
Coexistence and Within-Host Evolution of Diversified Lineages of Hypermutable in Long-term Cystic Fibrosis Infections
Patients with cystic fibrosis (CF) are often colonized by a single clone of the common, widespread bacterium Pseudomonas aeruginosa, resulting in chronic airway infections. Long-term persistence of the bacteria involves the emergence and selection of multiple phenotypic variants. Among these are “mutator” variants characterized by increased mutation rates resulting from the inactivation of DNA repair systems. The genetic evolution of mutators during the course of chronic infection is poorly understood, and the effects of hypermutability on bacterial population structure have not been studied using genomic approaches. We evaluated the genomic changes undergone by mutator populations of P. aeruginosa obtained from single sputum samples from two chronically infected CF patients, and found that mutators completely dominated the infecting population in both patients. These populations displayed high genomic diversity based on vast accumulation of stochastic mutations. Our results are in contrast to the concept of a homogeneous population consisting of a single dominant clone; rather, they support a model of populations structured by diverse subpopulations that coexist within the patient. Certain genes involved in adaptation were highly and convergently mutated in both lineages, suggesting that these genes were beneficial and potentially responsible for the co-selection of mutator alleles.
Published in the journal:
. PLoS Genet 10(10): e32767. doi:10.1371/journal.pgen.1004651
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004651
Summary
Patients with cystic fibrosis (CF) are often colonized by a single clone of the common, widespread bacterium Pseudomonas aeruginosa, resulting in chronic airway infections. Long-term persistence of the bacteria involves the emergence and selection of multiple phenotypic variants. Among these are “mutator” variants characterized by increased mutation rates resulting from the inactivation of DNA repair systems. The genetic evolution of mutators during the course of chronic infection is poorly understood, and the effects of hypermutability on bacterial population structure have not been studied using genomic approaches. We evaluated the genomic changes undergone by mutator populations of P. aeruginosa obtained from single sputum samples from two chronically infected CF patients, and found that mutators completely dominated the infecting population in both patients. These populations displayed high genomic diversity based on vast accumulation of stochastic mutations. Our results are in contrast to the concept of a homogeneous population consisting of a single dominant clone; rather, they support a model of populations structured by diverse subpopulations that coexist within the patient. Certain genes involved in adaptation were highly and convergently mutated in both lineages, suggesting that these genes were beneficial and potentially responsible for the co-selection of mutator alleles.
Introduction
The opportunistic pathogen Pseudomonas aeruginosa is found in many environments and can cause acute or chronic infections in a range of hosts from protozoans to plants to humans [1], [2]. In particular, patients with cystic fibrosis (CF) are highly susceptible to chronic colonization by P. aeruginosa, which is frequently fatal because of a persistent inflammatory response leading to gradual decline of lung function [3], [4]. In most cases, following a period of recurrent colonizations, a single strain of P. aeruginosa becomes predominant and persists for the rest of the patient's life [5], [6]. Genetic adaptation has been shown to play a major role in successful establishment of long-term chronic P. aeruginosa infections of CF patients, and natural selection acts on these bacteria in CF airways to accommodate the fixation of mutations that cause beneficial phenotypic changes [7], [8], [9]. The selected phenotypes display traits that differ from those of environmental isolates but are common in populations found in CF patients, suggesting repeatable patterns of long-term adaptation to the CF lung [10], [11], [12].
A trait frequently observed in chronic infections is an increased mutation rate leading to a mutator phenotype [13], [14]. P. aeruginosa from chronically infected CF airways was the first natural model to reveal a high proportion of mutators in contrast to reported proportions in acute infections [13]. Hypermutability in CF P. aeruginosa is due primarily to inactivation of the mismatch repair system (MRS) through lost function of the antimutator mutS and mutL genes [15], and 36–54% of CF patients have been shown to be infected by mutator isolates [13], [16], [17], [18], [19]. Theoretical and experimental approaches have attempted to explain the selection of MRS-mutators as the result of co-selection (hitchhiking) with linked beneficial mutations [20], [21], [22], [23], [24], [25], and their overrepresentation as a consequence of high recombination rates [26]. Mutators have been linked to the development of antibiotic resistance both in vitro and in vivo [13], [27], [28], [29], [30], [31], and have been reported to enhance genetic adaptation to CF airways through increased accumulation of new mutations [32]. However, comparisons between mutators and normo-mutators did not reveal any association between hypermutability and a particular distribution of mutations among genes, even for antibiotic resistance-related genes [32]. No study to date has linked hypermutability in CF adaptation to any specific adaptive mutation [17], [32], [33], nor to any key adaptive trait in the transition to a chronic state of infection [33].
Our previous studies demonstrated a role of MRS deficiency in the acquisition of CF-related phenotypes under in vitro conditions such as mucoid conversion [34], lasR inactivation [35], [36], and enhanced adaptability in biofilms [37] – all hallmarks of P. aeruginosa chronic airway infection. We also reported the ability of MRS deficiency to bias mutagenic pathways toward DNA simple sequence repeats (SSRs), which gave specific mutational spectra under both in vitro [34], [38] and in vivo conditions [18], [39]. In view of the widespread effect of hypermutability on the process of adaptation to the CF lung [32], it is important to elucidate the evolution of MRS-mutator strains in the course of CF chronic lung infections.
Previous genome analyses of longitudinally collected P. aeruginosa from CF patients demonstrated intra-patient genomic diversity of clonal isolates, suggesting that within-host P. aeruginosa population dynamics are driven by clonal competition (clonal interference) and/or niche specialization (adaptive radiation) [18], [40], [41], [42]. To further investigate these processes, Chung et al. compared the genomes of pairs of randomly selected contemporary isolates sampled from three chronically infected adult CF patients, and found that the pairs were differentiated by 1, 54, and 344 SNPs, respectively. In the latter case, both isolates were mutators [43].
Although mutators are frequently found in CF infections, no whole-genome studies have focused on the within-host evolution of mutators. Similarly, diversity of within-patient pathogen populations is relevant to planning of clinical intervention strategies, elucidation of transmission networks, and understanding of evolutionary processes, but no study to date has involved genome sequencing of a sufficiently large collection of P. aeruginosa isolates taken from the same patient at the same time point to facilitate an in-depth analysis of population diversity.
We combined two distinct strategies for a genome-wide analysis of P. aeruginosa MRS-mutators: (i) a longitudinal analysis of two separate clonal lineages of mutators obtained from two CF patients; (ii) a within-host population analysis of a large collection of isolates obtained from a single sputum sample from each of the patients, to provide a snapshot of mutator population structure in the CF lung at a single time point. Whole-genome sequencing of 27 P. aeruginosa isolates allowed us to quantify the nature and extent of the genomic changes of MRS-mutator clones and provide a panorama of the high genomic diversity that shapes the structure of P. aeruginosa mutator populations during long-term adaptation to the CF airway environment.
Results
P. aeruginosa sample collection
To quantitatively describe the evolutionary processes of MRS-deficient strains during chronic airway infections, we performed longitudinal and cross-sectional analyses of clonal P. aeruginosa isolates collected from two CF patients, referred to here as CFA and CFD (Figure 1, and Materials and Methods). The cross-sectional study included 90 isolates obtained from a single sputum sample from each patient. These large collections were used to investigate the clonal genomic diversity within mutator populations in a single host at a single time point. Two different non-epidemic P. aeruginosa strains were collected from geographically distant locations, Argentina (CFA) and Denmark (CFD), in which different therapeutic protocols are applied. We were thus able to analyze two independent mutator populations whose evolutionary histories were presumably subjected to common as well as patient-specific selective pressures.

The collection from CFA included: (i) Two sequential isolates obtained in 2004 (CFA_2004/01) and 2007 (CFA_2007/01). These isolates were characterized as MRS-mutators because they harbored missense mutations in the mutS and mutL genes and showed increased mutation rate (Table 1 and Text S1). (ii) A collection of 90 P. aeruginosa isolates obtained from a single sputum sample in 2010 (CFA_2010).

The collection from CFD included: (i) One normo-mutable isolate obtained in 1991 (CFD_1991/01). (ii) Two sequential MRS-deficient mutators from 1995 (CFD_1995/01) and 2002 (CFD_2002/01) (Table 1 and Text S1) that harbored the same mutS missense mutation [33]. (iii) A collection of 90 P. aeruginosa isolates obtained from a single sputum sample in 2011 (CFD_2011).
The CFA and CFD collections covered periods of 6 and 20 yrs in the patients' lives, respectively. Based on previous studies indicating a doubling time of 115 min for P. aeruginosa in sputum [44], we estimated that ∼36,500 and ∼91,400 duplication events occurred between the first and last isolates collected from CFA and CFD, respectively. Genotypic characterization of the two collections by various molecular methods (Materials and Methods) showed that each patient was chronically infected by a single, unrelated P. aeruginosa clone that persisted throughout the study period. The hexadecimal codes [45] for the SNP patterns of the CFA and CFD isolates analyzed are 2C32 and 249A, respectively.
