
The DAF-16 FOXO Transcription Factor Regulates to Modulate Stress Resistance in , Linking Insulin/IGF-1 Signaling to Protein N-Terminal Acetylation
What are the mechanisms used by animals to cope with stressful environments that inflict damage or restrict essential processes such as growth, development, and reproduction? One strategy is changes in physiology that increase stress resistance, and an extreme version of this strategy is diapause, an alternative developmental state that is enduring and stress resistant. In the nematode C. elegans, stress tolerance and entry into a diapause state called dauer larvae are mediated by the conserved insulin/IGF-1 pathway. Specifically, the FOXO transcription factor DAF-16 promotes stress tolerance and dauer larvae development. However, the targets of DAF-16 that mediate these processes remain largely elusive. Using an unbiased forward genetic screen to discover new mediators of stress tolerance, we identified natc-1, a novel target of DAF-16 and the insulin/IGF-1 pathway. natc-1 encodes a conserved subunit of the N-terminal acetyltransferase C (NAT) complex. The NatC complex modifies target proteins by acetylating the N-terminus. We demonstrated that natc-1 mediates diapause entry and stress tolerance. Furthermore, we elucidated regulation of NatC by demonstrating that natc-1 is a direct transcriptional target that is repressed by DAF-16. These findings may be relevant to other animals because both the insulin/IGF-1 signaling pathway and the NAT system are conserved during evolution.
Published in the journal:
. PLoS Genet 10(10): e32767. doi:10.1371/journal.pgen.1004703
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004703
Summary
What are the mechanisms used by animals to cope with stressful environments that inflict damage or restrict essential processes such as growth, development, and reproduction? One strategy is changes in physiology that increase stress resistance, and an extreme version of this strategy is diapause, an alternative developmental state that is enduring and stress resistant. In the nematode C. elegans, stress tolerance and entry into a diapause state called dauer larvae are mediated by the conserved insulin/IGF-1 pathway. Specifically, the FOXO transcription factor DAF-16 promotes stress tolerance and dauer larvae development. However, the targets of DAF-16 that mediate these processes remain largely elusive. Using an unbiased forward genetic screen to discover new mediators of stress tolerance, we identified natc-1, a novel target of DAF-16 and the insulin/IGF-1 pathway. natc-1 encodes a conserved subunit of the N-terminal acetyltransferase C (NAT) complex. The NatC complex modifies target proteins by acetylating the N-terminus. We demonstrated that natc-1 mediates diapause entry and stress tolerance. Furthermore, we elucidated regulation of NatC by demonstrating that natc-1 is a direct transcriptional target that is repressed by DAF-16. These findings may be relevant to other animals because both the insulin/IGF-1 signaling pathway and the NAT system are conserved during evolution.
Introduction
The ability to cope with fluctuating environmental stresses is critical for animal survival. Environmental stresses include a wide range of factors such as extremes in temperature, oxidation, and metal availability. A stress response might promote tolerance against a specific challenge or provide broad-spectrum resistance, and a critical question in this field is how specific stress responses mediate resistance to one or more forms of environmental challenge?
The nematode Caenorhabditis elegans is an important model system for studies of stress resistance. In response to stresses such as high temperature, low nutrient availability, and high population density, developing larvae will adopt an alternative L3 stage called dauer that is stress resistant [1]. Studies of dauer formation led to the discovery of an insulin/IGF-1 signaling pathway as a critical regulator of this stress response [2]. Loss-of-function mutations in daf-2 and age-1 cause dauer constitutive phenotypes, whereas loss-of-function mutations in daf-16 cause dauer defective phenotypes [1]. daf-2 encodes an insulin/IGF-1 receptor homolog, and age-1 encodes a phosphatidylinositol-3-OH kinase catalytic subunit homolog [3], [4]. In addition to mediating a developmental switch in larvae, this pathway functions throughout the life of the animal to mediate stress resistance, since daf-2 loss-of-function mutations cause increased tolerance to multiple stresses and an extended lifespan [3], [5], [6]. These daf-2 mutant phenotypes are suppressed by mutations in daf-16, indicating that daf-16 is a major downstream effector of the insulin/IGF-1 signaling pathway that is negatively regulated by daf-2 activity. daf-16 encodes a FOXO transcription factor [7], [8]. Because DAF-16 plays a central role in promoting longevity and stress tolerance, a major goal has been to identify and characterize DAF-16 transcriptional targets [9]–[17]. Although several genes have been demonstrated to be directly regulated by DAF-16, the understanding of how DAF-16 target genes mediate stress resistance and longevity remains fragmentary. The insulin/IGF-1 signaling pathway regulates stress tolerance, metabolism, and longevity in multiple species including mammals [18]–[22]. Thus, the identification of physiologically significant DAF-16 targets may suggest strategies for promoting longevity and stress tolerance in C. elegans and higher eukaryotes.
The metal zinc is a nutrient that is essential for all organisms and plays many roles in biological systems. Zinc functions in signal transduction pathways and contributes to protein structure and activity [23]–[25]. Zinc deficiency and excess both cause a wide spectrum of defects, demonstrating the importance of zinc homeostasis [26]–[31]. Zinc deficiency appears to be deleterious due to the reduced function of many zinc-requiring proteins and signaling events [25]. The mechanisms underlying excess zinc toxicity are not well characterized; excess zinc may displace other physiological metals or bind to low-affinity sites, leading to altered or decreased protein function [32]. In addition to zinc, several other metals are toxic in excess, including cadmium, nickel, and copper [33]–[35]. To characterize mechanisms of metal stress resistance, we performed a forward genetic screen for mutations that caused resistance to high levels of dietary zinc [36]. We reasoned that tolerance to high zinc could be caused by two general mechanisms: (1) mutations might affect zinc metabolism and reduce the accumulation of toxic zinc, or (2) mutations might cause alterations that promote growth and survival in the presence of high levels of zinc. We identified haly-1, which encodes the enzyme histidine ammonia lyase that metabolizes histidine [37]. Loss-of-function mutations in haly-1 do not affect zinc accumulation but rather cause an increase in histidine levels resulting in increased chelation of zinc and nickel, and chelation by histidine reduces the toxicity of these metals [37]. Notably, haly-1 mutations have not been demonstrated to cause resistance to other stressors. By contrast, mutations in daf-2 and age-1, members of the insulin/IGF-1 pathway, cause resistance to a broad-spectrum of stressors including the metals cadmium and copper, oxidative stress, and heat stress [5], [35], [38].
Here we describe natc-1, a new gene that was discovered in the screen for worms that are resistant to high zinc toxicity, which encodes the C. elegans N-α-acetyltransferase 35. N-α-acetyltransferases (NATs) are highly conserved among eukaryotes and function as protein complexes to transfer the acetyl group of acetyl coenzyme A to the α-amino group of the first amino acid of a target protein. natc-1 encodes an auxiliary subunit of the NatC complex which acetylates proteins that begin with the amino acids Met-Leu, Met-Phe, Met-Ile, and Met-Trp [39], [40]. natc-1 mutations caused resistance to multiple metals, oxidation, and heat, indicating that natc-1 modulates broad-spectrum stress resistance, similar to mutations in insulin/IGF-1 signaling genes. Interestingly, the natc-1 promoter contains an evolutionarily conserved DAF-16 binding site, and DAF-16 binds natc-1 in vivo [11], [41], [42]. We demonstrated that natc-1 is transcriptionally repressed by DAF-16 activity and that natc-1 interacts with genes in the insulin/IGF-1 signaling pathway to mediate stress resistance and dauer formation. These results indicate that natc-1 is directly regulated by DAF-16 and functions as a downstream effector of the insulin/IGF-1 signaling pathway. Supporting this model, mutations in natc-1 that increase stress resistance are epistatic to daf-16 mutations. These results provide novel insights into the transcriptional regulation of natc-1 by the insulin/IGF-1 signaling pathway and the biological function of protein N-terminal acetylation in mediating stress resistance. Furthermore, our data elucidate a new mechanism used by the insulin/IGF-1 signaling pathway to mediate stress tolerance and dauer formation.
Results
Mutations in natc-1 caused resistance to high levels of dietary zinc
We performed a forward genetic screen to identify mutant strains that are resistant to the growth arrest and lethality caused by high levels of dietary zinc [36]. Nineteen mutations were identified and positioned in the genome using a genome-wide map of single nucleotide polymorphism (SNP) markers [36]. Here we focus on two of these mutations, am134 and am138, that caused significant resistance to dietary zinc toxicity (Figure 1A,B). Both mutations displayed tightest linkage to the same SNP, pkP5513, positioned at +0.1 map units on chromosome V (Figure 1C). Three factor mapping experiments indicated that am138 is positioned between dpy-11 and unc-42, a 325 kilobase pair interval that contains pkP5513 (Figure 1C) [36].

