
Break-Induced Replication Requires DNA Damage-Induced Phosphorylation of Pif1 and Leads to Telomere Lengthening
Telomeres are the ends of linear eukaryotic chromosomes maintained by an enzyme called telomerase. Non-telomeric DNA ends are often generated as a result of broken replication forks and are usually repaired by break-induced replication (BIR) or homologous recombination to avoid genomic instability. However, telomerase can interfere with the repair by adding a new telomere to a broken DNA end, or the break can be ligated to a telomere, thereby inducing genome re-arrangements that are often found in human genetic disorders and cancer. To understand how cells avoid erroneous repair, we studied cdc9-1 yeast mutant cells that generate broken replication forks with high frequency. We discovered that, in cells with DNA damage, a helicase called Pif1 is phosphorylated and this phosphorylation enables Pif1 not only to inhibit telomerase at broken DNA ends but also stimulate the break repair by BIR, which in turn leads to additional telomere lengthening. Thus, a new regulatory pathway stimulates accurate break repair by BIR and at the same time promotes telomerase activity at telomeres.
Published in the journal:
. PLoS Genet 10(10): e32767. doi:10.1371/journal.pgen.1004679
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004679
Summary
Telomeres are the ends of linear eukaryotic chromosomes maintained by an enzyme called telomerase. Non-telomeric DNA ends are often generated as a result of broken replication forks and are usually repaired by break-induced replication (BIR) or homologous recombination to avoid genomic instability. However, telomerase can interfere with the repair by adding a new telomere to a broken DNA end, or the break can be ligated to a telomere, thereby inducing genome re-arrangements that are often found in human genetic disorders and cancer. To understand how cells avoid erroneous repair, we studied cdc9-1 yeast mutant cells that generate broken replication forks with high frequency. We discovered that, in cells with DNA damage, a helicase called Pif1 is phosphorylated and this phosphorylation enables Pif1 not only to inhibit telomerase at broken DNA ends but also stimulate the break repair by BIR, which in turn leads to additional telomere lengthening. Thus, a new regulatory pathway stimulates accurate break repair by BIR and at the same time promotes telomerase activity at telomeres.
Introduction
Replication is a major source of nuclear DNA damage under normal mitotic growth conditions, i.e. in the absence of drugs or irradiation [1]. Studies in both prokaryotes and eukaryotes suggest that replication fork movement rates are heterogeneous, with slower fork movement, fork pausing and breaks at “hard-to-replicate” regions of the genome [2]. Stalled or broken replication forks are recognized as sites of DNA damage and activate a DDR (DNA damage response). The recognition of DNA damage leads to activation of a signaling cascade: phosphorylation is used to transduce the signal from the sensor kinases ATR and ATM (Mec1 and Tel1, respectively, in budding yeast) to the adaptors (Rad9 and Mrc1 in yeast), and then to the effector kinases Chk1 and Chk2 (Chk1 and Rad53/Dun1 in yeast), which in turn induce cell cycle arrest and DNA repair [3].
Broken replication forks generate a one-ended DSB that can be repaired by different mechanisms relying on DNA homology between the broken end and unbroken sister chromatid: homologous recombination, synthesis dependent strand annealing, or break-induced replication (BIR). During BIR, DSB processing by exo - and endonucleases generates a 3′ssDNA overhang that enables the homologous recombination machinery (Rad51/52/54/55/57) to invade a homologous sequence of the unbroken sister chromatid to re-establish the DNA synthesis [4]. Unlike conventional DNA replication, BIR employs a conservative replication mode [5], [6] and requires the Polδ subunit Pol32 and the 5′→3′ helicase Pif1 [7], [8].
Replication of telomeres, the ends of eukaryotic chromosomes, involves the enzyme telomerase. The core telomerase complex of S. cerevisiae includes an RNA component TLC1 [9] and the reverse transcriptase Est2 [10], [11]. Telomerase-dependent telomere synthesis is tightly coupled to conventional DNA replication and occurs in the S/G2 phase of the cell cycle [12], [13]. Telomerase can also add a telomere to a non-telomeric DNA end [14], [15], such as a DSB or a broken replication fork, thereby leading to a terminal deletion [16]. To prevent de novo telomere addition, the Pif1 helicase is phosphorylated in cells with DNA damage and the phosphorylated form of the protein inhibits telomerase at broken DNA ends [17]. Pif1 helicase is also a negative regulator of telomerase at telomeres under normal growth conditions [16]. Therefore, cells lacking Pif1 possess both longer telomeres and elevated frequencies of de novo telomere addition to DSBs [16], [18].
Although telomerase is inhibited at broken DNA ends through the phosphorylation of Pif1 by the DNA damage signaling machinery, as yet it remains unknown if any modulation of telomere synthesis in response to DNA damage takes place. Here we report that the DNA damage-induced phosphorylation of Pif1 is required not only for the inhibition of telomerase at DSBs but also for the function of Pif1 in BIR. In turn, activation of BIR in cells with DNA damage leads to telomere lengthening which may provide an additional layer of telomere protection against the DNA repair machinery [19]. Thus, the DNA damage-induced phosphorylation of Pif1 promotes potentially telomere-stabilizing telomere extension while repressing deleterious de novo telomere addition events at DNA breaks, thereby funneling the broken DNA into appropriate genome-preserving repair pathways.
Results
Telomerase-dependent telomere addition is increased in cdc9-1 cells
During DNA replication, incomplete DNA ligation via partially abrogated ligase function may cause DNA discontinuities. In S. cerevisiae, CDC9 encodes a replicative DNA ligase essential for yeast cell viability [20]. Like other temperature sensitive cdc9 mutants, cdc9-1 cells grow normally at 23°C but at 36°C arrest as large-budded cells with the nuclei at the bud necks [21]. The cdc9-1 arrest is temporarily relieved in cdc9-1 rad9Δ cells, which at the non-permissive temperature undergo one or two cell divisions before losing viability [22].