MRS-deficient mutators are prevalent among CFA and CFD intra-patient P. aeruginosa populations
The proportion of MRS-deficient mutators present in each patient was determined based on the rifampicin mutation frequency of the 90 P. aeruginosa isolates of the CFA_2010 and CFD_2011 panels. All the CFA_2010 isolates showed mutation frequencies (1.7×10−5–7.6×10−6) consistent with a strong mutator phenotype. Similarly, in the CFD_2011 panel, strong mutator isolates (1.4×10−5–8.3×10−6) comprised ∼94% of the population. The remaining 6% showed mutation frequencies close to those observed in the prototypic wild-type normo-mutable strain PAO1 (3×10−8–1×10−7). These findings were supported by the observed prevalence of mutators in 90 isolates obtained ≥6 months later from new sputum samples (CFA_2011 and CFD_2012); 100% of CFA and 90% of CFD isolates displayed a strong mutator phenotype. To our knowledge, these are the highest proportions of mutators reported to date in large intra-patient populations of P. aeruginosa isolated from CF patients. A previous study, which analyzed sets of 40 isolates per sputum sample from 10 CF patients, reported proportions of 27% or less [8]. These findings indicate that P. aeruginosa can persist in chronic airway infections without the mutator phenotype, but in certain CF populations, such as those described here, MRS-deficient isolates may be prevalent and dominate the entire infecting population.
Genomic evolution of P. aeruginosa mutator isolates from CF chronic infections
To analyze the genomic evolution of CFA and CFD P. aeruginosa mutator lineages, we performed whole-genome sequencing of 13 CFA and 14 CFD isolates. From the CFA collection, we selected and sequenced the initial isolate CFA_2004/01, the intermediate CFA_2007/01, and 11 mutator isolates chosen randomly from the CFA_2010 population (CFA_2010/01, CFA_2010/11, CFA_2010/26, CFA_2010/31, CFA_2010/32, CFA_2010/40, CFA_2010/43, CFA_2010/72, CFA_2010/78, CFA_2010/82, CFA_2010/87). From the CFD collection, we selected and sequenced the initial normo-mutable isolate CFD_1991/01, the intermediate mutators CFD_1995/01 and CFD_2002/01, and 11 isolates from the CFD_2011 population consisting of five normo-mutators (CFD_2011/04, CFD_2011/11, CFD_2011/45, CFD_2011/57, CFD_2011/95) and six randomly selected mutators (CFD_2011/27, CFD_2011/28, CFD_2011/33, CFD_2011/34, CFD_2011/83, CFD_2011/94). The reads of CFA_2004/01 and CFD_1991/01 were assembled de novo, yielding genomes of 6,294,248 bp and 6,313,855 bp, respectively (Table S1), which were used as references in subsequent analyses. The sequences of the remaining CFA and CFD isolates were aligned against the corresponding references to assess the genetic changes accumulated in the two mutator lineages during the infection process (Table S2).
Both lineages accumulated a high number of mutations during their evolution in CF airways in comparison with previously reported normo-mutable CF isolates such as the DK2 and PA14 clones [18], [40]. The CFA collection had a total of 2,578 single-nucleotide polymorphisms (SNPs) and 544 1 - to 10-bp insertion/deletion mutations (microindels). The CFD collection had a total of 5,710 SNPs and 1,078 microindels (Table S3). We applied Bayesian statistical analysis to infer time-measured phylogenies [46], resulting in estimated mutation rates of 106 SNPs/yr (4.2×10−9 SNPs/bp per generation) for CFA and 89 SNPs/yr (3.2×10−9 SNPs/bp per generation) for CFD. These findings indicate a mutation rate ∼40-fold higher than that (2.6 SNPs/yr) reported previously for normo-mutable isolates obtained from CF chronic infections [18].
Genetic diversity generation in P. aeruginosa intra-patient mutator populations
SNPs were used as phylogenetic markers to perform a maximum-parsimony reconstruction of the evolutionary relationship of CFA and CFD isolate groups, and to evaluate temporal changes in their population genetic structure (Figure 2). Alleles of P. aeruginosa reference strain PAO1 were used to root the trees. In both cases, essentially all SNPs (>99.5%) supported single phylogenetic trees. Interestingly, high genetic diversity was observed in both CFA and CFD P. aeruginosa intra-patient populations. CFA_2010 and CFD_2011 contemporary clones were grouped together into three and four distinguishable clusters, respectively: Clusters II, III, and IV for CFA (Figure 2A) and Clusters I, IV, V, and VI for CFD (Figure 2B) trees. Clusters were composed of genetically similar isolates, whereas more extensive genetic dissimilarities were observed between clusters. The branch lengths among clusters differed substantially, indicating uneven mutational loads in coexisting P. aeruginosa intra-patient populations.

All CFD normo-mutable isolates were grouped together in Cluster IV (Figure 2B). In spite of the normo-mutable phenotype, this cluster shared common branches with the intermediate mutator isolates CFD_1995/01 and CFD_2002/01 and with their contemporary mutator clones (branches D, F, and H) (Figure 2B). These findings suggest that the normo-mutators arose from a mutator population of branch J at some point. Cluster IV showed the highest accumulation of mutations during the infection process, despite the low mutation frequencies of its members (Figure 2B and Table S3).
The estimated time points of the most recent common ancestor (MRCA) of the CFA and CFD populations were 2002 and 1988, respectively. Interestingly, each of these estimates coincided with the year at which the patient was diagnosed as chronically infected (Figure 1). This finding indicates that the divergent sub-lineages coexisted for many years – the same as the colonization period of the patient (Figure 2).
The question arose whether the genetic diversity observed in the two CF intra-patient populations was associated with differing repertoires of mutated genes among contemporary isolates, or whether most mutations occurred in common genes among the clones. To distinguish between these possibilities, we first determined the set of genes of each CFA and CFD isolate that were altered by nonsynonymous SNPs and microindels. Minimum Spanning Trees (MSTs) were then constructed to illustrate the relationships among contemporary isolates and their corresponding ancestors based on the number of distinctive mutated genes. The CFA_2010 intra-patient P. aeruginosa mutator population (Figure 3A) was distributed in three main clusters, whereas the CFD_2011 population (Figure 3B) showed four clusters with all normo-mutable isolates grouped together. The structures of the two MSTs indicate high genetic diversification and a scenario in which even contemporary CFA and CFD mutator populations diversified through mostly different evolutionary pathways.

Pathoadaptive genes and evidence for parallel evolution in mutator populations
We classified the SNPs according to their distribution in coding and noncoding regions and their effect in translation, to assess the selective forces acting on the CFA and CFD P. aeruginosa lineages. Most of the SNPs in CFA and CFD were found to occur within coding regions, and 58.0% and 60.2% (respectively) corresponded to missense mutations (Figure S1). The rates of nonsynonymous to synonymous mutations (dN/dS) were 0.68 for CFA and 0.79 for CFD lineages. Thus, most of the SNPs that became fixed in the P. aeruginosa mutator lineages were neutral mutations, with most of each genome showing a signature of purifying selection and/or genetic drift (dN/dS<1; P = 5.2×10−56 and P = 1.5×10−47, respectively).
However, it is conceivable that positively selected genes remain “hidden” among the much larger number of genes that have accumulated mutations by genetic drift. Evolving populations of P. aeruginosa presumably accumulate adaptive mutations in response to the human host environment in which they propagate. We would therefore expect to observe parallelism in the adaptive genetic routes of the different lineages. To confirm such convergent evolution, we attempted to identify genes that underwent parallel mutation in the 10 sub-lineages (four CFA sub-lineages and six CFD sub-lineages) that coexisted over many years (Figure 2). We analyzed our dataset by selecting those genes that were independently mutated in at least half of the parallel evolving sub-lineages (see Materials and Methods). Forty genes were found to be frequently mutated across the sub-lineages (Table 2), suggesting that the parallel mutation of these genes was due to positive selection for mutations. Consistently, the signature for selection for SNPs accumulated in the 40 genes (dN/dS = 0.97) was significantly higher than the ratio obtained for SNPs affecting all other genes (dN/dS = 0.75; P = 0.029 by Fisher's exact test), suggesting that these mutations were positively selected during evolution. Analysis of the 40 genes was further focused on those that were non-synonymously mutated in at least half of the 10 sub-lineages (Figure 4). Several of these genes were associated with functions related to CF host adaptation. In particular, ftsI, ampC, fusA1, mexY, PA1874, and PA0788 are involved in resistance to antibiotics commonly used in CF therapies, i.e., betalactams, aminoglycoside, quinolones, chloramphenicol, trimethoprim, and imipenem [29], [47], [48], [49], [50], [51], [52], [53]. Even though the GacA/GacS system is required for activation of genes involved in chronic persistence, gacS mutants are prone to generate stable and stress-tolerant small colony variants (SCVs) when growing in biofilms, exposed to stress factors [54], [55], or in vivo [56], suggesting that the absence of GacS may confer some additional advantage for persistence in the CF lung. fusA1 encodes the elongation factor EF-G1A, which confers resistance to the antibiotic argyrin in P. aeruginosa [57], [58]. Chung et al. recently reported independent fusA1 mutations in two CF patients and suggested that these mutations are involved in regulation of virulence through a ppGpp-dependent stringent response [43]. On the other hand, the gene pslA is involved in biofilm formation [59], and cupC3 is associated with motility/attachment [60]. Lack of motility is a trait frequently observed in isolates from chronically colonized patients, and may give P. aeruginosa a survival advantage in chronic CF infection by enabling it to resist phagocytosis and conserve energy [61].