To identify the gene affected by these mutations, we performed whole genome sequencing using DNA from the am134 and am138 mutant strains. Candidate mutations in the mapping interval were identified by comparing the am134 and am138 DNA sequence to the wild-type DNA sequence. The am134 and am138 strains both contained candidate mutations in the predicted open reading frame T23B12.4, suggesting that these mutations caused resistance to zinc toxicity. The predicted T23B12.4 protein is homologous to human N-α-acetyltransferase 35, an auxiliary subunit of the NatC complex. Thus, we named this gene natc-1. The mutation in the am134 mutant strain, which was induced with the mutagen ENU, is a C to T substitution that changes codon 691 from arginine (CGA) to stop (TGA). This nonsense mutation is predicted to truncate 110 amino acids from the NATC-1 protein (Figure 2A,B). The mutation in the am138 mutant strain, which was induced by ENU mutagenesis, is a 186 base pair deletion that eliminates portions of exons 1 and 2 and all of intron one (Figure 2A,B). This deletion eliminates the codons for amino acids 13–59, and the mutated open reading frame is predicted to have a frame shift and encounter a stop at the new codon 15. The loss of coding sequences and early truncation suggest natc-1(am138) is likely to be a strong loss-of-function or null allele.

To test the hypothesis that mutations in natc-1 cause resistance to zinc toxicity, we analyzed the natc-1(ok2062) deletion mutation that was generated by the C. elegans knock out consortium [43]. natc-1(ok2062) is a 1,108 base pair deletion that eliminates a portion of exon 3, intron 3, exon 4, intron 4, and a portion of exon 5 (Figure 2A,B). natc-1(ok2062) mutant animals displayed significant resistance to zinc toxicity, similar to natc-1(am134) and natc-1(am138) mutant animals (Figure 1B). This result supports the hypothesis that mutations in natc-1 cause resistance to zinc toxicity.
To independently test the hypothesis that the am134 mutation in natc-1 causes resistance to zinc toxicity, we determined whether a wild-type version of natc-1 could rescue this phenotype. We generated five independently derived transgenic strains containing extrachromosomal arrays with wild-type copies of natc-1 in the background of natc-1(am134). All the transgenic strains displayed a significant decrease in zinc resistance when compared to their non-transgenic siblings. This is indicative of a more wild-type phenotype and rescue activity (Figure 1C, Figure S1A). To determine if an intact natc-1 open reading frame is necessary for rescue activity, we generated transgenic animals containing a natc-1 locus that encodes a mutant protein with a 112 amino acid deletion in the background of natc-1(am134). These transgenic animals did not display rescue of the mutant phenotype, indicating that the rescue activity of the natc-1 locus requires an intact open reading frame (Figure 1C, Figure S1B). Together, these results demonstrated that natc-1 is the gene affected by am134 and am138 (reference allele), and that mutations in natc-1 caused resistance to zinc toxicity.
natc-1 encodes N-α-acetyltransferase 35, an auxiliary subunit of the NatC complex
To characterize the products generated from the natc-1 locus, we analyzed natc-1 mRNA. The C. elegans expressed sequence tag (EST) project isolated multiple cDNAs corresponding to natc-1, and we determined the DNA sequence of three independently derived cDNAs. The cDNA sequences were used to infer the mRNA sequence from exon 3 to the 3′ end, including the position of the polyA tail 330 nucleotides downstream of the TGA stop codon. To characterize the 5′ end of the transcript, we conducted a 5′ RACE experiment that showed the natc-1 mRNA contains a 22 nucleotide splice leader 1 (SL1) sequence that begins 14 base pairs upstream of the start codon. Together, the analysis of cDNAs and 5′ RACE indicated that the natc-1 mRNA contains 8 exons and defined the complete predicted open reading frame (Figure 2A).
The predicted NATC-1 protein contains 799 amino acids. To determine the expression pattern and sub-cellular localization of NATC-1, we generated transgenic natc-1(am138) animals expressing NATC-1 protein fused to green fluorescent protein (GFP) under the control of the native natc-1 promoter. Live animals were imaged with confocal fluorescence microscopy, and NATC-1::GFP was detected in a wide range of cells and tissues in a pattern that suggests cytoplasmic localization (Figure 3). NATC-1::GFP was detected throughout development from early larval stages through late adulthood. To confirm that the expression pattern of NATC-1::GFP is representative of the expression pattern of endogenous NATC-1, we demonstrated that the extrachromosomal array expressing NATC-1::GFP rescued the natc-1(am138) zinc-resistance phenotype (Figure 1C, Figure S1C).

Comparison of NATC-1 protein sequence to databases using the method of BLAST revealed that NATC-1 is most similar to N-α-acetyltransferase 35 proteins, which are auxiliary subunits of the NatC complex [44]. Figure 2B shows an alignment of C. elegans NATC-1 with Drosophila melanogaster and human proteins; C. elegans NATC-1 is 24% identical to human NAA35, suggesting that it may have similar biochemical functions. NATC-1 is an auxiliary subunit of the NatC complex, and C. elegans B0238.10 (NATC-2) is the predicted catalytic subunit [45]. The NatC complex catalyzes the acetylation of the N-termini of translating proteins (Figure 2C). The NatC complex specifically acetylates translating proteins that begin with Met-Ile, Met-Leu, Met-Trp, or Met-Phe [39], [40]. To identify predicted NatC target proteins, we conducted a bioinformatic analysis using the fully sequenced C. elegans genome. Approximately 4,300 proteins have Ile, Leu, Trp, or Phe in amino acid position two. These proteins represent ∼17% of the C. elegans proteome and are candidates to be acetylated by the NatC complex.
The zinc content of natc-1 mutant animals was similar to wild-type worms
Zinc resistance displayed by natc-1 mutant animals could be explained by two general models: (1) natc-1 mutant animals have decreased levels of zinc, perhaps as a result of reduced zinc uptake or increased zinc excretion and (2) natc-1 mutant animals have normal levels of zinc but increased tolerance to high zinc toxicity. To distinguish between these possibilities, we used inductively coupled plasma mass spectrometry (ICP-MS) to measure total animal zinc content. Synchronized populations of animals were cultured with NAMM, harvested, and analyzed for zinc content. The total animal zinc content of natc-1(am134), natc-1(am138), and natc-1(ok2062) mutant animals was not consistently different from wild-type animals when cultured with or without supplemental zinc (Figure S2). These results indicate that mutations in natc-1 cause resistance to zinc toxicity by increasing the ability of the animal to tolerate excess zinc that results from a high zinc diet rather then by reducing zinc accumulation.
natc-1(am138) caused zinc resistance in multiple genetic backgrounds
Mutations in haly-1 that cause resistance to high zinc toxicity were identified in the same genetic screen as mutations in natc-1 [37]. The mechanism of action of mutations in haly-1 appears to be accumulation of histidine, which is hypothesized to reduce high zinc toxicity by chelation of the ion. To determine if mutations in natc-1 may cause resistance to high zinc toxicity by a similar mechanism, we analyzed natc-1(am138);haly-1(am132) double mutant animals. If natc-1(am138) and haly-1(am132) cause zinc resistance by affecting the same pathway or process, then the resistance to high zinc toxicity phenotypes might not be additive. Interestingly, natc-1(am138);haly-1(am132) double mutant animals displayed enhanced resistance to high zinc toxicity compared to natc-1(am138) or haly-1(am132) single mutant animals (Figure S3). This result suggests that resistance to high zinc toxicity caused by natc-1 mutations may be mechanistically distinct from that caused by haly-1 mutations.
Mutations in natc-1 increased resistance to a wide range of stressors
To determine if natc-1 causes resistance to additional metals, we cultured wild-type animals and natc-1(am138) animals on NAMM plates supplemented with cadmium, nickel, or copper. Concentrations that caused ∼50% sterility of wild-type animals were chosen for each metal to maximize the sensitivity of the assay. natc-1 mutant animals displayed improved growth and development compared to wild-type animals when cultured with 200 µM zinc, 20 µM cadmium, 50 µM nickel, or 300 µM copper (Figure 4A–D). These data suggest that mutations in natc-1 cause resistance to toxicity induced by both physiological and non-physiological metal ions.

To determine if mutations in natc-1 cause resistance to stressors in addition to metal ions, we analyzed heat stress. Wild-type and natc-1 mutant animals were cultured at 35°C and survival times were monitored. natc-1(am138) animals displayed a significant 19% extension of survival compared to wild-type animals (Figure 5A, Table 1). These data suggest that mutations in natc-1 cause resistance to heat toxicity. To analyze oxidative stress, we cultured animals with 40 mM paraquat and monitored survival time. natc-1(am138) animals displayed a significant 40% extension of survival compared to wild-type animals (Figure 5B, Table 1). Taken together, these data suggest that mutations in natc-1 cause resistance to a broad-spectrum of stressors, including high levels of multiple metal ions, oxidative damage, and high heat.