It has been reported previously that cdc9-1 mutants have longer telomeres [23]. When grown at 22°C, the cdc9-1 mutant strain possessed a bulk telomere length similar to CDC9 cells (Figure 1A). However, when cdc9-1 yeast were propagated at an intermediate temperature, 26°C, after about 80 generations the telomeres were distinctly and reproducibly elongated (mean telomere length 450 bp) compared to the isogenic CDC9 strain (mean telomere length 350 bp) (Figure 1A). At this semi-permissive temperature (26°C), cdc9-1 cells showed only a very mild growth defect phenotype and possessed a slightly smaller colony size (Figure 1B).

The telomere length phenotype in cdc9-1 cells could have arisen from any of the following possibilities: (i) increased telomerase action, (ii) increased telomeric recombination, (iii) impaired telomere processing/shortening. To distinguish between these possibilities, we compared the dynamics of telomere length changes in response to cdc9-1 in the presence and absence of telomerase. The double heterozygous diploids CDC9/cdc9-1 EST2/est2Δ and CDC9/cdc9-1 TLC1/tlc1Δ were sporulated and germinated spores grown into single colonies (about 20 generations) at 22°C. The four progeny spores from the tetrads were subsequently grown on plates at 26°C for a further ∼40 generations (two streaks) for telomere length analysis. As expected, in the presence of telomerase, the cdc9-1 cells possessed longer telomeres than CDC9 cells (Figure 1C). However, in cells lacking Est2 or TLC1, the cdc9-1 mutation had no effect on telomere length: both CDC9 and cdc9-1 cells lacking either essential telomerase component showed similar rates of telomere shortening. Thus, the telomere lengthening in response to cdc9-1 was dependent on functional telomerase and was not a result of impaired telomere erosion or end processing.
Constitutive activation of the DNA damage signaling leads to telomere lengthening in cdc9-1 cells
FACS analysis indicated that non-synchronized populations of haploid cdc9-1 yeast were enriched for cells with 2n DNA content (Figure 2A), suggestive of a cell cycle progression delay in late S-phase or G2, perhaps due to checkpoint activation. In response to DNA damage, a central component of the DNA damage signaling pathway, Rad53, is activated by phosphorylation [24]. In cdc9-1 cells at 26°C, but not 22°C, a shift in Rad53 mobility upon SDS-PAGE characteristic of Rad53 phosphorylation in response to DNA damage was observed (Figure 2B). Therefore, at the semi-permissive temperature the cdc9-1 mutation leads to DDR activation.

We tested the genetic dependency of the telomere lengthening in cdc9-1 cells at 26°C on known DNA damage signaling network components. Diploid strains heterozygous for both CDC9/cdc9-1 and each gene of interest were sporulated and the spores germinated at 22°C. In the case of MEC1 or RAD53, the cells were also heterozygous for SML1/sml1Δ in order to suppress the known lethality of mec1Δ or rad53Δ with sml1Δ in the haploid progeny [25]. Progeny with different genotypes were propagated at 26°C for 4 re-streaks (∼80 generations) to allow telomere length to reach the stable length characteristic of each genotype.
Telomere elongation in cdc9-1 cells was retained in a tel1Δ or chk1Δ background, in the replication checkpoint deficient mutant mrc1-AQ [26] and in an sml1Δ dun1Δ background where sml1Δ suppresses the effect of dun1Δ on S-phase progression [27]. In contrast, the telomere lengthening in cdc9-1 cells was dependent on RAD50, MEC3, RAD24 (to some extent), MEC1, RAD9, and RAD53 (Figure 2C).
Cdc17 is a catalytic subunit of the DNA polymerase Polα that plays a pivotal role in the coordination of telomere synthesis and conventional DNA replication [28]. Like cdc9-1, a temperature sensitive mutation, cdc17-1, results in longer telomeres [23], [29]. However, cdc17-1 did not exhibit constitutive activation of DDR and the cdc17-1 induced telomere lengthening was independent of the DNA damage checkpoint (Figure S1). Therefore, we conclude that the telomere elongation phenotype of cdc9-1 at 26°C is not a defect related to the coordination of lagging strand synthesis at telomeres, but rather depends on activation of the central DNA damage checkpoint network (RAD50, MEC3, RAD24, MEC1, RAD9, and RAD53) but not on TEL1, MRC1, DUN1 or CHK1. Thus, the effect of cdc9-1 on telomere synthesis is likely to be indirect, via activation of the DNA damage signaling which then leads to telomere lengthening.
Phosphorylation of nPif1 via DNA damage signaling machinery is required for the cdc9-1 induced telomere lengthening
We used a candidate approach to identify a potential link between the activation of the DNA damage response and telomere lengthening in cdc9-1 cells based on (i) a previously reported role for the candidate proteins in telomere metabolism; (ii) the candidate proteins are regulated by the DNA damage signaling machinery. A set of candidates came from the report that activation of DDR leads to dynamic changes in telomere architecture [30]. The yeast telomeric chromatin components Rap1, Sir2/3/4, Rif1, and Rif2 (which regulate telomerase action on a telomere in cis [31]), and the Ku70/80 complex (which is required for telomerase localization in the nucleus and at the telomeres [32]) re-localize away from telomeres in a Rad9-dependent manner [30]. Such re-localization during DDR activation in cdc9-1 could lead to longer telomeres. We analyzed the effect of the cdc9-1 mutation on telomere length in cells deleted for RIF1, RIF2, SIR2, SIR3, SIR4, YKU70, or YKU80. The telomere length increase observed in cdc9-1 cells did not depend on RIF1 or RIF2 but was moderately decreased in the absence of the Sir proteins, and no telomere elongation was observed in either yku70 or yku80 backgrounds (Figure S2). Therefore, the telomere elongation observed in cdc9-1 cells is dependent on Ku70/80 and at least partially dependent on Sir proteins. The Ku70/80 dependence is in accord with the known requirement of Ku70/80 in telomerase recruitment [32], while the SIR-dependence could reflect known roles in telomere architecture and telomerase regulation in cis, or as yet unexplored role(s) in BIR-dependent telomere replication.