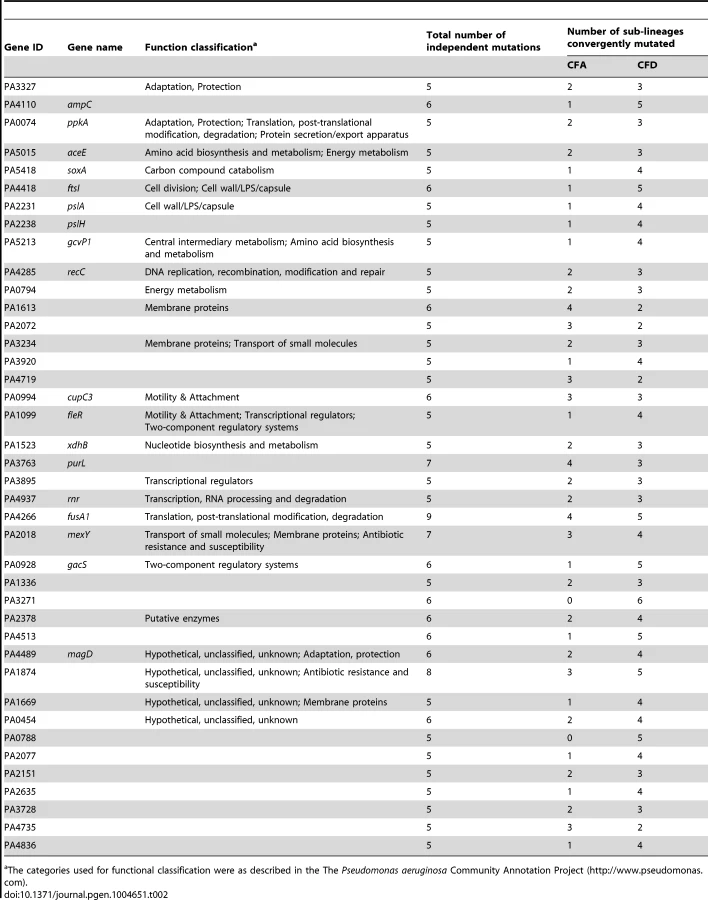
Alterations in several genes related to bacterial catabolism (e.g., aceE, gcvP1, soxA, xdhB, PA0794) were also observed, suggesting that the inactivation of certain metabolic functions may be a common trait related to CF host adaptation (see below).
The concurrent alteration of specific genes or functions related to adaptation to the CF airway environment provides strong evidence for parallel evolution not only across CFA and CFD lineages, but also across intra-patient coexisting sub-lineages. Certain genes were convergently but exclusively mutated among CFD sub-lineages, e.g., ampC (beta-lactamase precursor), PA0788 (penicillin binding protein), and PA3271 (two-component sensor). These findings suggest the occurrence of in-host parallel evolutionary processes resulting from specific selective pressures from differential antibiotic treatments.
Mutations in the global regulators mucA, algT, rpoN, and lasR are related primarily to adaptation to the CF airway environment [10], [17], [33], [62], [63], [64]. However, our analyses did not reveal such mutations because they arose in the ancestral isolates before diversification into sub-lineages (Table S4). The entire population from CFA had a mutation in mucA, whereas the population from CFD had mutations in lasR and rpoN (Table S4). These findings indicate that mutations in these regulator genes were specifically fixed in the respective bacterial populations during early in-host evolution.
Accumulation of mutations in MRS genes and the emergence of a subpopulation with reduced mutation rate
MRS genes in the CFA panel
To investigate the molecular basis for the strong mutator phenotypes observed in CFA_2010 and CFD_2011 clones, we analyzed the sequences of the MRS genes mutS and mutL from the whole-genome sequence data and looked for potential alterations.
Mutations in both MutS (T247P) and MutL (H469R) proteins were found in the ancestral isolate CFA_2004/01 (Table 1). This same combination of mutations (termed allelic combination SL1) was also present in the later isolate CFA_2007/01 and in seven of the 11 CFA_2010 clones. The remaining four CFA_2010 isolates showed two additional missense mutations in MutS (L794P and W160R) and one in MutL (L389S). These two new MRS allelic combinations were termed SL2 and SL3, respectively (Table 1). SL1, SL2, and SL3 were distributed in different phylogenetic clusters (Clusters I, IVa, and IVb for SL1, Cluster II for SL2, Cluster III for SL3) (Table 1 and Figure 2A), demonstrating the coexistence of multiple polymorphisms in the MRS genes as a reflection of the observed genomic diversity.
We further sequenced the mutS and mutL genes in 19 additional CFA_2010 isolates to extend our analysis to 30% of the population (30 isolates). Fourteen of these newly sequenced isolates carried the ancestral SL1 combination, five carried SL3, and none carried SL2, leading to final prevalences of 70% for SL1, 3% for SL2, and 27% for SL3. These observations suggest that SL1 was predominant and persisted over time among the within-patient coexisting isolates.
The logical next question was whether the different mutations observed in mutS and mutL in CFA_2010 isolates were able to alter the function of their respective gene products, resulting in the mutator phenotype. To address this point, we attempted to restore a wild-type normo-mutable phenotype from SL1, SL2, or SL3 mutator strains by complementation with a plasmid carrying the wild-type mutS or mutL gene (Materials and Methods). Our results indicated that the ancestral mutation in MutL (H469R), but not those in MutS, was responsible for the mutator phenotype observed in isolates that carried not only the SL1 combination but also SL2 and SL3 (Text S1). Although mutS has been shown to be the main target for acquisition of a stable hypermutor phenotype [15], [65], mutations in mutL are a frequent cause of MRS deficiency in P. aeruginosa CF isolates [17], [66].
MRS genes in the CFD panel
Analysis of the CFD mutator collection revealed that every CFD_2011 isolate, whether mutator or normo-mutator, harbored the same ancestral frameshift mutation in mutS (−CG1551) (Table 1). The normo-mutable isolates (grouped in Cluster IV) also harbored a +CC334 insertion (Table 1) in a 5-bp G∶C simple sequence repeat (SSR) of mutS. This insertion generates a premature stop codon at position 347 bp from the ATG start codon. In addition, the +CC334 insertion produced an ATG codon at 1822 bp, resulting in restoration of the frameshift now coding for a C-terminal peptide of 248 amino acids, which correspond to MutS functionally essential domains [67], [68] (File S1). This finding suggested that the +CC334 insertion is associated with the reduced mutation frequency in normo-mutators. To test this concept, we cloned the mutS+CC334−CG1551 allele into the pMC5 plasmid (Table S5) and performed complementation assays in mutS-deficient strain PAO1 (Materials and Methods). Nonetheless, the above allele did not revert the mutator phenotype, and we found no extra copies of a functional mutS allele in the normo-mutable genomes.
We next examined the mutational spectra of CFD_2011 isolates in Cluster IV. The proportion of transversions showed a significant increase (P≤0.05 in two-tailed T-test adjusted by Bonferroni correction), from 5.8% in contemporary mutators belonging to Cluster V and VI (with the ancestral mutS−CG1551 allele) to 10.6% in normo-mutators grouped in Cluster IV (with the mutS+CC334−CG1551 allele) (Figure S2).
We sequenced the mutS gene in 19 additional CFD_2011 mutator isolates (similarly to the studies of the CFA population) and found that all of them (100%) carried the −CG1551 mutation in mutS, indicating that the proportion of clones with reduced mutation frequency remains low in the CFD population (∼6%).
Mutational snapshot of SSRs in P. aeruginosa mutator populations
Our previous in vitro studies showed that, in a MRS-deficient background, G∶C SSRs constitute hotspots capable of biasing mutagenesis toward a specific genetic pathway [34], [38]. Our recent studies of P. aeruginosa PACS2 and epidemic DK2 strains demonstrated the same phenomenon at a genome-wide level in CF in vivo chronic airway infection [18], [39].
The present study design allows examination of such a genome-wide effect of biased mutagenesis in large populations obtained at single time points, and observation of SSR instability in coexisting isolates.
Analysis of types of mutations occurring in both the CFA_2010 and CFD_2011 isolates revealed a mutational spectrum typical of MRS-deficient strains. Transitions (∼80%) and small indels (1–4 bp) (∼16%) were the most frequently observed mutations in both collections (Figure 5C). The most prevalent transition was G∶C→A∶T, accounting for 65% and 62% of total transitions in CFA and CFD lineages, respectively. Of the 1–4 bp indels,>80% were located within a homopolymeric SSR, and ∼75% were located specifically in G∶C SSRs (Figure 5A). In contrast, indels in A∶T SSRs accounted in average for 5.2% and 6.7% of the 1–4 bp indels in CFA and CFD isolates, respectively (Figure 5A).