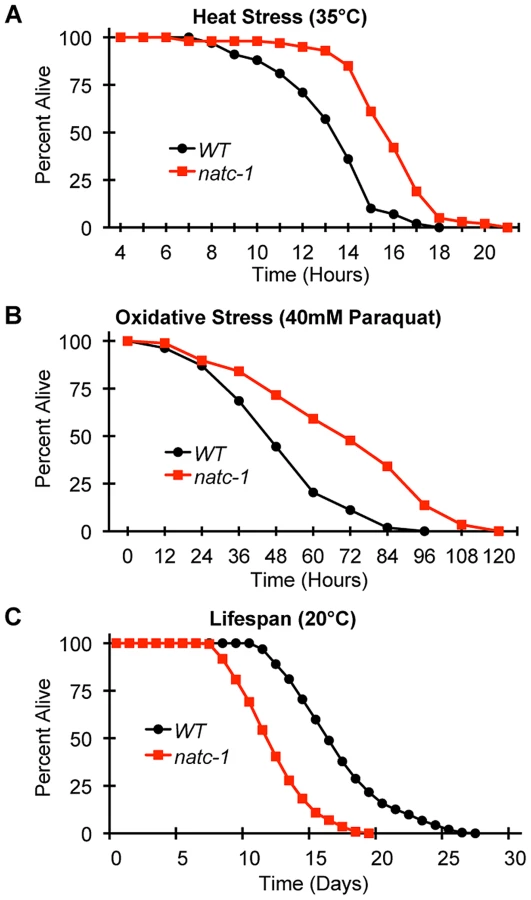

One hypothesis that might explain the stress resistance phenotype is that natc-1(lf) mutations stimulate the unfolded protein response. To investigate this hypothesis, we used the method of qRT-PCR to analyze the mRNA levels of the stress-induced genes hsp-4, hsp-6, and hsp-16.2. Wild-type and natc-1(am138) animals did not display statistically significant differences in mRNA levels for these genes (p>0.05), suggesting that loss of natc-1 activity does not stimulate the unfolded protein response. Furthermore, gst-4 mRNA levels, which are induced by oxidative stress and proteosomal dysfunction [46] were not significantly altered in natc-1(am138) mutants compared to wild type (p>0.05).
To determine how natc-1 activity affects longevity, we analyzed the lifespan of wild-type and natc-1(am138) mutant animals. natc-1(am138) animals displayed a significant 31% reduction in mean lifespan compared to wild-type animals (Figure 5C, Table 1). These data suggest that natc-1 is a lifespan assurance gene when animals are cultured at an optimal temperature with abundant food, conditions that minimize stress.
natc-1 mRNA appears to be directly repressed by the DAF-16 transcription factor
To analyze the regulation of natc-1, we used qRT-PCR to monitor the level of natc-1 mRNA. Because natc-1(lf) mutations cause zinc resistance, we examined the transcriptional response to high dietary zinc. natc-1 mRNA levels were not affected by 200 µM supplemental dietary zinc (p>0.05), suggesting that natc-1 transcription is not regulated to promote zinc tolerance. To further analyze regulation, we examined the insulin/IGF-1 signaling pathway because it plays a pivotal role in stress resistance in C. elegans [2]. daf-2 encodes the insulin receptor that functions to inhibit dauer formation and stress resistance; daf-2(e1370) is a partial loss-of-function mutation that causes a temperature-sensitive dauer constitutive (Daf-c) phenotype in larvae and an increased stress resistance phenotype in adults [6], [35], [38]. daf-16 encodes a FOXO transcription factor that is a crucial downstream target that is negatively regulated by the DAF-2 pathway; daf-16(mu86) is a null mutation that causes a dauer defective (Daf-d) phenotype in larvae and a reduced stress resistance phenotype in adults [7]. Interestingly, Lee et al. (2003) used bioinformatic techniques to identify a putative DAF-16 binding site (TTGTTTAC) positioned 90 base pairs upstream of the predicted start codon of the natc-1 locus [11] (Figure 6A). This predicted DAF-16 binding site is evolutionarily conserved in natc-1 homologues in Caenorhabditis briggsae and Drosophila melanogaster, suggesting it is functionally important [11]. We analyzed the genomic locus of human NAA35, the homolog of C. elegans NATC-1, and identified four predicted DAF-16 binding sites, consistent with the model that these binding sites might be conserved during evolution (Figure S4). Furthermore, Gerstein et al. (2010) and Riedel et al. (2013) used the method of chromatin immunoprecipitation followed by massively parallel DNA sequencing to demonstrate that DAF-16 protein interacts with the natc-1 locus in vivo [41], [42] (G. Ruvkun and C. Riedel, personal communication) (Figure 6A).

Based on these observations, we hypothesized that natc-1 transcription is directly regulated by binding of the DAF-16 transcription factor. According to this model, altering DAF-16 activity is predicted to alter natc-1 mRNA levels. When cultured in standard laboratory conditions, worms display a low level of DAF-16 activity because the insulin/IGF-1 pathway is strongly activated [47]. Therefore, comparing daf-16(lf) animals to control animals would not be highly informative. To test our hypothesis, we analyzed natc-1 mRNA levels in daf-2(e1370) mutant animals, since daf-2 mutant animals have been demonstrated to have increased DAF-16 nuclear localization and activity [48]. These daf-2(lf) phenotypes are similar to the consequences of food deprivation or stress, suggesting daf-2(lf) mutant worms have initiated a starvation or stress response [1]. daf-2 mutant animals displayed a ∼2-fold decrease in natc-1 mRNA levels compared to wild-type animals (Figure 6B). To confirm that this change was statistically significant, we analyzed six independent biological replicates of both wild-type and daf-2 RNA. These data are consistent with the model that DAF-16 represses transcription of natc-1, since DAF-16 activity is increased in daf-2 mutant animals. To directly test the function of daf-16, we analyzed daf-16;daf-2 double mutant animals; the decrease in natc-1 mRNA levels was abrogated in these animals, demonstrating that daf-16 is necessary for the regulation of natc-1 (Figure 6B). To confirm that daf-16 mutant animals do not contain daf-16 activity, we analyzed daf-16 transcript levels by qRT-PCR; daf-16 transcripts were detected in wild-type animals but were undetectable in the daf-16;daf-2 double mutant animals. These data suggest that DAF-16 is a transcriptional repressor of natc-1 and natc-1 is an effector of the insulin/IGF-1 signaling pathway that functions downstream of DAF-16.
The NatC complex mediates insulin/IGF-1 signaling during dauer formation
The insulin/IGF-1 signaling pathway mediates entry into an alternative third larval stage called dauer that has distinctive metabolic and developmental features that promote longevity and stress resistance [3], [8], [49]. To test the function of natc-1 in insulin/IGF-1 signaling, we analyzed dauer larvae formation in natc-1(am138) animals. A single mutation in natc-1 did not cause a Daf-c phenotype (Figure 6C). However, natc-1(am138) strongly enhanced dauer formation in the daf-2(e1370) background, compared to the daf-2(e1370) single mutant animals (Figure 6C). To determine if the daf-2(e1370);natc-1(am138) Daf-c phenotype was daf-16 dependent, we analyzed daf-16(mu86);daf-2(e1370);natc-1(am138) triple mutant animals for dauer formation at 25°C. None of the daf-16;daf-2;natc-1 triple mutant animals displayed dauer formation (N = 111), indicating that daf-16 is required for this Daf-c phenotype. Together, these data demonstrated that natc-1 was necessary to inhibit dauer formation, although the effect was only observed in a sensitive genetic background.
We hypothesized that the enhancement of the Daf-c phenotype caused by a mutation in the natc-1 auxiliary subunit reflects the reduction or loss of the acetylation activity of the NatC complex. To test this hypothesis, we analyzed the function of the predicted catalytic subunit of the NatC complex, B0238.10, which we named natc-2. We used the method of feeding RNAi to reduce natc-1 and natc-2 activity in a daf-2(e1370) mutant background. A significant increase in dauer formation was observed compared to control RNAi (Figure 6D). These data indicate that the catalytic subunit encoded by natc-2 is necessary to inhibit dauer formation, suggesting that the acetylation activity of the NatC complex mediates insulin/IGF-1 signaling.
natc-1 functions as a downstream effector of the insulin/IGF-1 signaling pathway to mediate stress resistance
Mutations in the insulin/IGF-1 receptor daf-2 cause increased longevity, while the natc-1(am138) mutation causes a shortened lifespan [50]. To further characterize the genetic interaction between natc-1 and daf-2, we analyzed the lifespan of wild-type, natc-1(am138), daf-2(e1370), and daf-2(e1370);natc-1(am138) animals. While natc-1(am138) shortens wild-type lifespan, natc-1(am138) had no effect on the daf-2(e1370) longevity phenotype (Figure S5, Table 1). This result suggests that daf-2 activity is necessary for the natc-1(am138) mutation to cause a reduction of lifespan.
To characterize the role of natc-1 in stress resistance mediated by the insulin/IGF-1 signaling pathway, we analyzed interactions between natc-1, daf-16, and daf-2 in response to heat and high zinc stress. Single mutations of natc-1 and daf-2 cause resistance to heat stress, and daf-2(e1370);natc-1(am138) double mutant animals displayed enhanced stress resistance compared to either single mutant animal (Figure 7A). One interpretation of this additivity is that natc-1 and daf-2 function in the same pathway, but neither single mutation maximizes the potential of the pathway to increase stress resistance; this is consistent with the fact that the daf-2(e1370) allele causes a partial loss-of-function. The alternative interpretation is that natc-1 and daf-2 function in parallel to mediate stress resistance.

Compared to wild-type animals, daf-16(mu86) animals displayed a mild sensitivity to heat stress (Figure 7B, Table 1). daf-16(mu86);natc-1(am138) double mutant animals displayed heat stress resistance similar to natc-1 single mutant animals. (Figure 7B, Table 1). These data indicate that natc-1 is epistatic to daf-16 with respect to heat stress resistance, consistent with the model that natc-1 is a downstream effector that is negatively regulated by daf-16.