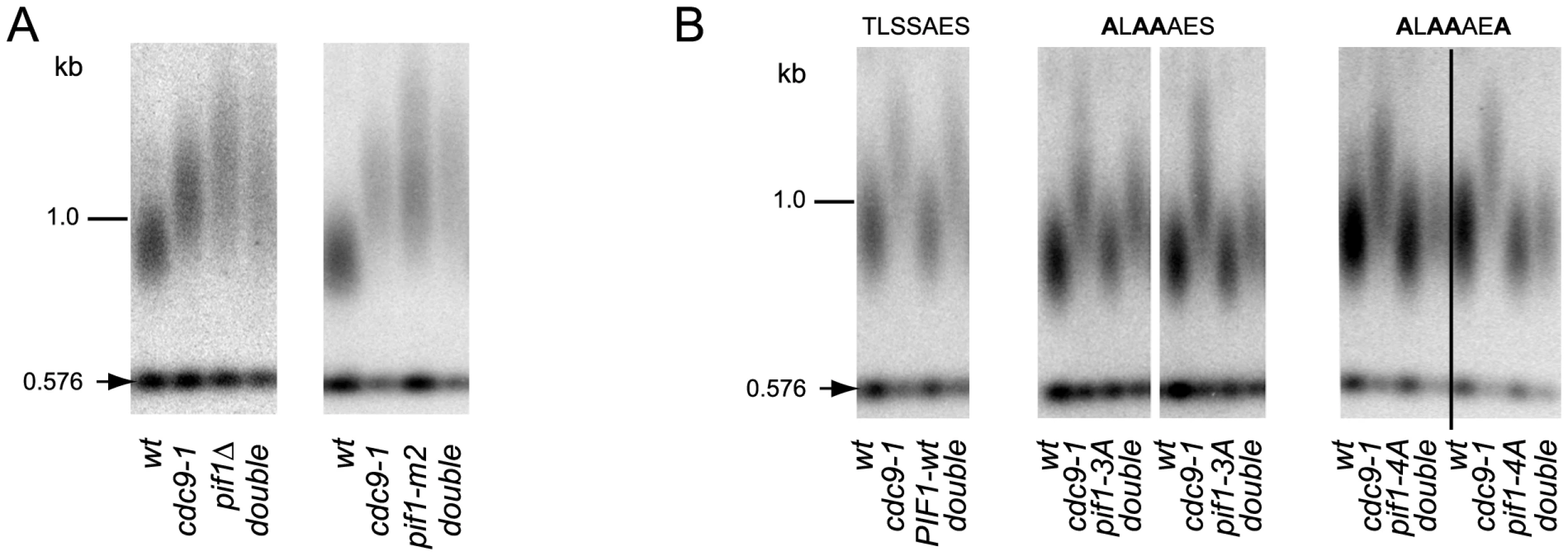
DNA damage-induced signaling is also known to regulate a nuclear form of the Pif1 helicase [17], a well characterized inhibitor of telomerase at both telomeres and DNA breaks [16]. Phosphorylation of nuclear Pif1 (nPif1) in response to DSBs is required for telomerase inhibition at broken DNA ends [17]. To query if nPif1 and its phosphorylation during the DDR were responsible for the telomere lengthening in cdc9-1 cells, we combined cdc9-1 with pif1Δ, pif1-m2 (nPif1 null [16]), or the previously reported PIF1 alleles pif1-3A and pif1-4A that abrogate Mec1-Rad53-dependent phosphorylation of the peptide TLSSAES [17]. Like cdc9-1, pif1Δ and pif1-m2 have longer telomeres but no additive telomere lengthening was observed in either cdc9-1 pif1Δ or cdc9-1 pif1-m2 (Figure 3A). Furthermore, both pif1-3A and pif1-4A mutations alleviated the cdc9-1 induced telomere lengthening, with slightly more pronounced effect in a pif1-4A background compared with pif1-3A (Figure 3B), suggesting a possibility of nPif1 phosphorylation playing a role in telomere elongation in cdc9-1 cells. Thus, the telomere lengthening in cdc9-1 cells is genetically dependent on the presence of the nuclear form of Pif1 and specifically on the phosphorylation of TLSSAES.
To test whether nPif1 was phosphorylated in cdc9-1 cells, we immunoprecipitated Pif1-4myc from CDC9 and cdc9-1 cells and treated a third of each sample with either CIP or λ phosphatase and compared their mobility by SDS-PAGE (Figure 4A). Indeed, nPif1 migration was retarded to a greater extent in cdc9-1 than in CDC9 cells and phosphatase treatment eliminated this mobility difference. In fact, phosphatase treatment resulted in faster mobility nPif1 species in both CDC9 and cdc9-1 cells (Figure 4A: compare lane 2 to lanes 4 and 6). Together, these findings suggest that nPif1 possesses a basal level of phosphorylation in wild-type cells, and that additional phosphorylation events occur in response to cdc9-1. Consistent with the DDR-dependent modulation of nPif1 activity at DSBs, nPif1 phosphorylation in response to cdc9-1 depended on MEC1 and RAD53 but not TEL1 or DUN1 (Figure 4B). To further probe nPif1phosphorylation, we used an antibody specific to the nPif1 phospho-regulatory locus (pT)LS(pS)AE (anti-P-Pif1 antibody) [17] and established its phosphatase-dependent recognition of nPif1 in cdc9-1 (but not CDC9) cells that was largely abrogated by the pif1-4A mutation (Figure 4C, top panel). In sml1Δ mec1Δ and sml1Δ rad53Δ backgrounds, the cdc9-1 dependent recognition of Pif1 with the anti-P-Pif1 antibody was considerably reduced (Figure 4D). Taken together, the data indicate that cdc9-1 activates MEC1-RAD53-dependent phosphorylation of nPif1 on TLSSAES (as well as on other positions on Pif1), and that the resulting telomere lengthening minimally requires TLSSAES phosphorylation.