Based on this strong skewing of MRS spectra toward small indels in G∶C SSRs, we selected homopolymeric G∶C SSRs of ≥6 bp, which were mutated in at least half of the coexisting isolates in both CFA and CFD lineages, and analyzed their mutational dynamics at the intra-population level. Eleven of these highly mutated G∶C SSRs harbored 2–5 distinct indel mutations accounting for independent mutational events (Figure 5B). A single SSR was observed to be either unaltered or modified by different indel mutations even in coexisting isolates from the same cluster. Analysis of these 11 G∶C SSRs in the normo-mutable CFD_1991/01 isolate showed no mutations. Using genome data available online, we evaluated the occurrence of indel mutations in these G∶C SSRs in 12 normo-mutable P. aeruginosa strains (PAO1, PA14, M18, NCGM2.S1, B136-33, RP73, 39016, PACSC2, 2192, C3719, DK2, LESB58; the latter five are normo-mutable isolates obtained from CF infections), whose genomes have been sequenced and are available in the Pseudomonas Genome Database (www.pseudomonas.com) [69]. Our survey revealed that these 12 strains harbored no indel mutations in the analyzed G∶C SSRs, even though large G∶C SSRs are considered to be “hotspots” for mutagenesis.
One of the identified homopolymers is located in a gene (PA4071/PADK2_03970) which has been previously suggested to be preferentially mutated in mutators and to represent a mutator-specific target of adaptive mutations [18].
These findings suggest a scenario in which MRS-deficient populations generates a vast of genetic diversity due to G∶C SSR instability. In this scenario, genes containing large G∶C SSRs constitute continual sources of genetic diversification primarily in mutator bacterial populations.
Global analysis of the catabolic capacities of CFA and CFD mutator lineages during long-term evolution in CF infections
We evaluated the dynamics of phenotypic changes in the 27 P. aeruginosa CFA and CFD isolates by determining global catabolic activities (the “catabolome”). Biolog phenotype microarrays were used to monitor the catabolic profiles of each isolate with various C and N sources (Table S7). The total catabolic functions in the isolates were greatly reduced (average reduction 73.5% and 63.8%, respectively) in comparison with those of the CFA_2004/01 and CFD_1991/01 ancestors (Figure 6). This extensive loss of functions led to homogeneous populations in both the CFA and CFD lineages, with slight catabolome variation among clones. Catabolic function reduction thus appears to be a phenotypic pattern shared by CFA and CFD mutator lineages. Accordingly, genes related to catabolism were convergently mutated in both the CFA and CFD lineages (Figure 4). This phenomenon may be partially responsible for the decreased catabolic phenotype.

Discussion
This study provides a complete panorama of the genomic diversity that shapes the structure of P. aeruginosa mutator populations during long-term adaptation to the CF airway environment. We combined a longitudinal study with an extensive cross-sectional approach, including multiple isolates obtained from single sputum samples, which allowed in-depth analysis of population diversity (Figure 1). We utilized P. aeruginosa panel collections from two chronically infected CF patients, CFA (Argentinian) and CFD (Danish), with time spans of 6 yrs and 20 yrs, respectively, from initial to later stages of chronic infection. Our comprehensive study design included whole-genome sequencing and high-throughput phenotypic approaches, calculation of mutation frequencies, phylogenetic estimation of time points of sub-lineage diversification, and analysis of mutS and mutL genes to obtain a wide-ranging depiction of hypermutability in CF.
We expected (and confirmed) that each of the two patients was infected by a single non-epidemic P. aeruginosa clone that did not present, during the initial stages of infection, the pathoadaptive mutations displayed by epidemic clones [42], [70], [71], [72]. We were therefore confident that our analysis addressed specific and independent in-host evolutionary processes. Mutator strains were highly prevalent in both patients, essentially dominating the populations. The proportion of mutators was ≥90% in the single time point 90-isolate collections, indicating that mutators, once selected, dominated the CFA and CFD infecting populations. The observed prevalence of within-patient mutators was much higher than the values reported in previous studies [8], [13]. These findings indicate that although P. aeruginosa may persist throughout the course of chronic infection without ever acquiring the mutator phenotype, mutator strains may become prevalent and even dominate the whole population under certain yet-unknown conditions. This concept is supported by the observation that two patients of different ages from geographically distant locations, infected with different non-epidemic P. aeruginosa clones and subjected to different therapeutic protocols, underwent overlapping evolutionary trajectories that led to complete domination of mutators.
Recent reports have demonstrated high diversity at the phenotypic level among P. aeruginosa populations from CF lung infections [8], [9], [73]. However, there have been no genome-wide studies of such diversity in bacterial populations from the same clinical samples. The global picture of genetic structure of intra-patient mutator populations in the present study reveals significant genomic diversity driven by high accumulation of mutations (Figures 2 and 3), reflected by the typical MRS spectra (Figure 5). The distribution and combination of thousands of mutations result in a unique genotype for every isolate, allowing long-term persistence in the CF airway environment. The observed genomic variation into the CFA and CFD lineages indicates that the population structure in each case was not determined by homogeneous single dominating clones, but occurred through multiple evolutionary genetic pathways that adapted equally to the CF airway environment and allowed the coexistence of diverse subpopulations for many years. We determined that this high genomic diversity, to an equal degree in the two patients, spread out from the establishment of chronic infection. Interestingly, MRS genes were also characterized by the coexistence of multiple polymorphisms. However, underlying the polymorphisms within the MRS genes, there is an ancestral mutation that was fixed in each CFA and CFD population and is apparently responsible for hypermutability. These findings suggest the existence of common selective forces acting on MRS inactivation in the two patients.
The long-term evolution of the P. aeruginosa CFA and CFD lineages was signed mainly by purifying selection and/or genetic drift. There are conflicting reports regarding whether genomic evolution of CF isolates shows signatures of positive selection [10] or (in contrast) genetic drift and/or purifying selection [32]. Our results strongly support the latter concept; i.e., that genomic signatures of purifying selection and/or genetic drift are not inherent consequences of mutators, but are characteristic of the genetic adaptation processes underlying P. aeruginosa persistence in chronic lung infections.
According to our observations, the large number of mutations were for the most part distributed randomly among the P. aeruginosa mutator genome (Figure 3). However, we also identified a group of genes that were convergently mutated in multiple genomes by independent mutational events (Table 2 and Figure 4). Most of these genes code for functions related to pathogenicity (e.g., antibiotic resistance, virulence, motility, attachment), suggesting that they were positively selected as beneficial mutations. We note that five of the genes (ampC, ftsI, fusA1, PA3271, PA2018) were also found to be mutated in isolates obtained from the epidemic DK2 clone [18], providing evidence of parallel evolution for certain specific traits among different P. aeruginosa lineages.
As we have reported recently for the PACS2 [39] and epidemic DK2 P. aeruginosa strains [18], the impact of hypermutability on the evolution of the CFA and CFD lineages is reflected by the high tendency of G∶C SSR-containing sequences to be mutated (Figure 5A). This finding confirms that genes which maintain G∶C SSRs in their coding region and/or in neighboring regulatory sequences are highly unstable in an MRS-deficient background and may be mutator-specific targets of adaptive mutations. This concept is extended here by the demonstration that large G∶C SSRs, as DNA sequences per se, are highly polymorphic in single time point populations, indicating that they are continual sources for diversification (Figure 5B). This SSR-driven diversity is not observed in genomes of other P. aeruginosa strains, even of normo-mutable CF clones. Our present and previous results [18], [34], [38], [39], taken together, demonstrate a clear association between MRS-deficiency and G∶C SSR instability, which exerts a global effect along the entire genome. The impact of hypermutability during evolution of P. aeruginosa in the CF airway environment is not simply a major, rapid acquisition of mutations in quantitative terms. In contrast with SNPs, indels that are not multiples of three produce frameshifts in the coding sequence of genes and thereby affect gene function. Indels in G∶C SSRs may play an important role in the evolutionary process and in relation to mutator competitiveness. Along this line, nine of the 11 analyzed G∶C SSRs (Figure 5B) were located in coding regions of the genome. Five of these genes are predicted to encode for hypothetical proteins with no assigned function. On the other hand, some G∶C SSRs were positioned in genes functionally related to transcriptional regulators (PA1490), adaptation-protection (PA1127), membrane proteins, and transport of small molecules (PA1626, PA2203).