To further analyze this pathway, we determined if daf-16 was necessary for natc-1(am138) to cause resistance to zinc toxicity. Attempts to analyze daf-2 and daf-2;natc-1 mutant animals for resistance to high zinc toxicity were not successful, since supplemental zinc caused a high rate of dauer formation in these mutant animals, precluding an analysis of growth rates (Figure S6). natc-1(am138) caused similar resistance to high zinc toxicity in both a wild-type and daf-16(mu86) background (Figure 7C), indicating that daf-16 function is not necessary for the natc-1(am138) zinc-resistance phenotype. These data support the model that natc-1 functions downstream of daf-16 to mediate zinc resistance, and together these results suggest that natc-1 is acting as a key downstream effector of the C. elegans insulin/IGF-1 signaling pathway (Figure 8A,B).

Discussion
natc-1 is a physiologically relevant effector of the insulin/IGF-1 signaling pathway that is likely to be directly regulated by DAF-16
The DAF-16 FOXO transcription factor is the key downstream target of the insulin/IGF-1 signaling pathway that promotes stress resistance and longevity [5], [35], [50]–. A critical goal in this field is to identify the functionally significant targets of DAF-16, since these genes are hypothesized to mediate stress resistance, nutrient utilization, and aging. A variety of approaches have been used to identify DAF-16 target genes, including bioinformatic, genomic, and reverse genetic techniques [9]–[17]. Lee et al. (2003) used the biochemically demonstrated DNA binding sequence of DAF-16 to bioinformatically identify predicted binding sites in the C. elegans genome. One of these sites is positioned 90 base pairs upstream of the natc-1 start codon, suggesting that DAF-16 binds the natc-1 promoter. A similar FOXO transcription factor binding site is present in the promoters of genes homologous to natc-1 in Caenorhabditis briggsae, Drosophila melanogaster, and humans, suggesting that this transcription factor binding site has been conserved during evolution and is functionally significant [11]. Gerstein et al. (2010) and Riedel et al. (2013) used the technique of chromatin immunoprecipitation followed by DNA sequencing to identify in vivo binding sites for DAF-16 (G. Ruvkun and C. Riedel, personal communication) [41], [42]. These studies independently demonstrated that DAF-16 binding is significantly enriched at the natc-1 locus, consistent with the hypothesis that DAF-16 occupies the predicted binding site.
Here we demonstrated that natc-1 is transcriptionally repressed by DAF-16. Worms cultured in standard laboratory conditions with abundant food have high activity of the DAF-2 insulin/IGF-1 receptor and low activity of DAF-16. By contrast, daf-2 loss-of-function mutant animals have high activity of DAF-16 and display enhanced stress resistance and extended longevity. daf-2(lf) mutant animals displayed reduced levels of natc-1 transcripts, and natc-1 transcripts were restored to WT levels in daf-16(lf);daf-2(lf) double mutant animals. These results suggest that daf-16 activity reduces the level of natc-1 transcripts, and daf-2 activity increases the level of natc-1 transcripts by negatively regulating daf-16. Lee et al. (2003) used the technique of Northern blotting to examine natc-1 transcript levels and did not detect a substantial difference between wild-type and daf-2(e1370) mutant animals [11]. By contrast, Riedel et al. (2013) used the technique of high-throughput sequencing of mRNA (mRNA-Seq) [42] and detected a significant decrease (∼2-fold) in natc-1 mRNA levels in daf-2(e1370) animals compared to wild-type animals (G. Ruvkun and C. Riedel, personal communication). A possible explanation of these findings is that the techniques qRT-PCR and RNA-Seq, which gave similar results, are more quantitative than Northern blotting and were able to detect a small but statistically significant difference in transcript levels. Based on the presence of an evolutionarily conserved DAF-16 binding site in the natc-1 promoter, the in vivo binding of DAF-16 to the natc-1 locus, and our observed alterations in natc-1 transcript levels caused by mutations in daf-2 and daf-16, we propose the model that DAF-16 is a direct transcriptional repressor of natc-1 (Figure 8B).
Our genetic analysis showed that natc-1 inhibits stress resistance and dauer formation and genetically interacts with the insulin/IGF-1 signaling pathway, suggesting that natc-1 is a physiologically relevant target of DAF-16. These results do not exclude the possibility that natc-1 might be regulated by an additional pathway(s) in parallel to the insulin/IGF-1 pathway; indeed, the natc-1 promoter has been reported to interact with multiple transcription factors such as PQM-1, SKN-1, and PHA-4, suggesting there are additional regulatory control mechanisms [41], [53]. natc-1 mutant animals were discovered in an unbiased forward genetic screen for resistance to the stress of high dietary zinc. Further analysis revealed that natc-1(lf) mutations cause resistance to a wide range of stressors, such as heat, oxidation, and multiple metals, similar to daf-2(lf) mutations. daf-2 functions upstream of daf-16 in a signaling pathway, and daf-16(lf) mutations are epistatic to daf-2(lf) mutations. By contrast, our model that natc-1 functions downstream of daf-16 predicts that the stress resistance caused by natc-1(lf) mutations is epistatic to daf-16(lf) mutations. Indeed, we observed that the resistance to heat and high levels of dietary zinc caused by natc-1(lf) mutations was epistatic to daf-16(lf) mutations. Thus, both molecular and genetic studies support the model that natc-1 is directly targeted by DAF-16 to promote stress resistance. Consistent with this model, we demonstrated that natc-1 inhibits dauer formation, since natc-1(lf) mutations enhanced the dauer constitutive phenotype of daf-2(lf) mutations.
Although a variety of DAF-16 target genes have been demonstrated to interact genetically with the insulin/IGF-1 signaling pathway, these target genes are primarily activated by daf-16, and loss-of-function of these target genes impairs the stress resistance mediated by daf-16 activity. For example DAF-16 promotes transcription of the superoxide dismutase sod-3 [5]. sod-3 functions to promote resistance to oxidative stress [54]. Thus, sod-3 represents a class of DAF-16 targets that are transcriptionally activated by DAF-16 and promote stress resistance (Figure 8A). To our knowledge, our results with natc-1 are the first demonstration of a direct DAF-16 target gene that is repressed by DAF-16 to promote stress resistance (Figure 8A). Thus, these studies make a novel contribution to the understanding of how the activity of the insulin/IGF-1 signaling pathway mediates stress resistance.
In addition to stress resistance, DAF-16 also promotes longevity [55]. Because mutations in natc-1 reduce longevity, DAF-16 likely regulates lifespan through a natc-1-independent mechanism.
The NatC complex functions in determining the balance between reproductive growth and stress resistance
N-terminal acetyltransferases (NATs) are multi-subunit enzymes that catalyze the transfer of the acetyl group of acetyl coenzyme A to the α-amino group of the first amino acid of a target protein. N-terminal acetylation is a widespread modification that affects the majority of eukaryotic proteins; for example, ∼80–85% of human proteins are N-terminally acetylated [56]. Eukaryotes possess multiple NAT complexes (NatA-NatE) that vary in subunit composition and substrate specificity [57]. NAT activity is relevant to human health, since mutations in a human NAT gene are associated with Ogden syndrome, an X-linked disorder that is lethal in infancy [58]. Whereas the biochemical activity of NAT enzymes is well characterized, the functional roles of these enzymes are only beginning to be explored.
The NatC complex is comprised of the catalytic subunit Naa30 and the auxiliary subunits Naa35 and Naa38, and it acetylates proteins with the N-terminal sequences Met-Leu, Met-Phe, Met-Ile, and Met-Trp [39], [40], [44], [59], [60]. Genetic studies in yeast revealed NatC complex subunits are necessary for dsRNA virus particle assembly and WT growth rate in media lacking a fermentable carbon source [61], [62]. The Arabidopsis thaliana Naa30 homolog was identified in a screen for photosynthesis altered mutants, and Naa30 mutants display decreased chloroplast density and slow growth [63]. The zebrafish Naa35 homolog is the embryonic growth-associated protein (EGAP), and knock down studies with morpholinos suggest that it is necessary for WT growth and development [64]. The rat Naa35 homolog was discovered based on increased expression in healing corneal epithelium, and it is highly expressed in developing rat cornea and skin [65]. In human cells, reducing the levels of NatC complex subunits causes reduced cell viability and p53-dependent apoptosis [66]. These results suggest that the NatC complex functions to promote growth and development in a wide range of organisms.
Our studies of C. elegans natc-1 make a unique contribution to understanding the biological function of NatC, since these results are the first molecular and genetic characterization of an animal with a NatC subunit mutation. Genetic studies demonstrated that mutations in natc-1 increased resistance to multiple environmental stressors including excess metal, heat, and oxidation. These results suggest that natc-1 activity reduces stress resistance. Environments with low food, high population density, and high temperatures promote formation of dauer larvae. Dauers are a stress resistant form that allows animals in unfavorable environmental conditions to suspend development, resist environmental stresses, and be prepared to resume reproductive development when conditions improve [1]. Dauer formation is mediated by the insulin/IGF-1 signaling pathway [1]. The capacity to form dauer larvae highlights the importance of maintaining a balance between promoting growth and reproduction and surviving environmental stressors. In a sensitized genetic background, natc-1 activity inhibited dauer formation. Because a developmental switch mediates the decision between dauer larvae and larvae destined for reproductive development, these findings indicate that natc-1 activity promotes a development fate characterized by growth and reproduction. These findings identify novel phenotypes associated with a subunit of the NatC complex.