Telomere lengthening in cdc9-1 requires functional components of the BIR pathway
In addition to its roles in regulation of telomerase at telomeres and DSBs [16], [17], nPif1 also has been reported to have a role in BIR [8]. The cdc9-1 mutation causes replicative ligase insufficiency which results in nicks as evidenced from accumulation of unligated nascent DNA strands [20], [33]. Passage of replication forks through these nicks would lead to broken replication forks [34] that would require homology-mediated repair, such as homologous recombination or BIR. To test if BIR had any role in telomere lengthening in cdc9-1 cells, we combined cdc9-1 with deletions in genes encoding the major components of BIR machinery, such as Rad51, Rad52, and Pol32. Indeed, we observed that RAD51, RAD52, and POL32 were required for the cdc9-1 induced telomere elongation (Figure 5A). Further, the cdc9-1 induced accumulation of G2 cells was retained in pif1-4A and pol32Δ backgrounds (Figure S3) despite the absence of telomere lengthening (see Figures 3B and 5A), suggesting that PIF1 and POL32 are downstream of the DDR activation in the pathway leading to telomere elongation in cdc9-1 cells. Because Pol32 and nPif1 are required specifically for BIR [7], [8] but not for homologous recombination, our data suggest that BIR plays a major role in telomere elongation in cdc9-1 yeast.

To explore this hypothesis further we tested if the PCSS complex (Psy3/Csm2/Shu1/Shu2) and Mph1 helicase, known to regulate Rad51 filament formation and post-invasion steps in BIR respectively [35], [36], [37], had an effect on telomere length in cdc9-1. Deletion of PSY3 partially suppressed telomere elongation in cdc9-1 cells (Figure 5B), consistent with the previously reported role for the PCSS complex as a positive regulator of Rad51 filament assembly on a processed DSB [35], [36]. Mph1 disrupts D-loops formed upon invasion of Rad51-covered ssDNA into a homologous sequence [37] and MPH1 overexpression from the GAL1 promoter inhibits BIR [38]. Overexpression of MPH1 in cdc9-1 cells resulted in alleviation of telomere elongation (Figure 5C) consistent with the hypothesis that telomere elongation is BIR-dependent. Thus, the telomere elongation in cdc9-1 is genetically dependent on core components of BIR machinery Rad51, Rad52, nPif1, Pol32 as well as on its regulators Mph1 and the PCSS complex.
The DNA damage induced phosphorylation of nPif1 on TLSSAES is required for its role in BIR
Since telomere elongation in response to cdc9-1 was dependent on both BIR and nPif1 phosphorylation on TLSSAES, we tested if the nPif1 TLSSAES phosphorylation was required for BIR using a well-established system of galactose-inducible HO endonuclease expression to generate a DSB [39] next to two selectable markers, each located on either side of the break (see Figure 6A for details). The KAN selectable marker on the centromere-proximal side of the break was further separated into two non-functional fragments on CHR VIIL and CHR IIR, such that a functional allele of KAN would be restored only when BIR initiation at CHR IIR provided the template for repair of DSB on CHR VIIL (Figure 6A). Consistent with previously reported data [8], PIF1 cells were more efficient at BIR than pif1-m2, and the pif1-4A mutants exhibited a loss of function similar to pif1-m2 (Figure 6B). Both pol32 and rad9 abrogated PIF1-dependent BIR (Figure 6B), suggesting that nPif1 requires functional Polδ and DDR to promote BIR. Since de novo telomere addition is upregulated in pif1-4A cells [17] and thus might compete with BIR for DSBs, we tested whether est2 or yku80 mutations, which disrupt de novo telomere addition [18], affected BIR in PIF1 and pif1-4A cells and found that the increased de novo telomere addition in pif1-4A did not impede BIR (Figure 6B). Therefore, the phosphorylation of nPif1 at the TLSSAES locus is required specifically for its role in BIR that is not affected by de novo telomere addition events that occur at DSBs.

Longer telomeres in cdc44-5 and rrm3Δ strains are further examples of BIR-dependent telomere elongation
Our hypothesis that cdc9-1 cells suffer from broken replication forks as a result of replication passing through nicks is based on (i) the known role of Cdc9 in replication as a ligase of newly synthesized DNA strands [20], (ii) observed accumulation of unligated nascent DNA strands in the cdc9-1 mutant [20], [33] which suggests nicks or single-stranded gaps, (iii) the established fact that replication through a nick results in a broken replication fork [34], and (iv) RAD9-dependent cell cycle arrest of cdc9-1 mutants at the non-permissive temperature [20], [21] which can be explained by the existence of broken replication forks. If the hypothesis that broken replication forks repaired by BIR contribute to telomere elongation is correct, then there should be mutations in other components of replication machinery that also result in longer telomeres due to increased breakage of replication forks and activation of a DDR and BIR. Rrm3 is a non-essential DNA helicase required for replication through “hard-to-replicate” regions and the loss of RRM3 results in a higher rate of fork breakage, constitutive activation of a DDR, and slight telomere elongation that is PIF1-dependent [40], [41]. cdc44-5 is a temperature-sensitive mutant allele of Replication Factor C which operates as a PCNA clamp loader. At 30°C, cdc44-5 mutants possess longer telomeres and accumulate cells in G2 [23]. We analyzed both rrm3Δ and cdc44-5 for constitutive activation of a DDR and telomere elongation, and the possible dependence of the latter on Pol32 and nPif1. The DDR was constitutively activated in both rrm3Δ and cdc44-5, with more pronounced Rad53 phosphorylation observed in cdc44-5 cells (Figure S4A). The slight telomere elongation in rrm3Δ was abolished by the pif1-4A mutation and there was no further increase in telomere length in rrm3Δ pol32Δ cells compared to either single mutant alone (Figure S4B). Combining cdc44-5 with pol32Δ resulted in a synthetic lethality (Figure S4C) suggesting that BIR might be constitutively active in cdc44-5 and required for cell viability (or that the combination of the two mutations could be too disruptive for conventional replication). The extensive telomere lengthening in cdc44-5 cells was strongly suppressed by the pif1-4A mutation (Figure S4D). Therefore, both rrm3Δ and cdc44-5 resemble cdc9-1 in their constitutive activation of DDR and longer telomere phenotype and, like cdc9-1, telomere elongation in rrm3Δ cells is dependent on the BIR components nPif1 and Pol32 and the long telomere phenotype in cdc44-5 is dependent on nPif1.