A small percentage (6%) of the CFD population has a reduced mutation frequency similar to that of normo-mutable strains. This subpopulation, which is grouped in a single cluster (Cluster IV in Figure 2), had the highest accumulation of mutations observed in the whole CFD collection (Table S3). This observation posed the question whether the mutational load of these clones is too heavy to continue supporting a mutator phenotype. However, the normo-mutable subpopulation carried the same mutS loss-of-function mutation (Table 1) as the mutator isolates. Phylogenetic analysis indicated that isolates from Cluster IV had arisen from a mutator subpopulation at some undetermined point in branch J (Figure 2). Our sequencing data suggested that the most feasible explanation is the emergence of secondary mutations, in genes not belonging to the MRS, that compensate for mutS hypermutability, since neither reversion of the original −CG1551 nor duplication of the mutS gene was observed in normo-mutable genomes. Although the new +CC334 mutation restored in part the mutS reading frame, this fact did not account for the reduction in mutation frequency observed in these clones. In a previous study, E. coli MRS-deficient populations in vitro evolved a compensation of the mutator phenotype based on secondary mutations in genes related to oxidative stress response which, although selected for different increasing-fitness traits, resulted in a reduction of the mutation rate [74]. Future studies will elucidate the mechanisms underlying the observed reduction in mutation frequency of these mutS-deficient P. aeruginosa strains.
How are these mutator populations maintained over extended periods of time, and even able to accumulate greater numbers of mutations? The chronically infected CF lung is a uniquely challenging habitat in which P. aeruginosa must cope with aggressive immune system responses, intense antibiotic therapies, and/or competition with other resident microorganisms. This environment presents stressful and variable conditions, and multiple mutations may occur that increase fitness. On the other hand, favorable nutritional conditions of CF airways [75], [76] may sustain the inactivation of certain functions that are no longer necessary for survival, particularly in this environment. In this regard, CF isolates have been reported to accumulate a high number of auxotrophies – higher than in other studied environments [77]. The results obtained here for CFA and CFD catabolomes indicate overall reduction of total catabolic activities (Figure 6). Reduction of catabolic functions appears to be part of a more general adaptive process of P. aeruginosa residing for long periods in the CF lung, because such reduction has also been observed in normo-mutable strains [42]. The large genome of P. aeruginosa may have the potential to undergo reductive evolution, with elimination of functions that are redundant and/or dispensable in the CF host environment, thereby buffering the heavy mutational load observed [78].
Based on these considerations, we hypothesize that, during long-term evolution of P. aeruginosa in CF airways, the increased availability of certain beneficial mutations, in combination with a whole-genomic signature of neutral evolution, provided favorable conditions to increase the percentage of mutators, until reaching a frequency at which mutators dominated both the CFA and CFD populations. P. aeruginosa adaptation to the CF lung is presumably manifested through selection of multiple genetic combinations [79], and hypermutability is consequently maintained over time as a constant source of genetic variation.
Materials and Methods
P. aeruginosa isolate collections
Clinical P. aeruginosa isolates were obtained from sputum samples from two CF patients at the Hospital de Niños Santísima Trinidad (Córdoba, Argentina) (patient CFA) and the Copenhagen CF Centre at Rigshospitalet (Copenhagen, Denmark) (patient CFD). Patient age at the time of the first isolate collection was 8 and 23 yrs, respectively. The onset of chronic infection with P. aeruginosa was 2001 and 1986, respectively. Chronic pulmonary infection was defined as the persistence of P. aeruginosa in sputum for 6 consecutive months, or less if the persistence was combined with presence of two or more precipitating antibodies against P. aeruginosa [33]. The criteria for choosing these patients were: (i) chronic airway infection by P. aeruginosa; (ii) absence of transmissible P. aeruginosa clones; (iii) presence of single clonal P. aeruginosa infecting lineages throughout the course of infection; (iv) appearance of MRS-deficient P. aeruginosa mutators during the course of infection.
Sputum samples were collected by expectoration during routine hospital visits, stored on ice, and processed immediately. Sputa were liquefied by addition of an equal volume of Sputolysin (Calbiochem), diluted, and plated onto Pseudomonas isolation agar (BD Biosciences), which promotes growth of P. aeruginosa and other Pseudomonas species. For cross-sectional analysis, 90 isolates were taken randomly from a single sputum sample. Isolates were maintained and stored at −70°C in glycerol stock solution. For assays, isolates were subcultured from the frozen stocks onto Luria Bertani (LB) agar plates, and inocula were prepared from overnight cultures in LB broth at 37°C with appropriate aeration.
Genotyping analysis
Genotyping analysis of the P. aeruginosa isolates was performed by Randomly Amplified Polymorphic DNA (RAPD), using primer 272 (Table S5) [5], [80]. Banding patterns from electrophoretic gels were analyzed by GelPro Analyzer V. 6.3 software. Epidemiological relatedness was evaluated by pulsed-field gel electrophoresis (PFGE) using SpeI enzyme as described previously [81]. DNA macrorestriction patterns were interpreted according to the criteria of Tenover et al. [82]; i.e., isolates with PFGE patterns differing by (i) 1–3 bands are closely related clones; (ii) 4–6 bands are possibly related clones; (iii) ≥7 bands belong to different strains. Clonal relatedness among the isolates selected for genome sequencing analysis was evaluated by single-nucleotide polymorphism (SNP) typing using AT biochips (Clondiag Chip Technologies, Germany) [83]. Strain assignment was performed by visual array analysis using a hexadecimal code as described previously [45].
Mutation frequency measurement
Mutation frequencies were estimated as described previously [13], [15]. In brief, five independent colonies of each isolate were grown overnight in 10 ml LB medium at 37°C with appropriate aeration. Cultures were collected by centrifugation at 3000 rpm for 10 min and resuspended in 1 ml LB medium. Serial ten-fold dilutions were plated on LB agar with and without 300 µg/ml rifampicin. After 36 h incubation (48 h for slow-growing strains) at 37°C, colonies were counted and the mean percentage of mutants was estimated. Strains were considered hypermutable if the mutation frequency was at least 20-fold higher than that of control strain PAO1 [13]. Frequencies were determined from two independent experiments.
Genome sequencing
Genomic libraries were prepared as described previously [42] and sequenced on an Illumina Hiseq2000 platform, generating 100 base paired-end reads using a multiplexed protocol. A total of 224 million paired-end reads were generated to yield average genomic coverage of 33–207 fold (all average coverage depths were ≥82× except for CFD287_2011/94 (33×) and CFD_2011/27 (52×)). Genome sequence reads were deposited in the European Nucleotide Archive (ENA/SRA ERP002379).
Illumina reads from isolates CFA_2004/01 and CFD_1991/01 were de novo assembled using Velvet software V. 1.2.07 [84] using the following settings: -ins_length 238 and 244 for CFA_2004/01 and CFD_1991/01, respectively; -ins_length_sd 54.4 and 50.3 for CFA_2004/01 and CFD_1991/01, respectively; -exp_cov 291.1 and 296.7 for CFA_2004/01 and CFD_1991/01, respectively; -cov_cutoff 10; -min_contig_lgth 500. Selected kmer sizes were 51 and 39 for CFA_2004/01 and CFD_1991/01, respectively. Each de novo assembly was used as a reference genome sequence to map reads from the remaining CFA and CFD genome sequences using Novoalign V. 2.08.02 (Novocraft Technologies) [85]. Pileups of the mapped reads were processed by SAMtools V. 0.1.7 [86]. SNPs were identified by the varFilter algorithm in SAMtools (samtools.pl varFilter -d 3 -D 10000), and only unambiguous SNPs with quality scores (Phred-scaled probability of sample reads used as homozygous reference) of ≥50 (i.e., P≤10−5) were retained. Read alignments surrounding all putative indels were realigned using GATK V. 1.0.5083 [87], and microindels were extracted from the read pileup using the following criteria: (i) quality score ≥500; (ii) root-mean-square (RMS) mapping quality ≥25; (iii) support by ≥20% of the covering reads. The false-negative rates obtained were 2% and 3% by in silico introduction of random base-substitutions and microindels (lengths 1–10 bp), respectively. All sites with putative polymorphisms in the pileup of reads from the reference were excluded to avoid false-positives resulting from errors in the reference assembly or general mapping errors. MUMmer3 [88] was used for whole-genome alignments. Mutations were described according to their relative gene positions in orthologs of P. aeruginosa reference strains PAO1, PA14, and LESB58 with completed genome sequences [69]. If no ortholog was found in the reference strains, the mutation was described by the GenBank ID of the homolog sequence (Genbank_ID:ORF_name: Position) or as “Not annotated”.
Phylogenetic trees based on the SNP mutations identified in each alignment were constructed using maximum-parsimony analysis as described previously [42].
For calculation of selection coefficients (dN/dS ratio), we assumed that codon usages were identical to those in strain PAO1, in which 25% of random mutations are synonymous [42], and that the probability of the observed number of nonsynonymous SNPs, given the expected number of SNPs, is calculated from the Poisson distribution.
Bayesian evolutionary analysis
Bayesian analysis of evolutionary rates was performed using BEAST V. 1.7.2 [46]. The BEAST program was run with the following user-determined settings: a lognormal relaxed molecular clock model, which allows rates of evolution to vary among the branches of the tree, and a HKY substitution model, which distinguishes between the rate of transitions and transversions and allows unequal base frequencies. Mutation rates were calculated from chains of 100 million steps, sampled every 5,000 steps. The first 10 million steps of each chain were discarded as a burn-in.