To determine how the NatC complex might execute these newly discovered functionalities, we bioinformatically identified predicted NatC target proteins in C. elegans. NatC might modulate processes such as stress resistance and dauer formation by acetylating groups of proteins with similar functionalities. To investigate this hypothesis, we performed a gene ontology (GO) analysis. Predicted NatC target proteins in C. elegans were enriched for 20 functional classes. To determine if NatC has an evolutionarily conserved preference for these functional classes, we used bioinformatics to predict NatC target proteins in humans. GO analyses of human NatC target proteins identified 11 functional classes that were enriched. Electron carrier and oxidoreductase activity were enriched in C. elegans and human GO analyses suggesting that the NatC complex may regulate a subset of proteins associated with these functionalities (Table S1). We further determined if predicted NatC target proteins were enriched for a specific cellular localization. GO analysis of NatC target proteins identified 4 cellular component terms enriched in C. elegans and 18 enriched in humans. NatC target proteins were enriched for mitochondrial localization in both C. elegans and humans (Table S2). These conserved functionalities and cellular components may inform future experimental efforts aimed at understanding how the NatC complex regulates physiology such as dauer entry and stress tolerance.
To identify protein targets of the NatC complex that might contribute to the mutant phenotype by having altered acetylation in natc-1(lf) mutant animals, we identified predicted protein targets that are implicated in zinc metabolism and stress resistance. Published genes implicated in C. elegans zinc metabolism include the cation diffusion facilitator (CDF) zinc transporters cdf-1, sur-7, cdf-2, and ttm-1 [67]–[70], the metallothioneins mtl-1 and mtl-2 [71], and haly-1 [37]. Of these seven genes, only cdf-2 is a putative target of the NatC complex. Mutations in cdf-2 affect zinc accumulation [69], whereas natc-1(lf) mutations do not alter zinc accumulation, suggesting that cdf-2 is not a critical target of the NatC complex in zinc resistance. Rather, we hypothesize that natc-1 mutations cause high zinc resistance by triggering mechanisms that allow animals to ameliorate the toxicity of high levels of zinc. This hypothesis is supported by the observation that natc-1 mutations cause resistance to a wide variety of stressors. Genes implicated in stress resistance include those encoding proteins involved in reactive oxygen species metabolism and dauer formation. The NatC complex is predicted to target several proteins that met these criteria; sod-1, sod-2, and sod-3 encode superoxide dismutases that increase oxidative stress resistance [54], mev-1 encodes cytochrome b, a subunit of the mitochondrial respiratory chain complex II, and frh-1 encodes a frataxin ortholog that promotes the oxidative stress response [72], [73]. Several predicted protein targets are encoded by genes that influence dauer formation; tph-1 encodes a tryptophan hydroxylase, daf-36 encodes an oxygenase, and cyp-35a3 encodes one of 42 cytochrome P450 proteins predicted to be targeted by the NatC complex [17], [74], [75]. Previous studies of stress responses and dauer formation have largely focused on the importance of transcriptional regulation. Our work suggests posttranslational modifications like N-terminal acetylation might also play an important role, and future proteomic analyses may help identify key effectors of stress tolerance and dauer formation.
Our findings are relevant to a general principle that organisms must balance growth and reproduction, which is facilitated by nutrient-rich environments, with stress resistance and quiescence, which are adaptive in nutrient poor and/or high stress environments. Plants that evolved in low-resource, stressful environments share a common set of traits, including relatively low rates of growth, photosynthesis, tissue turnover, and nutrient absorption [76]–[78]. This has been named the “stress resistance syndrome” (SRS), since these plants are resistant to a wide spectrum of physiologic stressors [79]. SRS may represent an adaptive strategy for coping with harsh environmental conditions [76], and this process has interesting analogies to dauer formation in C. elegans. Both dauer formation and SRS represent organisms balancing growth and stress tolerance to promote survival. The NatC complex may have an evolutionarily conserved role in mediating this balance between growth and stress tolerance. Consistent with this hypothesis, loss-of-function of the catalytic subunit of the Arabidopsis NatC complex causes decreased photosynthetic activity [63], an SRS trait. Additionally, our genetic studies demonstrate that loss-of-function of natc-1 promotes dauer formation and inhibits reproductive development, a trait analogous to SRS. Therefore, we propose that the NatC complex may have evolutionarily conserved functions in maintaining the balance between promoting growth and reproduction and resisting stressful environmental conditions.
Regulation of natc-1
Given that NatC functions to mediate the balance between growth and stress resistance, which is finely tuned by environmental conditions, it is important to determine how the activity of NatC is regulated. However, little is known about the regulation of these enzymes. In yeast, the natc-1 homolog (MAK10) protein levels are glucose repressible, but it was not established whether regulation occurs at the level of RNA or protein [61]. Here we demonstrated that natc-1 is negatively regulated at the level of transcription by DAF-16. The insulin/IGF-1 signaling pathway responds to environmental cues, such as nutrient availability, temperature, and dauer pheromone, by regulating the activity of DAF-16. Therefore, our findings establish a direct link between environmental sensing mediated by the insulin/IGF-1 signaling pathway and protein N-terminal acetylation mediated by NatC. These results make a new contribution to understanding NatC regulation in several ways. First, NAT subunits have not previously been reported to be regulated at the level of transcription. Second, this is a novel demonstration that a NAT complex is regulated by the insulin/IGF-1 signaling pathway, linking an environmental sensing pathway to the regulation of protein N-terminal acetylation (Figure 8A,B). NatC complexes appear to have an evolutionarily conserved role in modulating growth and stress resistance, and our findings suggest that they may also have a conserved role in responding to the insulin/IGF-1 signaling pathway.
Mechanisms of specific and broad-spectrum stress resistance
We have molecularly characterized two genes identified by screening for mutant animals that display increased tolerance to excess dietary zinc [36]: natc-1 and haly-1 [37]. These are the only genes that have been demonstrated to cause resistance to excess dietary zinc in an animal, and mutations in these two genes appear to act by very different mechanisms. First, these genes encode proteins with distinct functions. haly-1 encodes an enzyme that metabolizes histidine, and haly-1 mutant animals display increased levels of histidine. natc-1 encodes a subunit of a protein N-terminal acetylation complex, suggesting that protein N-terminal acetylation of many proteins is altered in these mutant animals, although this prediction has not been biochemically tested. Second, the spectrum of stress resistance caused by mutations in these two genes is distinct. Increased histidine appears to chelate and detoxify excess zinc and nickel, but the effect is quite specific since haly-1 mutant animals are not resistant to the toxicity caused by other metals [37]. By contrast, here we demonstrated that natc-1 mutations cause broad-spectrum stress resistance, including resistance to multiple metals, heat, and oxidation. Consistent with the model that these genes function by distinct mechanisms, the resistance to excess zinc toxicity caused by mutations of natc-1 and haly-1 was additive.
These findings raise a general question about stress resistance; how does a mutation in a single gene such as natc-1 promote resistance to a broad-spectrum of stresses? (1) One possibility is that the single-gene mutation results in a cascade of events that changes the activity of many proteins in the cell. In this model, each specific change in activity might mediate resistance to only one or a small number of stressors. For example, changes in haly-1 activity only mediate resistance to zinc and nickel. However, the combination of many different changes in activity could mediate broad-spectrum resistance. This is an attractive model for daf-2 mutant animals, which display broad-spectrum stress resistance and are documented to have changes in the expression of many genes as a result of the regulation of the DAF-16 transcription factor. This model is also attractive for natc-1, since this enzyme is predicted to mediate the N-terminal acetylation of many different proteins. (2) An alternative model is that diverse environmental stresses converge on a single type of important molecular damage. For example, heat, oxidation, and excess metals may all cause toxicity as a result of similar damage, such as protein unfolding. In this model, changing the activity of a single gene might confer broad-spectrum stress resistance by enhancing tolerance to the major form of cellular damage. These two basic models represent extremes of a continuum, and are not mutually exclusive. Our results document that resistance to high zinc toxicity can be increased by mutations that cause specific or broad-spectrum stress resistance, contributing to a conceptual framework for understanding stress resistance.
Materials and Methods
General methods and strains
C. elegans strains were cultured at 20°C on nematode growth medium (NGM) seeded with E. coli OP50 unless otherwise noted [80]. The wild-type C. elegans strain and parent of all mutant strains was Bristol N2. The following mutations were used: daf-16(mu86) [7] is a strong loss-of-function or null mutation of the DAF-16 forkhead transcription factor; daf-2(e1370) [6] is a partial loss-of-function mutation of the DAF-2 insulin/IGF-1 receptor; haly-1(am132) [37] is a strong loss-of-function or null mutation of the HALY-1 histidine ammonia lyase. natc-1(am134) and natc-1(am138) were identified in a genetic screen for resistance to high zinc toxicity [36], backcrossed four times to wild type, and are described here; natc-1(ok2062) was obtained from the C. elegans knockout consortium [43] and backcrossed four times to wild type. The back crossing procedure replaced ∼94% of the genome of mutant strains with wild-type DNA that has not been exposed to chemical mutagenesis, minimizing background mutations. Double mutant animals were generated by standard methods, and genotypes were confirmed by PCR or DNA sequencing.