Discussion
Here we report that the DNA damage-induced phosphorylation of nPif1, previously found to be critical for its role in inhibiting telomerase action at DSBs [17], is also required for functional BIR which in turn promotes telomere elongation.
The role of the replicative ligase Cdc9 in DNA replication is very well understood and its functional insufficiency in cdc9-1 cells at the non-permissive temperature of 37°C leads to accumulation of unligated nascent DNA strands and RAD9-dependent cell cycle arrest [20], [21], [22], [33]. Growth of cdc9-1 yeast at the semi-permissive temperature of 26°C predicts residual post-replication nicks: replication nicks are known to lead to DSBs as a result of broken replication forks [34]. We used the cdc9-1 mutant allele to study how cells cope with such replication-coupled DSBs.
We found that the longer telomere length in cdc9-1 cells stems from the activation of BIR and depended upon Rad51, Rad52, Pol32, and nPif1. The cdc9-1 induced telomere elongation was also dependent on telomerase and therefore could not be attributed solely to recombination involving telomeric DNA. We postulate that the Rad51 - and Rad52-dependence of telomere elongation in cdc9-1 stems from constitutive activation of BIR, a homology-based mechanism of DSB repair, which in turn affects telomerase-dependent telomere elongation by either permitting a window of opportunity for telomerase at telomeres (i.e. when BIR forks reach chromosome ends) or via sequestration of nPif1 to BIR forks and removal of Pif1-dependent inhibition of telomerase access to telomeres.
BIR might not be the primary pathway of repair for broken replication forks due to its increased mutagenesis rate [42], [43]. A broken replication fork is likely to be met by a fork coming from the opposite direction so that a two-ended DSB is generated and repaired via homologous recombination as suggested by Cortés-Ledesma and Aguilera [34]. However, BIR might be more common in yeast sub-telomeric regions containing genome duplications and repetitive sequences [44], particularly if no replication origin has been fired in the region between a broken fork and the telomere downstream. BIR shares many similarities with conventional replication [45], but the unique requirements for Pol32 and nPif1 [7], [8] as well as the use of conservative replication synthesis [5], [6] suggest structural and functional differences between the replication forks in conventional replication and BIR. Because telomerase-dependent telomere lengthening is coupled to the passage of replication forks through telomeres [12], [13] this coupling could be different during BIR and result in longer telomeres (Figure 7). In another scenario, two rounds of replication fork passage through a telomere within the same S-phase might be responsible for the longer telomeres in cdc9-1 cells. First, a sub-telomeric origin is activated and a conventional replication fork passes through a telomere followed by a potential telomere elongation round. If a BIR fork moving towards telomere is established and if a previously replicated terminal fragment is not included in the repair, the BIR fork will pass through the same telomere creating another opportunity for telomerase to add more telomeric repeats. Therefore, two replication forks and potentially two rounds of telomerase action may take place and could account for the telomere elongation in cdc9-1 cells. Either scenario would predict that BIR could affect telomere lengthening in cis but not in trans, i.e. only the telomeres passed by BIR forks would undergo additional lengthening. An alternative explanation of the telomere elongation in cdc9-1 cells may stem from the effect of the DNA damage response on the regulation of telomerase by nPif1. In cells with no DNA damage, nPif1 inhibits telomerase at telomeres. However, during a DDR nPif1 localizes to the sites of damage and repair [8], [17]. The involvement of nPif1 in DSB repair may reduce its availability at all telomeres and result in a relief of the usual Pif1-dependent inhibition of telomerase at telomeres. This latter model would predict that the activation of BIR on a subset of chromosomes would affect all the telomeres to the same extent, i.e. the regulation would occur in trans.

Compromised DNA replication affects telomere maintenance through DNA damage signaling
The DDR-dependence of telomere elongation in cdc9-1 cells differed from the telomere elongation observed in cdc17-1 cells, which is not DDR-dependent (Figure S1). We suggest that the effect of cdc9-1 on telomere length is indirect, i.e. it is not due to uncoordinated chromosome replication and telomerase-dependent telomere lengthening. Instead, the DDR in cdc9-1 cells is required for successful repair of broken replication forks via BIR that in turn causes additional telomere lengthening. We propose that this mechanism might explain previous reports that rrm3 mutants, which suffer from replication fork pausing and breakage, also show constitutive activation of Rad53 and longer telomeres [40], [46]. A combination of constitutive damage signaling and telomere lengthening is also seen in cdc44-5 cells. It is unknown whether the telomere lengthening in these mutants is DDR-dependent (rrm3 cells require RAD53 for their viability [46]), but the telomere elongation in both mutants was dependent on the DNA damage induced phosphorylation of nPif1. Pol32 was also required for longer telomeres in rrm3 cells and the synthetic lethality between cdc44-5 and pol32Δ suggests a possibility that BIR is critical for cdc44-5 cell viability. In the genome-wide screens for S. cerevisiae deletion mutants causing telomere lengthening or shortening phenotypes, many genes that are relevant to DNA metabolism through replication, repair or chromatin structure have been reported [47], [48]. We suggest that like cdc9-1, the telomere length effects of some of these mutations may result from constitutive generation of DNA damage.