Identification of genes subject to convergent evolution
To identify genes subject to convergent evolution, we picked out those that were mutated in at least half of the in parallel evolving sub-lineages CFA I–IV and CFD I–VI. The sub-lineages/clusters were defined as follows (see Figure 2). CFA lineage: Cluster I: branches Z, Y; Cluster II: branch A; Cluster III: branches C, D, E, F, G; Cluster IV: branches N, J, T, U, R, S, O, Q, K, I, M. CFD lineage: Cluster I: branch A2; Cluster II: branch C; Cluster III: branch E; Cluster IV: branches J, I, L, M, N, K, P, O; Cluster V: branches U, T, W, V, X; Cluster VI: branches R, G, Q. For example, gene aceE was hit by independent mutations in CFA branch J (sub-lineage CFA-IV), CFA branch Y (sub-lineage CFA-I), CFD branch C (sub-lineage CFD-II), CFD branch E (sub-lineage CFD-III), and CFD branch T (sub-lineage CFD-V). To rule out the possibility that high accumulation of mutations resulted from particularly large gene sizes, mutations from large genes (≥10 kb) were excluded from the analysis.
Minimum spanning trees (MSTs)
MSTs for the set of mutated genes within each CFA and CFD lineage were obtained using Prim's algorithm (Prim, 1957) as implemented in the Info-Gen program [89]. In this model, nodes represent an isolate's set of mutated genes, the distance between a pair of nodes is shown as the number of distinctive mutated genes, and nodes are connected in such a way that the sum of the distances is minimized (Table S6).The network connects each genotype to all other genotypes through a pathway of mutated genes. The CFA_2004/01 and CFD_1991/01 genomes were used as starting points of the network of the corresponding MSTs for each lineage. The number of mutated genes along the network was used as a measure of divergence between two given genotypes.
Sequence analysis of the mutS and mutL genes
Genomic DNA of P. aeruginosa CF isolates was extracted using a DNA Isolation Kit (Qiagen). Primers used for PCR amplification and DNA sequencing are listed in Table S5. PCR amplifications were performed with the following conditions: 8 min at 95°C, 33 cycles of 1 min at 94°C, 1 min 20 sec at 60°C, 2 min at 72°C, and a final extension of 10 min at 72°C. PCR products were cleaned with a Gel Purification Kit (Qiagen), and both strands were sequenced directly using the same PCR primers (DNA Sequencing Facility, Univ. of Chicago, IL, USA). To score mutations within the gene, sequencing results were compared with the corresponding gene sequence of strain PAO1 (www.pseudomonas.com) using the BLAST program of the NCBI database (www.ncbi.nlm. nih.gov/blast/).
Construction of plasmids pMC5-MutS−CG1551 and pMC5-MutS+CC344−CG1551
Two constructs of plasmid pMC5-MutS [90], which contains the full coding region of the mutS gene from strain PAO1, were generated by introducing −CG1551 and +CC334−CG1551 mutations to produce plasmids pMC5-MutS−CG1551 and pMC5-MutS+CC344−CG1551, respectively. To introduce these mutations, the mutS genes from CFD_2011/27 and CFD_2011/11 isolates were amplified by PCR using oligonucleotides mutS-for1 and mutS-rev4 (Table S5), ligated to the pGEM-T Easy vector (Promega), and cloned in the NdeI/EcoRI restriction sites of pMC5-mutS, in which the mutS gene has been digested with NdeI/EcoRI. Both plasmids were propagated on E. coli DH5α (Invitrogen) transformed by heat shock by standard procedures.
Complementation assays with pMC5-MutS, pMC5-MutL, pMC5-MutS−CG1551, and pMC5-MutS+CC344−CG1551 plasmids
To evaluate the genetic basis of the mutator phenotype, plasmids pMC5-mutS [90] and pMC5-mutL [91] were successively transferred into the mutator CFA and CFD isolates. To analyze +CC344−CG1551 mutations in the mutS gene, plasmids pMCS-MutS−CG1551 and pMCS-MutS+CC344−CG1551 were independently transferred into reference strain MPAO1MS. All plasmids were transferred by electroporation as described by Choi and Schweizer [92], and transformed strains were selected on LB agar plates supplemented with 100 mg/ml gentamicin. Complementation was checked by the rifampin test described above. For each strain, complementation was confirmed for three independent transformed colonies.
Catabolic analysis by Biolog Phenotype MicroArrays
Phenotype MicroArrays PM1 and PM3B (Biolog; Hayward, CA, USA) were performed according to the manufacturer's instructions [93], [94]. Export of Omnilog data was performed using Omnilog OL-FM/Kin software V. 1.20.02 (Biolog). Phenotypes were determined based on the parameter “ave area” (the area beneath the respiration curve of reduced tetrazolium vs. time). For comparison of clinical isolates, data were exported after 72 h incubation. Data analysis and statistical analysis were performed using R Project V. 2.10.0 (http://www.R-project.org). The R packages used for analysis were “bioDist” (B. Ding, R. Gentleman, V. Carey). Total catabolic function was calculated as described previously [95].
Statistical analyses
Statistical analyses were performed using a two-tailed T-test adjusted by Bonferroni correction. Differences with P≤0.05 were considered statistically significant.
Ethics statements
The P. aeruginosa isolates were obtained from sputum samples from one CF patient at the Hospital de Niños Santísima Trinidad (Córdoba, Argentina) (patient CFA) and one CF patient at the Copenhagen CF Centre at Rigshospitalet (Copenhagen, Denmark) (patient CFD), as byproducts of the routine established for bacterial typing and antimicrobial susceptibility testing. I.e., sputum sampling was not performed for the purposes or intent of the present study; isolates recovered from these sputa were simply derivatives of routine CF patient therapeutic controls. The therapeutic treatments of the two patients were not modified in any way as a consequence of the results obtained in this study. The research protocols followed in this study were approved and reviewed by the Ethics Committee of the Hospital de Niños Santísima Trinidad, Córdoba, Argentina and the local ethics committee, Region Hovedstaden, Copenhagen, Denmark (H-A-141 and H-1-2013-032). Both of the patients gave informed consent.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. BodeyGP, BolivarR, FainsteinV, JadejaL (1983) Infections caused by Pseudomonas aeruginosa. Rev Infect Dis 5 : 279–313.
2. MorrisonAJJr, WenzelRP (1984) Epidemiology of infections due to Pseudomonas aeruginosa. Rev Infect Dis 6 Suppl 3: S627–642.
3. GovanJR, DereticV (1996) Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol Rev 60 : 539–574.
4. LyczakJB, CannonCL, PierGB (2002) Lung infections associated with cystic fibrosis. Clin Microbiol Rev 15 : 194–222.
5. MahenthiralingamE, CampbellME, FosterJ, LamJS, SpeertDP (1996) Random amplified polymorphic DNA typing of Pseudomonas aeruginosa isolates recovered from patients with cystic fibrosis. J Clin Microbiol 34 : 1129–1135.
6. RömlingU, WingenderJ, MullerH, TümmlerB (1994) A major Pseudomonas aeruginosa clone common to patients and aquatic habitats. Appl Environ Microbiol 60 : 1734–1738.
7. MartinC, IchouMA, MassicotP, GoudeauA, QuentinR (1995) Genetic diversity of Pseudomonas aeruginosa strains isolated from patients with cystic fibrosis revealed by restriction fragment length polymorphism of the rRNA gene region. J Clin Microbiol 33 : 1461–1466.
8. MowatE, PatersonS, FothergillJL, WrightEA, LedsonMJ, et al. (2011) Pseudomonas aeruginosa population diversity and turnover in cystic fibrosis chronic infections. Am J Respir Crit Care Med 183 : 1674–1679.
9. WorkentineML, SibleyCD, GlezersonB, PurighallaS, Norgaard-GronJC, et al. (2013) Phenotypic heterogeneity of Pseudomonas aeruginosa populations in a cystic fibrosis patient. PLoS One 8: e60225.
10. SmithEE, BuckleyDG, WuZ, SaenphimmachakC, HoffmanLR, et al. (2006) Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci U S A 103 : 8487–8492.
11. HuseH, KwonT, ZlosnikJ, SpeertD, MarcotteE, et al. (2010) Parallel Evolution in Pseudomonas aeruginosa over 39,000 Generations in vivo. MBio 1: e00199–00110.
12. WongA, KassenR (2011) Parallel evolution and local differentiation in quinolone resistance in Pseudomonas aeruginosa. Microbiology 157 : 937–944.
13. OliverA, CantónR, CampoP, BaqueroF, BlázquezJ (2000) High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288 : 1251–1254.
14. OliverA, MenaA (2010) Bacterial hypermutation in cystic fibrosis, not only for antibiotic resistance. Clin Microbiol Infect 16 : 798–808.
15. OliverA, BaqueroF, BlázquezJ (2002) The mismatch repair system (mutS, mutL and uvrD genes) in Pseudomonas aeruginosa: molecular characterization of naturally occurring mutants. Mol Microbiol 43 : 1641–1650.
16. CiofuO, RiisB, PresslerT, PoulsenHE, HøibyN (2005) Occurrence of hypermutable Pseudomonas aeruginosa in cystic fibrosis patients is associated with the oxidative stress caused by chronic lung inflammation. Antimicrob Agents Chemother 49 : 2276–2282.
