Aspergillus research community. Nucleic Acids Res 38: D420–427.
47. YuJH, HamariZ, HanKH, SeoJA, Reyes-DomínguezY, et al. (2004) Double-joint PCR: a PCR-based molecular tool for gene manipulations in filamentous fungi. Fungal Genet Biol 41 : 973–981.
48. YangL, UkilL, OsmaniA, NahmF, DaviesJ, et al. (2004) Rapid production of gene replacement constructs and generation of a green fluorescent protein-tagged centromeric marker in Aspergillus nidulans. Eukaryot Cell 3 : 1359–1362.
49. SzewczykE, NayakT, OakleyCE, EdgertonH, XiongY, et al. (2006) Fusion PCR and gene targeting in Aspergillus nidulans. Nat Protoc 1 : 3111–3120.
50. ShaabanM, PalmerJM, El-NaggarWA, El-SokkaryMA, HabibE-SE, et al. (2010) Involvement of transposon-like elements in penicillin gene cluster regulation. Fungal Genet Biol 47 : 423–432.
51. MillerBL, MillerKY, TimberlakeWE (1985) Direct and indirect gene replacements in Aspergillus nidulans. Mol Cell Biol 5 : 1714–1721.
52. TsitsigiannisDI, ZarnowskiR, KellerNP (2004) The lipid body protein, PpoA, coordinates sexual and asexual sporulation in Aspergillus nidulans. J Biol Chem 279 : 11344–11353.
53. van den EntF, LöweJ (2006) RF cloning: a restriction-free method for inserting target genes into plasmids. J Biochem Biophys Methods 67 : 67–74.
54. BuschS, SchwierEU, NahlikK, BayramO, HelmstaedtK, et al. (2007) An eight-subunit COP9 signalosome with an intact JAMM motif is required for fungal fruit body formation. Proc Natl Acad Sci U S A 104 : 8089–8094.
55. ChoJ, YunS, JangY, ChaM, KwonN, et al. (2003) Identification and cloning of jipA encoding a polypeptide that interacts with a homolog of yeast Rad6, UVSJ in Aspergillus nidulans. J Microbiol 41 : 46–51.
56. VojtekAB, HollenbergSM, CooperJA (1993) Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell 74 : 205–214.
57. GietzRD, SchiestlRH (2007) Frozen competent yeast cells that can be transformed with high efficiency using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2 : 1–4.
58. DongH, RenS, ZhangB, ZhouY, Puig-BasagoitiF, et al. (2008) West Nile virus methyltransferase catalyzes two methylations of the viral RNA cap through a substrate-repositioning mechanism. J Virol 82 : 4295–4307.
59. SchäggerH (2006) Tricine-SDS-PAGE. Nat Protoc 1 : 16–22.
60. Momany M (2001) Cell biology of the duplication cycle in fungi. In: Talbot N, editor. Molecular and cellular biology of filamentous fungi: a practical approach: Oxford University Press, USA. pp. 119–124.
61. KatzJE, DlakićM, ClarkeS (2003) Automated identification of putative methyltransferases from genomic open reading frames. Mol Cell Proteomics 2 : 525–540.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 1
Nejčtenější v tomto čísle
- Function and Regulation of , a Gene Implicated in Autism and Human Evolution
- An Insertion in 5′ Flanking Region of Causes Blue Eggshell in the Chicken
- Comprehensive Methylome Characterization of and at Single-Base Resolution
- Susceptibility Loci Associated with Specific and Shared Subtypes of Lymphoid Malignancies