The dependency of the cdc9-1 induced telomere lengthening on the DNA damage signaling is likely to be more complex than the requirement of the signaling kinases for nPif1 phosphorylation. The checkpoint activation is also necessary for the cell cycle arrest to ensure the completion of BIR [43], and therefore is also required for BIR fork passage through telomeres before the cells can enter mitosis. The signaling is also required for re-localization of the telomere bound proteins Sir3 and Ku in response to DNA damage [30]. This event might be relevant to the cdc9-1 induced telomere lengthening as the latter was reduced in the sir mutants and abolished in the yku70 and yku80 backgrounds (Figure S2). The DNA damage induced changes in telomeric chromatin may affect telomerase access to telomeres so that telomerase is recruited to a larger number of telomeres and/or it is allowed longer time to elongate each telomere during S-phase, thereby increasing telomere length in cells with constitutive DNA damage.
Implications of nPif1 regulation by DNA damage signaling pathway for the preservation of genomic integrity
Our findings suggest that the DDR-induced nPif1 phosphorylation does not only inhibit telomerase at DSBs as previously demonstrated, but it also plays a pivotal role in promoting BIR at broken replication forks and DSBs. Healing a broken DNA end by de novo telomere addition leads to global re-arrangements or partial loss of genetic material, thereby contributing to genome instability. Therefore nPif1 regulation directed at telomerase inhibition at DSBs [17] and facilitation of DSB repair via BIR (see Figure 6B) serve together to ensure genome repair and preservation. At the same time, the increased telomerase activity on telomeres as a result of an active BIR may have additional biological significance. For example, the telomere elongation in response to DNA damage could promote recruitment of additional telomere-associated chromatin factors (Rap1, Rif1, Sir proteins, etc.) that could in turn protect shorter telomeres against the DNA repair machinery activated in response to DNA damage [19]. In addition, the telomere lengthening via BIR could promote repair of critically short telomeres that have previously eroded and activated BIR in sub-telomeric regions. Since BIR leads to telomere lengthening, it may boost telomerase-dependent elongation of critically short telomeres involved in BIR. This mechanism could act in addition to the Tel1-mediated signaling which has been reported to increase telomerase recruitment on a shortened telomere [49], [50]. On the other hand, longer telomeres are harder to replicate as telomeric chromatin causes replication fork stalling [33], [41] and broken replication forks within telomeric repeats would result in truncated telomeres. Truncated telomeres after DSBs or as a result of active trimming [51] could therefore benefit from repair by telomerase activity. Thus, dynamic telomere length adjustments in response to changing extracellular and intracellular environment could have evolved to minimize genomic instability and maximize cell fitness and survival.
In summary, we uncovered that telomerase-dependent telomere elongation is modulated in response to DNA damage. Additional elongation occurs as a result of active BIR that requires Mec1-Rad53 dependent phosphorylation of nPif1. Therefore, nPif1 is a multifunctional regulator of telomere synthesis: it inhibits telomerase at telomeres in cells with no DNA damage, whereas in cells with DNA damage, its phosphorylation impairs telomerase action at DSBs and at the same time promotes telomere synthesis via BIR.
Materials and Methods
Yeast strains are described in Table S1.
Telomere length analysis (teloblots)
For genetic analysis of telomere length in different mutants, teloblot analysis was performed on a minimum of 3 tetrads, often 4-6 tetrads, that originated from at least 2 independently constructed parental strains. Relevant heterozygous diploids of each genotype were sporulated and progeny of at least four tetrads (at least two coming from each of the two diploids) were passaged for ∼80 generations at either 26°C (tetrads containing cdc9-1 mutants) or 30°C (tetrads containing rrm3 or cdc44-5 mutants) on YPD agar (or YPGal agar if induction of GAL1 - promoter was required), to equilibrate telomere length. Yeast genomic DNA was purified, digested with KpnI, and resolved on 0.85% w/v agarose gels. Southern blotting and hybridization were performed as described previously [33]. The random-primer (Prime-It II Kit, Stratagene) radiolabeled probe KL1 (recognizes the telomere proximal 650 bp of Y′ repeats) was used for hybridization in the KpnI experiments. In cases where slight variability in mutant behavior was observed, more than one tetrad (or clone) for the given combination of mutations was included in the corresponding figures (see Figure 3B for cdc9-1 pif1-3A, Figure 5B for cdc9-1 psy3, Figure S1 for cdc17-1 rad53 sml1, and Figure S4 for cdc44-5 pif1-4A).
Immunoblotting
Yeast protein extracts were prepared using TCA precipitation [52]. Proteins were resolved on SDS-PAGE and transferred to PVDF membrane. Immunological detection was performed using ECL+ kit (GE Healthcare) according to the manufacturer's instructions. Mouse monoclonal α-myc 9E10 (Covance), goat polyclonal α-Rad53 yC-19 (Santa Cruz), and rabbit polyclonal α - VIDFYL(pT)LS(pS)AE (custom made, QCB) antibodies were used for Pif1-4myc, endogenous Rad53, and phosphorylated Pif1 detection respectively.