17. FelizianiS, LujánAM, MoyanoAJ, SolaC, BoccoJL, et al. (2010) Mucoidy, quorum sensing, mismatch repair and antibiotic resistance in Pseudomonas aeruginosa from cystic fibrosis chronic airways infections. PLoS One 5.
18. MarvigRL, JohansenHK, MolinS, JelsbakL (2013) Genome analysis of a transmissible lineage of Pseudomonas aeruginosa reveals pathoadaptive mutations and distinct evolutionary paths of hypermutators. PLoS Genet 9: e1003741.
19. WaineDJ, HoneybourneD, SmithEG, WhitehouseJL, DowsonCG (2008) Association between hypermutator phenotype, clinical variables, mucoid phenotype, and antimicrobial resistance in Pseudomonas aeruginosa. J Clin Microbiol 46 : 3491–3493.
20. TaddeiF, RadmanM, Maynard-SmithJ, ToupanceB, GouyonPH, et al. (1997) Role of mutator alleles in adaptive evolution. Nature 387 : 700–702.
21. CoxEC, GibsonTC (1974) Selection for high mutation rates in chemostats. Genetics 77 : 169–184.
22. SniegowskiPD, GerrishPJ, LenskiRE (1997) Evolution of high mutation rates in experimental populations of E. coli. Nature 387 : 703–705.
23. MaoEF, LaneL, LeeJ, MillerJH (1997) Proliferation of mutators in A cell population. J Bacteriol 179 : 417–422.
24. TenaillonO, ToupanceB, Le NagardH, TaddeiF, GodelleB (1999) Mutators, population size, adaptive landscape and the adaptation of asexual populations of bacteria. Genetics 152 : 485–493.
25. GiraudA, MaticI, TenaillonO, ClaraA, RadmanM, et al. (2001) Costs and benefits of high mutation rates: adaptive evolution of bacteria in the mouse gut. Science 291 : 2606–2608.
26. DenamurE, MaticI (2006) Evolution of mutation rates in bacteria. Mol Microbiol 60 : 820–827.
27. OliverA, LevinBR, JuanC, BaqueroF, BlázquezJ (2004) Hypermutation and the preexistence of antibiotic-resistant Pseudomonas aeruginosa mutants: implications for susceptibility testing and treatment of chronic infections. Antimicrob Agents Chemother 48 : 4226–4233.
28. MaciáMD, BlanquerD, TogoresB, SauledaJ, PérezJL, et al. (2005) Hypermutation is a key factor in development of multiple-antimicrobial resistance in Pseudomonas aeruginosa strains causing chronic lung infections. Antimicrob Agents Chemother 49 : 3382–3386.
29. HenrichfreiseB, WiegandI, PfisterW, WiedemannB (2007) Resistance mechanisms of multiresistant Pseudomonas aeruginosa strains from Germany and correlation with hypermutation. Antimicrob Agents Chemother 51 : 4062–4070.
30. PlasenciaV, BorrellN, MaciáMD, MoyaB, PérezJL, et al. (2007) Influence of high mutation rates on the mechanisms and dynamics of in vitro and in vivo resistance development to single or combined antipseudomonal agents. Antimicrob Agents Chemother 51 : 2574–2581.
31. FerroniA, GuillemotD, MoumileK, BernedeC, Le BourgeoisM, et al. (2009) Effect of mutator P. aeruginosa on antibiotic resistance acquisition and respiratory function in cystic fibrosis. Pediatr Pulmonol 44 : 820–825.
32. MenaA, SmithEE, BurnsJL, SpeertDP, MoskowitzSM, et al. (2008) Genetic adaptation of Pseudomonas aeruginosa to the airways of cystic fibrosis patients is catalyzed by hypermutation. J Bacteriol 190 : 7910–7917.
33. CiofuO, MandsbergLF, BjarnsholtT, WassermannT, HøibyN (2010) Genetic adaptation of Pseudomonas aeruginosa during chronic lung infection of patients with cystic fibrosis: strong and weak mutators with heterogeneous genetic backgrounds emerge in mucA and/or lasR mutants. Microbiology 156 : 1108–1119.
34. MoyanoAJ, LujánAM, ArgarañaCE, SmaniaAM (2007) MutS deficiency and activity of the error-prone DNA polymerase IV are crucial for determining mucA as the main target for mucoid conversion in Pseudomonas aeruginosa. Mol Microbiol 64 : 547–559.
35. SmaniaAM, SeguraI, PezzaRJ, BecerraC, AlbesaI, et al. (2004) Emergence of phenotypic variants upon mismatch repair disruption in Pseudomonas aeruginosa. Microbiology 150 : 1327–1338.
36. LujánAM, MoyanoAJ, SeguraI, ArgarañaCE, SmaniaAM (2007) Quorum-sensing-deficient (lasR) mutants emerge at high frequency from a Pseudomonas aeruginosa mutS strain. Microbiology 153 : 225–237.
37. LujánAM, MaciáMD, YangL, MolinS, OliverA, et al. (2011) Evolution and adaptation in Pseudomonas aeruginosa biofilms driven by mismatch repair system-deficient mutators. PLoS One 6: e27842.
38. MoyanoAJ, SmaniaAM (2009) Simple sequence repeats and mucoid conversion: biased mucA mutagenesis in mismatch repair-deficient Pseudomonas aeruginosa. PLoS One 4: e8203.
39. MoyanoAJ, FelizianiS, Di RienzoJA, SmaniaAM (2013) Simple Sequence Repeats Together with Mismatch Repair Deficiency Can Bias Mutagenic Pathways in Pseudomonas aeruginosa during Chronic Lung Infection. PLoS One 8: e80514.
40. CramerN, KlockgetherJ, WrasmanK, SchmidtM, DavenportCF, et al. (2011) Microevolution of the major common Pseudomonas aeruginosa clones C and PA14 in cystic fibrosis lungs. Environ Microbiol 13 : 1690–1704.
41. SmithEE, SimsEH, SpencerDH, KaulR, OlsonMV (2005) Evidence for diversifying selection at the pyoverdine locus of Pseudomonas aeruginosa. J Bacteriol 187 : 2138–2147.
42. YangL, JelsbakL, MarvigRL, DamkiaerS, WorkmanCT, et al. (2011) Evolutionary dynamics of bacteria in a human host environment. Proc Natl Acad Sci U S A 108 : 7481–7486.
43. ChungJC, BecqJ, FraserL, Schulz-TrieglaffO, BondNJ, et al. (2012) Genomic variation among contemporary Pseudomonas aeruginosa isolates from chronically infected cystic fibrosis patients. J Bacteriol 194 : 4857–4866.
44. YangL, HaagensenJA, JelsbakL, JohansenHK, SternbergC, et al. (2008) In situ growth rates and biofilm development of Pseudomonas aeruginosa populations in chronic lung infections. J Bacteriol 190 : 2767–2776.
45. WiehlmannL, WagnerG, CramerN, SiebertB, GudowiusP, et al. (2007) Population structure of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 104 : 8101–8106.
46. DrummondAJ, SuchardMA, XieD, RambautA (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29 : 1969–1973.
47. GodfreyAJ, BryanLE, RabinHR (1981) beta-Lactam-resistant Pseudomonas aeruginosa with modified penicillin-binding proteins emerging during cystic fibrosis treatment. Antimicrob Agents Chemother 19 : 705–711.
48. KohlerT, Michea-HamzehpourM, HenzeU, GotohN, CurtyLK, et al. (1997) Characterization of MexE-MexF-OprN, a positively regulated multidrug efflux system of Pseudomonas aeruginosa. Mol Microbiol 23 : 345–354.
49. LivermoreDM (1995) beta-Lactamases in laboratory and clinical resistance. Clin Microbiol Rev 8 : 557–584.
50. MineT, MoritaY, KataokaA, MizushimaT, TsuchiyaT (1999) Expression in Escherichia coli of a new multidrug efflux pump, MexXY, from Pseudomonas aeruginosa. Antimicrob Agents Chemother 43 : 415–417.
51. PooleK (2005) Aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother 49 : 479–487.
52. VettorettiL, PlesiatP, MullerC, El GarchF, PhanG, et al. (2009) Efflux unbalance in Pseudomonas aeruginosa isolates from cystic fibrosis patients. Antimicrob Agents Chemother 53 : 1987–1997.
53. ZhangL, MahTF (2008) Involvement of a novel efflux system in biofilm-specific resistance to antibiotics. J Bacteriol 190 : 4447–4452.
54. DaviesJA, HarrisonJJ, MarquesLL, FogliaGR, StremickCA, et al. (2007) The GacS sensor kinase controls phenotypic reversion of small colony variants isolated from biofilms of Pseudomonas aeruginosa PA14. FEMS Microbiol Ecol 59 : 32–46.
55. SallKM, CasabonaMG, BordiC, HuberP, de BentzmannS, et al. (2014) A gacS Deletion in Pseudomonas aeruginosa Cystic Fibrosis Isolate CHA Shapes Its Virulence. PLoS One 9: e95936.