Metal resistance assays
Hermaphrodites were cultured on NGM, and one embryo was transferred to a 35×10 mm Petri dish containing NAMM [36] supplemented with zinc sulfate (ZnSO4), cadmium chloride (CdCl2), nickel chloride (NiCl2), or copper chloride (CuCl2) and 5× concentrated E. coli OP50 as a food source. Dishes were analyzed daily until progeny were observed or the animal died, except Figure S3 where dishes were analyzed only until day 6. Animals that generated one or more live progeny were scored as “fertile adults.” To determine the metal concentration for these assays, we generated dose response curves of fertility for each metal using wild-type animals and selected the concentration that caused ∼50% of wild-type animals to fail to display fertility (Figure 4).
Determination of zinc content via inductively coupled plasma mass spectrometry
Large populations of animals were cultured on NAMM supplemented with 0 or 200 µM zinc sulfate (ZnSO4). The animals were desiccated to determine dry weight, and total zinc content was determined by ICP-MS as described by Murphy et al. (2011) [37].
Imaging of NATC-1::GFP fusion protein
Live transgenic animals were immobilized using levamisole in phosphate buffered saline (PBS) and mounted onto a thin pad of ∼7.5% agarose. More than 100 transgenic animals were analyzed, and representative images are presented. All images were captured on a PerkinElmer spinning disk confocal microscope utilizing Volocity imaging software.
Heat stress, oxidative stress, and lifespan assays
Gravid adult hermaphrodites were bleached to obtain embryos. Embryos were allowed to hatch in M9 buffer to synchronize at the L1 stage and cultured at 15°C on NGM. For heat stress assays, animals were shifted to 35°C on day 1 of adulthood. Animals were analyzed hourly for spontaneous or provoked motility and pharyngeal pumping; animals displaying none of these traits were scored as dead. Animals were briefly exposed to room temperature (24–25°C) for scoring. For the experiment shown in Figure 7A, animals were cultured continually at 35°C until hourly scoring began at 12 hours; summary statistics were not calculated in this case because some data points were not collected.
For oxidative stress assays, day 3 adults were transferred to NGM dishes supplemented with 40 mM methyl viologen dichloride hydrate (paraquat), fed E. coli OP50 and cultured at 20°C. We analyzed day 3 adults to avoid the high frequency of matricidal hatching in response to oxidative stress displayed by younger adults. Animals were scored every 12 hours for survival.
For lifespan assays, L4 animals were cultured on NGM at 20°C (defined as day 0) and fed E. coli OP50. Adult hermaphrodites were transferred to fresh Petri dishes every day until the cessation of progeny production and analyzed every day for survival.
In heat stress, oxidative stress and lifespan assays, animals that displayed matricidal hatching or a vulval-bursting phenotype were omitted from the analysis.
Dauer formation assays
To analyze dauer formation, we transferred embryos to NGM with E. coli OP50 at 15–25°C until adult animals began to lay embryos, approximately 3–5 days depending on the temperature. Animals were scored as dauer or non-dauer using a dissecting microscope based on the visible radial constriction phenotype [81]. To determine the effect of zinc, we conducted this assay on NAMM supplemented with zinc sulfate (ZnSO4).
To analyze genetic regulation of dauer formation, we performed feeding RNAi as described by Kamath et al. (2001) with minor modifications [82]. Briefly, daf-2(e1370) P0 hermaphrodites and F1 progeny were incubated at 20°C and continuously fed RNAi expressing bacteria. F1 progeny were scored as dauer or non-dauer after approximately 4 days. We used the empty vector control (L4440) and clones encoding dsRNA corresponding to T23B12.4 (natc-1) and B0238.10 (natc-2) from the Ahringer RNAi Library [83]. The DNA sequence of each clone was confirmed by standard methods.
Plasmid generation and transgenic rescue
Plasmid pJM5 is pBlueScript SK+ (Stratagene) containing a 3,356 base pair fragment of C. elegans genomic DNA from fosmid WRM067bF02 that extends 139 base pairs upstream of the predicted natc-1 start codon and 395 base pairs downstream of the predicted stop codon. To generate pJM6, we modified pJM5 by digestion with BstEII (New England Biolabs) and religation resulting in a 382 base pair deletion that removes parts of exons 2 and 3 and all of intron 2. The resulting pJM6 mutant open reading frame is predicted to truncate at amino acid 33 in a premature stop codon (TAG). To generate the Pnatc-1::NATC-1::GFP::unc-54 3′UTR translational fusion protein construct (pJM8), we inserted the natc-1 genomic locus (without the stop codon) into pBlueScript SK+ with the GFP coding sequence and the unc-54 3′ UTR. The DNA sequence of each plasmid was confirmed by standard methods.
Transgenic animals were generated by injecting natc-1(am134) hermaphrodites with pJM5 or pJM6, and natc-1(am138) hermaphrodites with pJM8. All injections were done with the dominant Rol marker pRF4 [84]. We selected independently derived Rol self progeny that transmitted the Rol phenotype. These transgenes formed extrachromosomal arrays, since the Rol phenotype was transmitted to only a sub-set of the self-progeny. To analyze transgenic rescue of the natc-1(am134) or natc-1(am138) resistance to high zinc toxicity phenotype, we calculated the fraction of transgenic animals on baseline and high zinc concentrations. nonRol animals were presumed to lack the extrachromosomal array and were thus non-transgenic. We defined rescue as a significant decrease in percentage of transgenic animals able to survive (and thus be quantified) on toxic zinc conditions (300 µM supplemental zinc) compared to the baseline zinc concentration (0 µM supplemental zinc), as described by Murphy et al. (2011) [37].
Bioinformatic analysis of targets of the NatC complex
To identify protein targets of the NatC complex, we wrote a custom Perl script that computationally identified predicted NatC targets in C. elegans and human protein databases from the National Center for Biotechnology Information (NCBI). Gene ontology (GO) analysis was performed using GOrilla [85] by comparing predicted NatC targets to the entire proteome for each species. We report significant GO functional terms (p<0.001) according to Eden et al. (2009) [85].
natc-1 mRNA analyses
Three natc-1 cDNA clones (EST) were obtained from the National Institutes of Genetics, Japan (yk194g4, yk262c3, and yk420a1). We determined the complete sequence of these cDNAs using standard techniques. These data were used to infer the mRNA sequence from exon 3 to the polyA tail attached 330 nucleotides downstream of the TGA stop codon. To experimentally define the 5′ end of the natc-1 mRNA, we used 5′ RACE System V2.0 (Invitrogen) according to the manufacturer's instructions. These data were used to infer the natc-1 mRNA sequence from the 22 nucleotide splice leader 1 (SL1) sequence that begins 14 base pairs upstream of the start codon to exon 3.
To generate synchronous populations of worms for RNA extraction, we treated gravid adult hermaphrodite animals with a mixture of bleach and 4M sodium hydroxide (NaOH) and cultured embryos overnight in M9 solution at 20°C, resulting in L1 stage arrest. L1 larvae were transferred to NGM plates at 20°C, fed E. coli OP50, and allowed to develop to the L4 stage (approximately 2 days). RNA isolation and cDNA synthesis were performed as described by Davis et al. (2009) [86]. Quantitative, real-time PCR was performed using an Applied Biosystems 7900HT Fast Real-Time PCR system and the Applied Biosystems SYBR Green Master Mix. mRNA fold change was calculated using the comparative CT method [87]. Forward and reverse amplification primers were: rps-23 5′ - aaggctcacattggaactcg and 5′ - aggctgcttagcttcgacac; mtl-1 5′-ggcttgcaagtgtgactgc and 5′-cctcacagcagtacttctcac; natc-1 5′-tcagctttacgggtccaatg and 5′-ccgaaaatgctctgtggttac; daf-16 5′ - gacggaaggcttaaactcaatg and 5′ - gagacagattgtgacggatcg.
Statistical analysis
All data were analyzed utilizing the two-tailed students t-test of samples with unequal variance unless otherwise specified. For binary data such as dauer entry and fertility, the Chi-squared test was utilized. P-values less than 0.05 were considered statistically significant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. HuPJ (2007) Dauer. WormBook 1–19 doi:10.1895/wormbook.1.144.1
2. BraeckmanBP, HouthoofdK, VanfleterenJR (2001) Insulin-like signaling, metabolism, stress resistance and aging in Caenorhabditis elegans. Mech Ageing Dev 122 : 673–693.
3. KimuraKD, TissenbaumHA, LiuY, RuvkunG (1997) daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 277 : 942–946 doi:10.1126/science.277.5328.942
4. MorrisJZ, TissenbaumHA, RuvkunG (1996) A phosphatidylinositol-3-OH kinase family member regulating longevity and diapause in Caenorhabditis elegans. Nature 382 : 536–539 doi:10.1038/382536a0
5. HondaY, HondaS (1999) The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. FASEB J 13 : 1385–1393.
6. RiddleDL, SwansonMM, AlbertPS (1981) Interacting genes in nematode dauer larva formation. Nature 290 : 668–671.
7. LinK, DormanJB, RodanA, KenyonC (1997) daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science 278 : 1319–1322.