Phosphatase treatments
Pif1-4myc was immunoprecipitated from cleared cell lysates at 4°C. Yeast cells were homogenized by bead beating in Lysis Buffer (25 mM HEPES (pH 7.5), 150 mM KCl, 1 mM EDTA, 1 mM EGTA, 0.1% v/v NP-40, 10% v/v glycerol) with phosphatase inhibitors (50 mM NaF, 50 mM sodium glycerophosphate) and protease inhibitors (Compete, EDTA-free protease inhibitors cocktail tablets, Roche). The lysates were cleared by centrifugation (14K rpm, 20′). Pif1-4Myc was immunoprecipitated by incubation of cleared lysates with 1∶150 dilution of α-myc antibody 9E10 (Covance) for 2 h followed by addition of Protein G agarose beads (Sigma) for an additional 2 h. Beads were washed 3×5′ in the Lysis Buffer and then rinsed in 1× phosphatase buffer supplied with either CIP or λ phosphatase (NEB). Phosphatase treatments were performed at room temperature in the corresponding buffers. Reactions were stopped by addition of SDS-PAGE sample buffer and 5′ boiling.
Break induced replication assay
Cells were patched on YPRaffinose plates and grown overnight. Cells were re-suspended in YP broth to OD∼1. Serial dilutions were plated on YPD and YPGalactose agar. In 3 days, YPGalactose plates were replica-plated onto minimal media without uracil and on YPD+G418. The frequency of BIR was scored as a ratio of G418R ura− colonies to the total number of cells plated (represented by the number of colonies on YPD plates). At least two independently constructed strains for every genotype were used and at least three repeats for each genotype were performed to calculate standard deviations.
Supporting Information
Zdroje
1. MyungK, DattaA, KolodnerRD (2001) Suppression of spontaneous chromosomal rearrangements by S phase checkpoint functions in Saccharomyces cerevisiae. Cell 104 : 397–408.
2. MirkinEV, MirkinSM (2007) Replication fork stalling at natural impediments. Microbiol Mol Biol Rev 71 : 13–35.
3. MeloJ, ToczyskiD (2002) A unified view of the DNA-damage checkpoint. Curr Opin Cell Biol 14 : 237–245.
4. AnandRP, LovettST, HaberJE (2013) Break-induced DNA replication. Cold Spring Harb Perspect Biol 5: a010397.
5. DonnianniRA, SymingtonLS (2013) Break-induced replication occurs by conservative DNA synthesis. Proc Natl Acad Sci U S A 110 : 13475–13480.
6. SainiN, RamakrishnanS, ElangoR, AyyarS, ZhangY, et al. (2013) Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature 502 : 389–392.
7. LydeardJR, JainS, YamaguchiM, HaberJE (2007) Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 448 : 820–823.
8. WilsonMA, KwonY, XuY, ChungWH, ChiP, et al. (2013) Pif1 helicase and Poldelta promote recombination-coupled DNA synthesis via bubble migration. Nature 502 : 393–396.
9. SingerMS, GottschlingDE (1994) TLC1: template RNA component of Saccharomyces cerevisiae telomerase. Science 266 : 404–409.
10. LendvayTS, MorrisDK, SahJ, BalasubramanianB, LundbladV (1996) Senescence mutants of Saccharomyces cerevisiae with a defect in telomere replication identify three additional EST genes. Genetics 144 : 1399–1412.
11. LingnerJ, HughesTR, ShevchenkoA, MannM, LundbladV, et al. (1997) Reverse transcriptase motifs in the catalytic subunit of telomerase. Science 276 : 561–567.
12. DiedeSJ, GottschlingDE (1999) Telomerase-mediated telomere addition in vivo requires DNA primase and DNA polymerases alpha and delta. Cell 99 : 723–733.
13. MarcandS, BrevetV, MannC, GilsonE (2000) Cell cycle restriction of telomere elongation. Curr Biol 10 : 487–490.
14. HarringtonLA, GreiderCW (1991) Telomerase primer specificity and chromosome healing. Nature 353 : 451–454.
15. MorinGB (1991) Recognition of a chromosome truncation site associated with alpha-thalassaemia by human telomerase. Nature 353 : 454–456.
16. SchulzVP, ZakianVA (1994) The saccharomyces PIF1 DNA helicase inhibits telomere elongation and de novo telomere formation. Cell 76 : 145–155.
17. MakovetsS, BlackburnEH (2009) DNA damage signalling prevents deleterious telomere addition at DNA breaks. Nat Cell Biol 11 : 1383–1386.
18. MyungK, ChenC, KolodnerRD (2001) Multiple pathways cooperate in the suppression of genome instability in Saccharomyces cerevisiae. Nature 411 : 1073–1076.
19. McEachernMJ, IyerS (2001) Short telomeres in yeast are highly recombinogenic. Mol Cell 7 : 695–704.
20. JohnstonLH, NasmythKA (1978) Saccharomyces cerevisiae cell cycle mutant cdc9 is defective in DNA ligase. Nature 274 : 891–893.
21. HartwellLH, MortimerRK, CulottiJ, CulottiM (1973) Genetic Control of the Cell Division Cycle in Yeast: V. Genetic Analysis of cdc Mutants. Genetics 74 : 267–286.
22. SchiestlRH, ReynoldsP, PrakashS, PrakashL (1989) Cloning and sequence analysis of the Saccharomyces cerevisiae RAD9 gene and further evidence that its product is required for cell cycle arrest induced by DNA damage. Mol Cell Biol 9 : 1882–1896.
23. AdamsAK, HolmC (1996) Specific DNA replication mutations affect telomere length in Saccharomyces cerevisiae. Mol Cell Biol 16 : 4614–4620.
24. SanchezY, DesanyBA, JonesWJ, LiuQ, WangB, et al. (1996) Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science 271 : 357–360.
25. ZhaoX, MullerEG, RothsteinR (1998) A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell 2 : 329–340.
26. OsbornAJ, ElledgeSJ (2003) Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev 17 : 1755–1767.
27. ZhaoX, RothsteinR (2002) The Dun1 checkpoint kinase phosphorylates and regulates the ribonucleotide reductase inhibitor Sml1. Proc Natl Acad Sci U S A 99 : 3746–3751.