56. NelsonLK, StantonMM, ElphinstoneRE, HelwerdaJ, TurnerRJ, et al. (2010) Phenotypic diversification in vivo: Pseudomonas aeruginosa gacS - strains generate small colony variants in vivo that are distinct from in vitro variants. Microbiology 156 : 3699–3709.
57. BieleckiP, LukatP, HuseckenK, DotschA, SteinmetzH, et al. (2012) Mutation in elongation factor G confers resistance to the antibiotic argyrin in the opportunistic pathogen Pseudomonas aeruginosa. Chembiochem 13 : 2339–2345.
58. NyfelerB, HoepfnerD, PalestrantD, KirbyCA, WhiteheadL, et al. (2012) Identification of elongation factor G as the conserved cellular target of argyrin B. PLoS One 7: e42657.
59. OverhageJ, SchemionekM, WebbJS, RehmBH (2005) Expression of the psl operon in Pseudomonas aeruginosa PAO1 biofilms: PslA performs an essential function in biofilm formation. Appl Environ Microbiol 71 : 4407–4413.
60. ValletI, OlsonJW, LoryS, LazdunskiA, FillouxA (2001) The chaperone/usher pathways of Pseudomonas aeruginosa: identification of fimbrial gene clusters (cup) and their involvement in biofilm formation. Proc Natl Acad Sci U S A 98 : 6911–6916.
61. MahenthiralingamE, CampbellME, SpeertDP (1994) Nonmotility and phagocytic resistance of Pseudomonas aeruginosa isolates from chronically colonized patients with cystic fibrosis. Infect Immun 62 : 596–605.
62. D'ArgenioDA, WuM, HoffmanLR, KulasekaraHD, DézielE, et al. (2007) Growth phenotypes of Pseudomonas aeruginosa lasR mutants adapted to the airways of cystic fibrosis patients. Mol Microbiol 64 : 512–533.
63. HoffmanLR, KulasekaraHD, EmersonJ, HoustonLS, BurnsJL, et al. (2009) Pseudomonas aeruginosa lasR mutants are associated with cystic fibrosis lung disease progression. J Cyst Fibros 8 : 66–70.
64. MartinDW, SchurrMJ, MuddMH, GovanJR, HollowayBW, et al. (1993) Mechanism of conversion to mucoidy in Pseudomonas aeruginosa infecting cystic fibrosis patients. Proc Natl Acad Sci U S A 90 : 8377–8381.
65. MontanariS, OliverA, SalernoP, MenaA, BertoniG, et al. (2007) Biological cost of hypermutation in Pseudomonas aeruginosa strains from patients with cystic fibrosis. Microbiology 153 : 1445–1454.
66. García-CastilloM, MáizL, MorosiniMI, Rodríguez-BañosM, SuarezL, et al. (2012) Emergence of a mutL mutation causing multilocus sequence typing-pulsed-field gel electrophoresis discrepancy among Pseudomonas aeruginosa isolates from a cystic fibrosis patient. J Clin Microbiol 50 : 1777–1778.
67. WuTH, MarinusMG (1999) Deletion mutation analysis of the mutS gene in Escherichia coli. J Biol Chem 274 : 5948–5952.
68. MiguelV, MontiMR, ArgarañaCE (2008) The role of MutS oligomers on Pseudomonas aeruginosa mismatch repair system activity. DNA Repair (Amst) 7 : 1799–1808.
69. WinsorGL, LamDK, FlemingL, LoR, WhitesideMD, et al. (2011) Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res 39: D596–600.
70. JelsbakL, JohansenHK, FrostAL, ThogersenR, ThomsenLE, et al. (2007) Molecular epidemiology and dynamics of Pseudomonas aeruginosa populations in lungs of cystic fibrosis patients. Infect Immun 75 : 2214–2224.
71. RauMH, HansenSK, JohansenHK, ThomsenLE, WorkmanCT, et al. (2010) Early adaptive developments of Pseudomonas aeruginosa after the transition from life in the environment to persistent colonization in the airways of human cystic fibrosis hosts. Environ Microbiol 12 : 1643–1658.
72. WinstanleyC, LangilleMG, FothergillJL, Kukavica-IbruljI, Paradis-BleauC, et al. (2009) Newly introduced genomic prophage islands are critical determinants of in vivo competitiveness in the Liverpool Epidemic Strain of Pseudomonas aeruginosa. Genome Res 19 : 12–23.
73. WilderCN, AlladaG, SchusterM (2009) Instantaneous within-patient diversity of Pseudomonas aeruginosa quorum-sensing populations from cystic fibrosis lung infections. Infect Immun 77 : 5631–5639.
74. TurrientesMC, BaqueroF, LevinBR, MartínezJL, RipollA, et al. (2013) Normal mutation rate variants arise in a Mutator (MutS) Escherichia coli population. PLoS One 8: e72963.
75. GalabertC, JacquotJ, ZahmJM, PuchelleE (1987) Relationships between the lipid content and the rheological properties of airway secretions in cystic fibrosis. Clin Chim Acta 164 : 139–149.
76. RoseMC, VoynowJA (2006) Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol Rev 86 : 245–278.
77. BarthAL, PittTL (1996) The high amino-acid content of sputum from cystic fibrosis patients promotes growth of auxotrophic Pseudomonas aeruginosa. J Med Microbiol 45 : 110–119.
78. RauMH, MarvigRL, EhrlichGD, MolinS, JelsbakL (2012) Deletion and acquisition of genomic content during early stage adaptation of Pseudomonas aeruginosa to a human host environment. Environ Microbiol 14 : 2200–2211.
79. LeeDG, UrbachJM, WuG, LiberatiNT, FeinbaumRL, et al. (2006) Genomic analysis reveals that Pseudomonas aeruginosa virulence is combinatorial. Genome Biol 7: R90.
80. MareghettiL, Marquet-van der MeeN, LoulergueJ, RollandJC, AA (1998) Pseudomonas aeruginosa from cystic fibrosis patients: study using whole cell RAPD and antibiotic susceptibility. Pathol Biol 46 : 319–324.
81. RömlingU, TümmlerB (2000) Achieving 100% typeability of Pseudomonas aeruginosa by pulsed-field gel electrophoresis. J Clin Microbiol 38 : 464–465.
82. TenoverFC, ArbeitRD, GoeringRV, MickelsenPA, MurrayBE, et al. (1995) Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typing. J Clin Microbiol 33 : 2233–2239.
83. MoralesG, WiehlmannL, GudowiusP, van DeldenC, TümmlerB, et al. (2004) Structure of Pseudomonas aeruginosa populations analyzed by single nucleotide polymorphism and pulsed-field gel electrophoresis genotyping. J Bacteriol 186 : 4228–4237.
84. ZerbinoDR, BirneyE (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18 : 821–829.
85. KrawitzP, RodelspergerC, JagerM, JostinsL, BauerS, et al. (2010) Microindel detection in short-read sequence data. Bioinformatics 26 : 722–729.
86. LiH, HandsakerB, WysokerA, FennellT, RuanJ, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25 : 2078–2079.
87. DePristoMA, BanksE, PoplinR, GarimellaKV, MaguireJR, et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43 : 491–498.
88. KurtzS, PhillippyA, DelcherAL, SmootM, ShumwayM, et al. (2004) Versatile and open software for comparing large genomes. Genome Biol 5: R12.
89. Balzarini MG. DRJA InfoGen versión 2013.FCA, Universidad Nacional de Córdoba, Argentina. URL http://www.info-gen.com.ar.
90. PezzaRJ, SmaniaAM, BarraJL, ArgarañaCE (2002) Nucleotides and heteroduplex DNA preserve the active conformation of Pseudomonas aeruginosa MutS by preventing protein oligomerization. Biochem J 361 : 87–95.
91. JacquelínDK, MartinaMA, ArgarañaCE, BarraJL (2008) Plasmid expression of mutS, -L and/or -H gene in Escherichia coli dam cells results in strains that display reduced mutation frequency. Mutat Res 637 : 197–204.
92. ChoiKH, SchweizerHP (2005) An improved method for rapid generation of unmarked Pseudomonas aeruginosa deletion mutants. BMC Microbiol 5 : 30.
93. BochnerBR (2009) Global phenotypic characterization of bacteria. FEMS Microbiol Rev 33 : 191–205.
94. BochnerBR (2003) New technologies to assess genotype-phenotype relationships. Nat Rev Genet 4 : 309–314.
95. CooperVS, LenskiRE (2000) The population genetics of ecological specialization in evolving Escherichia coli populations. Nature 407 : 736–739.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 10
Nejčtenější v tomto čísle
- The Master Activator of IncA/C Conjugative Plasmids Stimulates Genomic Islands and Multidrug Resistance Dissemination
- A Splice Mutation in the Gene Causes High Glycogen Content and Low Meat Quality in Pig Skeletal Muscle
- Keratin 76 Is Required for Tight Junction Function and Maintenance of the Skin Barrier
- A Role for Taiman in Insect Metamorphosis