8. OggS, ParadisS, GottliebS, PattersonGI, LeeL, et al. (1997) The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 389 : 994–999 doi:10.1038/40194
9. MurphyCT (2006) The search for DAF-16/FOXO transcriptional targets: Approaches and discoveries. Exp Gerontol 41 : 910–921 doi:10.1016/j.exger.2006.06.040
10. YuH, LarsenPL (2001) DAF-16-dependent and independent expression targets of DAF-2 insulin receptor-like pathway in Caenorhabditis elegans include FKBPs. Journal of Molecular Biology 314 : 1017–1028 doi:10.1006/jmbi.2000.5210
11. LeeSS, KennedyS, TolonenAC, RuvkunG (2003) DAF-16 target genes that control C. elegans life-span and metabolism. Science 300 : 644–647 doi:10.1126/science.1083614
12. OokumaS, FukudaM, NishidaE (2003) Identification of a DAF-16 transcriptional target gene, scl-1, that regulates longevity and stress resistance in Caenorhabditis elegans. Curr Biol 13 : 427–431.
13. McElweeJ, BubbK, ThomasJH (2003) Transcriptional outputs of the Caenorhabditis elegans forkhead protein DAF-16. Aging Cell 2 : 111–121.
14. MurphyCT, McCarrollSA, BargmannCI, FraserA, KamathRS, et al. (2003) Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424 : 277–283 doi:10.1038/nature01789
15. Wook OhS, MukhopadhyayA, DixitBL, RahaT, GreenMR, et al. (2005) Identification of direct DAF-16 targets controlling longevity, metabolism and diapause by chromatin immunoprecipitation. Nat Genet 38 : 251–257 doi:10.1038/ng1723
16. YuRX, LiuJ, TrueN, WangW (2008) Identification of Direct Target Genes Using Joint Sequence and Expression Likelihood with Application to DAF-16. PLoS ONE 3: e1821 doi:10.1371/journal.pone.0001821.s008
17. JensenVL, SimonsenKT, LeeY-H, ParkD, RiddleDL (2010) RNAi screen of DAF-16/FOXO target genes in C. elegans links pathogenesis and dauer formation. PLoS ONE 5: e15902 doi:10.1371/journal.pone.0015902
18. FinchCE, RuvkunG (2001) The genetics of aging. Annual review of genomics and human genetics 2 : 435–462.
19. GarofaloRS (2002) Genetic analysis of insulin signaling in Drosophila. Trends Endocrinol Metab 13 : 156–162.
20. NakaeJ, BiggsWH, KitamuraT, CaveneeWK, WrightCVE, et al. (2002) Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat Genet 32 : 245–253 doi:10.1038/ng890
21. HolzenbergerM, DupontJ, DucosB, LeneuveP, GéloënA, et al. (2003) IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 421 : 182–187 doi:10.1038/nature01298
22. BlüherM, KahnBB, KahnCR (2003) Extended longevity in mice lacking the insulin receptor in adipose tissue. Science 299 : 572–574 doi:10.1126/science.1078223
23. YamasakiS, Sakata-SogawaK, HasegawaA, SuzukiT, KabuK, et al. (2007) Zinc is a novel intracellular second messenger. The Journal of Cell Biology 177 : 637–645 doi:10.1083/jcb.200702081
24. MurakamiM, HiranoT (2008) Intracellular zinc homeostasis and zinc signaling. Cancer Science 99 : 1515–1522 doi:10.1111/j.1349-7006.2008.00854.x
25. ValleeBL, FalchukKH (1993) The biochemical basis of zinc physiology. Physiol Rev 73 : 79–118.
26. HambidgeM (2000) Human zinc deficiency. J Nutr 130 : 1344S–9S.
27. ChowanadisaiW, KelleherSL, LönnerdalB (2005) Zinc deficiency is associated with increased brain zinc import and LIV-1 expression and decreased ZnT-1 expression in neonatal rats. J Nutr 135 : 1002–1007.
28. HambidgeKM, KrebsNF (2007) Zinc deficiency: a special challenge. J Nutr 137 : 1101–1105.
29. MaverakisE, FungMA, LynchPJ, DrazninM, MichaelDJ, et al. (2007) Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol 56 : 116–124 doi:10.1016/j.jaad.2006.08.015
30. KohJY, SuhSW, GwagBJ, HeYY, HsuCY, et al. (1996) The role of zinc in selective neuronal death after transient global cerebral ischemia. Science 272 : 1013–1016.
31. NiesDH (2007) Biochemistry. How cells control zinc homeostasis. Science 317 : 1695–1696 doi:10.1126/science.1149048
32. FinneyLA, O'HalloranTV (2003) Transition metal speciation in the cell: insights from the chemistry of metal ion receptors. Science 300 : 931–936 doi:10.1126/science.1085049
33. PophamJD, WebsterJM (1979) Cadmium toxicity in the free-living nematode, Caenorhabditis elegans. Environ Res 20 : 183–191.
34. WangD, WangY (2008) Nickel sulfate induces numerous defects in Caenorhabditis elegans that can also be transferred to progeny. Environ Pollut 151 : 585–592 doi:10.1016/j.envpol.2007.04.003
35. BarsyteD, LovejoyDA, LithgowGJ (2001) Longevity and heavy metal resistance in daf-2 and age-1 long-lived mutants of Caenorhabditis elegans. FASEB J 15 : 627–634 doi:10.1096/fj.99-0966com
36. BruinsmaJJ, SchneiderDL, DavisDE, KornfeldK (2008) Identification of Mutations in Caenorhabditis elegans That Cause Resistance to High Levels of Dietary Zinc and Analysis Using a Genomewide Map of Single Nucleotide Polymorphisms Scored by Pyrosequencing. Genetics 179 : 811–828 doi:10.1534/genetics.107.084384
37. MurphyJT, BruinsmaJJ, SchneiderDL, CollierS, GuthrieJ, et al. (2011) Histidine Protects Against Zinc and Nickel Toxicity in Caenorhabditis elegans. PLoS Genet 7: e1002013 doi:10.1371/journal.pgen.1002013.t001
38. LithgowGJ, WhiteTM, MelovS, JohnsonTE (1995) Thermotolerance and extended life-span conferred by single-gene mutations and induced by thermal stress. Proc Natl Acad Sci USA 92 : 7540–7544.
39. PolevodaB, NorbeckJ, TakakuraH, BlombergA, ShermanF (1999) Identification and specificities of N-terminal acetyltransferases from Saccharomyces cerevisiae. EMBO J 18 : 6155–6168 doi:10.1093/emboj/18.21.6155
40. PolevodaB, ShermanF (2000) Nalpha -terminal acetylation of eukaryotic proteins. J Biol Chem 275 : 36479–36482 doi:10.1074/jbc.R000023200
41. GersteinMB, LuZJ, Van NostrandEL, ChengC, ArshinoffBI, et al. (2010) Integrative Analysis of the Caenorhabditis elegans Genome by the modENCODE Project. Science 330 : 1775–1787 doi:10.1126/science.1196914
42. RiedelCG, DowenRH, LourencoGF, KirienkoNV, HeimbucherT, et al. (2013) DAF-16 employs the chromatin remodeller SWI/SNF to promote stress resistance and longevity. Nat Cell Biol 15 : 491–501 doi:10.1038/ncb2720
43. MoermanDG, BarsteadRJ (2008) Towards a mutation in every gene in Caenorhabditis elegans. Brief Funct Genomic Proteomic 7 : 195–204 doi:10.1093/bfgp/eln016
44. PolevodaB, ShermanF (2001) NatC Nalpha-terminal acetyltransferase of yeast contains three subunits, Mak3p, Mak10p, and Mak31p. J Biol Chem 276 : 20154–20159 doi:10.1074/jbc.M011440200
45. RuanJ, LiH, ChenZ, CoghlanA, CoinLJM, et al. (2008) TreeFam: 2008 Update. Nucleic Acids Res 36: D735–D740 doi:10.1093/nar/gkm1005
46. KahnNW, ReaSL, MoyleS, KellA, JohnsonTE (2008) Proteasomal dysfunction activates the transcription factor SKN-1 and produces a selective oxidative-stress response in Caenorhabditis elegans. Biochem J 409 : 205–213 doi:10.1042/BJ20070521
47. LinK, HsinH, LibinaN, KenyonC (2001) Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat Genet 28 : 139–145 doi:10.1038/88850
48. LeeRY, HenchJ, RuvkunG (2001) Regulation of C. elegans DAF-16 and its human ortholog FKHRL1 by the daf-2 insulin-like signaling pathway. Curr Biol 11 : 1950–1957.
49. MaloneEA, ThomasJH (1994) A screen for nonconditional dauer-constitutive mutations in Caenorhabditis elegans. Genetics 136 : 879–886.
50. KenyonC, ChangJ, GenschE, RudnerA, TabtiangR (1993) A C. elegans mutant that lives twice as long as wild type. Nature 366 : 461–464 doi:10.1038/366461a0
51. MurakamiS, JohnsonTE (1996) A genetic pathway conferring life extension and resistance to UV stress in Caenorhabditis elegans. Genetics 143 : 1207–1218.