28. QiH, ZakianVA (2000) The Saccharomyces telomere-binding protein Cdc13p interacts with both the catalytic subunit of DNA polymerase alpha and the telomerase-associated est1 protein. Genes Dev 14 : 1777–1788.
29. CarsonMJ, HartwellL (1985) CDC17: an essential gene that prevents telomere elongation in yeast. Cell 42 : 249–257.
30. MartinSG, LarocheT, SukaN, GrunsteinM, GasserSM (1999) Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell 97 : 621–633.
31. WellingerRJ, ZakianVA (2012) Everything you ever wanted to know about Saccharomyces cerevisiae telomeres: beginning to end. Genetics 191 : 1073–1105.
32. PfingstenJS, GoodrichKJ, TaabazuingC, OuenzarF, ChartrandP, et al. (2012) Mutually exclusive binding of telomerase RNA and DNA by Ku alters telomerase recruitment model. Cell 148 : 922–932.
33. MakovetsS, HerskowitzI, BlackburnEH (2004) Anatomy and dynamics of DNA replication fork movement in yeast telomeric regions. Mol Cell Biol 24 : 4019–4031.
34. Cortes-LedesmaF, AguileraA (2006) Double-strand breaks arising by replication through a nick are repaired by cohesin-dependent sister-chromatid exchange. EMBO Rep 7 : 919–926.
35. BernsteinKA, ReidRJ, SunjevaricI, DemuthK, BurgessRC, et al. (2011) The Shu complex, which contains Rad51 paralogues, promotes DNA repair through inhibition of the Srs2 anti-recombinase. Mol Biol Cell 22 : 1599–1607.
36. SasanumaH, TawaramotoMS, LaoJP, HosakaH, SandaE, et al. (2013) A new protein complex promoting the assembly of Rad51 filaments. Nat Commun 4 : 1676.
37. PrakashR, SatoryD, DrayE, PapushaA, SchellerJ, et al. (2009) Yeast Mph1 helicase dissociates Rad51-made D-loops: implications for crossover control in mitotic recombination. Genes Dev 23 : 67–79.
38. Luke-GlaserS, LukeB (2012) The Mph1 helicase can promote telomere uncapping and premature senescence in budding yeast. PLoS One 7: e42028.
39. RudinN, HaberJE (1988) Efficient repair of HO-induced chromosomal breaks in Saccharomyces cerevisiae by recombination between flanking homologous sequences. Mol Cell Biol 8 : 3918–3928.
40. IvessaAS, LenzmeierBA, BesslerJB, GoudsouzianLK, SchnakenbergSL, et al. (2003) The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol Cell 12 : 1525–1536.
41. IvessaAS, ZhouJQ, SchulzVP, MonsonEK, ZakianVA (2002) Saccharomyces Rrm3p, a 5′ to 3′ DNA helicase that promotes replication fork progression through telomeric and subtelomeric DNA. Genes Dev 16 : 1383–1396.
42. DeemA, KeszthelyiA, BlackgroveT, VaylA, CoffeyB, et al. (2011) Break-induced replication is highly inaccurate. PLoS Biol 9: e1000594.
43. VasanS, DeemA, RamakrishnanS, ArguesoJL, MalkovaA (2014) Cascades of genetic instability resulting from compromised break-induced replication. PLoS Genet 10: e1004119.
44. LitiG, LouisEJ (2005) Yeast evolution and comparative genomics. Annu Rev Microbiol 59 : 135–153.
45. LydeardJR, Lipkin-MooreZ, SheuYJ, StillmanB, BurgersPM, et al. (2010) Break-induced replication requires all essential DNA replication factors except those specific for pre-RC assembly. Genes Dev 24 : 1133–1144.
46. TorresJZ, SchnakenbergSL, ZakianVA (2004) Saccharomyces cerevisiae Rrm3p DNA helicase promotes genome integrity by preventing replication fork stalling: viability of rrm3 cells requires the intra-S-phase checkpoint and fork restart activities. Mol Cell Biol 24 : 3198–3212.
47. AskreeSH, YehudaT, SmolikovS, GurevichR, HawkJ, et al. (2004) A genome-wide screen for Saccharomyces cerevisiae deletion mutants that affect telomere length. Proc Natl Acad Sci U S A 101 : 8658–8663 Epub 2004 May 8625.
48. GatbontonT, ImbesiM, NelsonM, AkeyJM, RuderferDM, et al. (2006) Telomere length as a quantitative trait: genome-wide survey and genetic mapping of telomere length-control genes in yeast. PLoS Genet 2: e35 Epub 2006 Mar 2017.
49. ChangM, ArnericM, LingnerJ (2007) Telomerase repeat addition processivity is increased at critically short telomeres in a Tel1-dependent manner in Saccharomyces cerevisiae. Genes Dev 21 : 2485–2494.
50. SabourinM, TuzonCT, ZakianVA (2007) Telomerase and Tel1p preferentially associate with short telomeres in S. cerevisiae. Mol Cell 27 : 550–561.
51. PickettHA, ReddelRR (2012) The role of telomere trimming in normal telomere length dynamics. Cell Cycle 11 : 1309–1315.
52. O'RourkeSM, HerskowitzI (1998) The Hog1 MAPK prevents cross talk between the HOG and pheromone response MAPK pathways in Saccharomyces cerevisiae. Genes Dev 12 : 2874–2886.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 10
Nejčtenější v tomto čísle
- The Master Activator of IncA/C Conjugative Plasmids Stimulates Genomic Islands and Multidrug Resistance Dissemination
- A Splice Mutation in the Gene Causes High Glycogen Content and Low Meat Quality in Pig Skeletal Muscle
- Keratin 76 Is Required for Tight Junction Function and Maintenance of the Skin Barrier
- A Role for Taiman in Insect Metamorphosis