52. MuñozMJ, RiddleDL (2003) Positive selection of Caenorhabditis elegans mutants with increased stress resistance and longevity. Genetics 163 : 171–180.
53. ZhongM, NiuW, LuZJ, SarovM, MurrayJI, et al. (2010) Genome-Wide Identification of Binding Sites Defines Distinct Functions for Caenorhabditis elegans PHA-4/FOXA in Development and Environmental Response. PLoS Genet 6: e1000848 doi:10.1371/journal.pgen.1000848.s017
54. HunterT, BannisterWH, HunterGJ (1997) Cloning, expression, and characterization of two manganese superoxide dismutases from Caenorhabditis elegans. J Biol Chem 272 : 28652–28659 doi:10.1074/jbc.272.45.28652
55. LibinaN, BermanJR, KenyonC (2003) Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell 115 : 489–502.
56. Van DammeP, HoleK, Pimenta-MarquesA, HelsensK, VandekerckhoveJ, et al. (2011) NatF Contributes to an Evolutionary Shift in Protein N-Terminal Acetylation and Is Important for Normal Chromosome Segregation. PLoS Genet 7: e1002169 doi:10.1371/journal.pgen.1002169.t002
57. StarheimKK, GevaertK, ArnesenT (2012) Protein N-terminal acetyltransferases: when the start matters. Trends Biochem Sci 37 : 152–161 doi:10.1016/j.tibs.2012.02.003
58. RopeAF, WangK, EvjenthR, XingJ, JohnstonJJ, et al. (2011) Using VAAST to identify an X-linked disorder resulting in lethality in male infants due to N-terminal acetyltransferase deficiency. Am J Hum Genet 89 : 28–43 doi:10.1016/j.ajhg.2011.05.017
59. UetzPP, GiotLL, CagneyGG, MansfieldTAT, JudsonRSR, et al. (2000) A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 403 : 623–627 doi:10.1038/35001009
60. RigautG, ShevchenkoA, RutzB, WilmM, MannM, et al. (1999) A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol 17 : 1030–1032 doi:10.1038/13732
61. LeeYJ, WicknerRB (1992) MAK10, a glucose-repressible gene necessary for replication of a dsRNA virus of Saccharomyces cerevisiae, has T cell receptor alpha-subunit motifs. Genetics 132 : 87–96.
62. TerceroJC, WicknerRB (1992) MAK3 encodes an N-acetyltransferase whose modification of the L-A gag NH2 terminus is necessary for virus particle assembly. J Biol Chem 267 : 20277–20281.
63. PesaresiP, GardnerNA, MasieroS, DietzmannA, EichackerL, et al. (2003) Cytoplasmic N-terminal protein acetylation is required for efficient photosynthesis in Arabidopsis. Plant Cell 15 : 1817–1832.
64. WenzlauJM, GarlPJ, SimpsonP, StenmarkKR, WestJ, et al. (2006) Embryonic growth-associated protein is one subunit of a novel N-terminal acetyltransferase complex essential for embryonic vascular development. Circ Res 98 : 846–855 doi:10.1161/01.RES.0000214539.86593.7a
65. YiXJ, LiXF, YuFS (2000) A novel epithelial wound-related gene is abundantly expressed in developing rat cornea and skin. Curr Eye Res 20 : 430–440.
66. StarheimKK, GromykoD, EvjenthR, RyningenA, VarhaugJE, et al. (2009) Knockdown of Human N -Terminal Acetyltransferase Complex C Leads to p53-Dependent Apoptosis and Aberrant Human Arl8b Localization. Mol Cell Biol 29 : 3569–3581 doi:10.1128/MCB.01909-08
67. BruinsmaJJ, JirakulapornT, MuslinAJ, KornfeldK (2002) Zinc ions and cation diffusion facilitator proteins regulate Ras-mediated signaling. Dev Cell 2 : 567–578.
68. YoderJH, ChongH, GuanK-L, HanM (2004) Modulation of KSR activity in Caenorhabditis elegans by Zn ions, PAR-1 kinase and PP2A phosphatase. EMBO J 23 : 111–119 doi:10.1038/sj.emboj.7600025
69. RohHC, CollierS, GuthrieJ, RobertsonJD, KornfeldK (2012) Lysosome-related organelles in intestinal cells are a zinc storage site in C. elegans. Cell Metab 15 : 88–99 doi:10.1016/j.cmet.2011.12.003
70. RohHC, CollierS, DeshmukhK, GuthrieJ, RobertsonJD, et al. (2013) ttm-1 Encodes CDF Transporters That Excrete Zinc from Intestinal Cells of C. elegans and Act in a Parallel Negative Feedback Circuit That Promotes Homeostasis. PLoS Genet 9: e1003522 doi:10.1371/journal.pgen.1003522.s004
71. Zeitoun-GhandourS, CharnockJM, HodsonME, LeszczyszynOI, BlindauerCA, et al. (2010) The two Caenorhabditis elegans metallothioneins (CeMT-1 and CeMT-2) discriminate between essential zinc and toxic cadmium. FEBS Journal 277 : 2531–2542 doi:10.1111/j.1742-4658.2010.07667.x
72. IshiiN, FujiiM, HartmanPS, TsudaM, YasudaK, et al. (1998) A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature 394 : 694–697 doi:10.1038/29331
73. Vázquez-ManriqueRP, González-CaboP, RosS, AzizH, BaylisHA, et al. (2006) Reduction of Caenorhabditis elegans frataxin increases sensitivity to oxidative stress, reduces lifespan, and causes lethality in a mitochondrial complex II mutant. The FASEB Journal 20 : 172–174 doi:10.1096/fj.05-4212fje
74. FielenbachN, AntebiA (2008) C. elegans dauer formation and the molecular basis of plasticity. Genes Dev 22 : 2149–2165 doi:10.1101/gad.1701508
75. RottiersV, MotolaDL, GerischB, CumminsCL, NishiwakiK, et al. (2006) Hormonal control of C. elegans dauer formation and life span by a Rieske-like oxygenase. Dev Cell 10 : 473–482 doi:10.1016/j.devcel.2006.02.008
76. Grime JP (1977) Evidence for the existence of three primary strategies in plants and its relevance to ecological and evolutionary theory. American Naturalist: 1169–1194.
77. ChapinFS (1980) The mineral nutrition of wild plants. Annual review of ecology and systematics 11 : 233–260.
78. BryantJP, ChapinFSIII, KleinDR (1983) Carbon/nutrient balance of boreal plants in relation to vertebrate herbivory. Oikos 357–368.
79. ChapinFSIII, AutumnK, PugnaireF (1993) Evolution of suites of traits in response to environmental stress. American Naturalist S78–S92.
80. BrennerS (1974) The genetics of Caenorhabditis elegans. Genetics 77 : 71–94.
81. CassadaRC, RussellRL (1975) The dauerlarva, a post-embryonic developmental variant of the nematode Caenorhabditis elegans. Dev Biol 46 : 326–342.
82. KamathRS, Martinez-CamposM, ZipperlenP, FraserAG, AhringerJ (2001) Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol 2: RESEARCH0002 doi:10.1186/gb-2000-2-1-research0002
83. KamathRS, AhringerJ (2003) Genome-wide RNAi screening in Caenorhabditis elegans. Methods 30 : 313–321.
84. MelloCC, KramerJM, StinchcombD, AmbrosV (1991) Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J 10 : 3959–3970.
85. EdenE, NavonR, SteinfeldI, LipsonD, YakhiniZ (2009) GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10 : 48 doi:10.1186/1471-2105-10-48
86. DavisDE, RohHC, DeshmukhK, BruinsmaJJ, SchneiderDL, et al. (2009) The Cation Diffusion Facilitator Gene cdf-2 Mediates Zinc Metabolism in Caenorhabditis elegans. Genetics 182 : 1015–1033 doi:10.1534/genetics.109.103614
87. SchmittgenTD, LivakKJ (2008) Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3 : 1101–1108 doi:10.1038/nprot.2008.73
88. GottliebS, RuvkunG (1994) daf-2, daf-16 and daf-23: genetically interacting genes controlling Dauer formation in Caenorhabditis elegans. Genetics 137 : 107–120.
89. HondaY, TanakaM, HondaS (2008) Modulation of longevity and diapause by redox regulation mechanisms under the insulin-like signaling control in Caenorhabditis elegans. Exp Gerontol 43 : 520–529 doi:10.1016/j.exger.2008.02.009
90. GemsD, SuttonAJ, SundermeyerML, AlbertPS, KingKV, et al. (1998) Two pleiotropic classes of daf-2 mutation affect larval arrest, adult behavior, reproduction and longevity in Caenorhabditis elegans. Genetics 150 : 129–155.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 10
Nejčtenější v tomto čísle
- The Master Activator of IncA/C Conjugative Plasmids Stimulates Genomic Islands and Multidrug Resistance Dissemination
- A Splice Mutation in the Gene Causes High Glycogen Content and Low Meat Quality in Pig Skeletal Muscle
- Keratin 76 Is Required for Tight Junction Function and Maintenance of the Skin Barrier
- A Role for Taiman in Insect Metamorphosis