
HIPPO Pathway Members Restrict SOX2 to the Inner Cell Mass Where It Promotes ICM Fates in the Mouse Blastocyst
Pluripotent stem cells can give rise to any cell type in the body, making them an attractive tool for regenerative medicine. Pluripotent stem cells can be derived from the mammalian embryo at the blastocyst stage or they can be created from mature adult cells by reprogramming. During reprogramming, SOX2 helps establish pluripotency, but it is not clear how SOX2 establishes pluripotency in the blastocyst. We evaluated where SOX2 is present, how SOX2 is regulated, and where SOX2 is active during blastocyst formation. Our data show that the roles and the regulation of SOX2 are unique compared to other pluripotency/reprogramming factors, such as OCT4 and NANOG. SOX2 marks pluripotent cells earlier than do other factors, but does not regulate pluripotency until several days later. Rather, the earlier role of SOX2 is to help establish the yolk sac lineage, which is essential for gestation.
Published in the journal:
. PLoS Genet 10(10): e32767. doi:10.1371/journal.pgen.1004618
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004618
Summary
Pluripotent stem cells can give rise to any cell type in the body, making them an attractive tool for regenerative medicine. Pluripotent stem cells can be derived from the mammalian embryo at the blastocyst stage or they can be created from mature adult cells by reprogramming. During reprogramming, SOX2 helps establish pluripotency, but it is not clear how SOX2 establishes pluripotency in the blastocyst. We evaluated where SOX2 is present, how SOX2 is regulated, and where SOX2 is active during blastocyst formation. Our data show that the roles and the regulation of SOX2 are unique compared to other pluripotency/reprogramming factors, such as OCT4 and NANOG. SOX2 marks pluripotent cells earlier than do other factors, but does not regulate pluripotency until several days later. Rather, the earlier role of SOX2 is to help establish the yolk sac lineage, which is essential for gestation.
Introduction
To create and use pluripotent stem cells, it is essential to understand the origins of pluripotency during normal development. During mouse blastocyst formation, pluripotent epiblast (EPI) cells are established by two cell fate decisions that segregate pluripotent progenitors from extraembryonic tissues [1], [2]. During the first cell fate decision, trophectoderm (TE) is segregated from inner cell mass (ICM) prior to blastocyst formation. During the second cell fate decision, the ICM is subdivided into EPI and primitive endoderm (PE) lineages after blastocyst formation. Recent studies have examined the roles and regulation of pluripotency genes, such as Oct4, Nanog, and Sox2, during establishment of EPI cells in the blastocyst [3]–[12], but aspects of the roles and regulation of Sox2 in the blastocyst are unresolved. For example, several studies reported that Sox2 is restricted to the ICM by the blastocyst stage [3], [13]–[15], but the molecular mechanisms regulating Sox2 expression in the blastocyst are unknown.
In addition to the unresolved mechanism by which Sox2 expression is patterned, the functional roles of Sox2 in the blastocyst are not yet clear. ES cells cannot be derived from embryos lacking zygotic (Z) Sox2 [5], indicating that Sox2 is essential for pluripotency. In ES cells, Sox2 is required for the expression of pluripotency genes, such as Oct4 and Nanog, and for the repression of TE genes [16]–[20]. Therefore, Sox2 might be required for initial expression of pluripotency genes and repression of TE genes in the ICM. However, the expression of pluripotency and TE genes in Sox2 Z null blastocysts has not yet been examined at the level of individual cells. Moreover, maternal (M) Sox2 is also thought to participate in blastocyst formation, which could partially compensate for loss of Z Sox2. RNAi knockdown of M and Z Sox2 in the zygote was reported to disrupt blastocyst formation [6]. However, RNAi knockdown embryos do not always phenocopy MZ null embryos [3], [21]. Because understanding the regulation and roles of SOX2 in the blastocyst is key to understanding the molecular regulation of preimplantation development and the establishment of pluripotency, we examined both the mechanisms that pattern SOX2, as well as the functional requirements for MZ Sox2 during development.
Results
SOX2 is restricted to ICM progenitors by HIPPO pathway members, and not by CDX2
Sox2 mRNA is enriched in ICM progenitors starting at the 16-cell stage [14], but the SOX2 protein expression pattern at this stage is unclear, as is the mechanism by which Sox2 is restricted to ICM progenitors. Using immunofluorescence and confocal microscopy, we observed that SOX2 is restricted to nuclei of ICM progenitors at the 16-cell stage and later (Fig. 1A; see Table S1 for wild-type embryo staging scheme). In morulae, a weaker signal was detected in the cytoplasm of outside cells, but this was also detected in embryos lacking MZ Sox2 (Fig. S1A), indicating that the cytoplasmic stain is non-specific. In the early blastocyst (E3.25–E3.5), SOX2 was detected in most ICM cells (Fig. 1A and Fig. S1B), and SOX2 did not colocalize with CDX2 in outside cells (n = 13 embryos; Fig. S1C). By contrast, NANOG and OCT4 are still detected in the TE at this stage (Fig. 1A and 2C) [22], [23]. Therefore SOX2 is a unique, early marker of ICM fate.
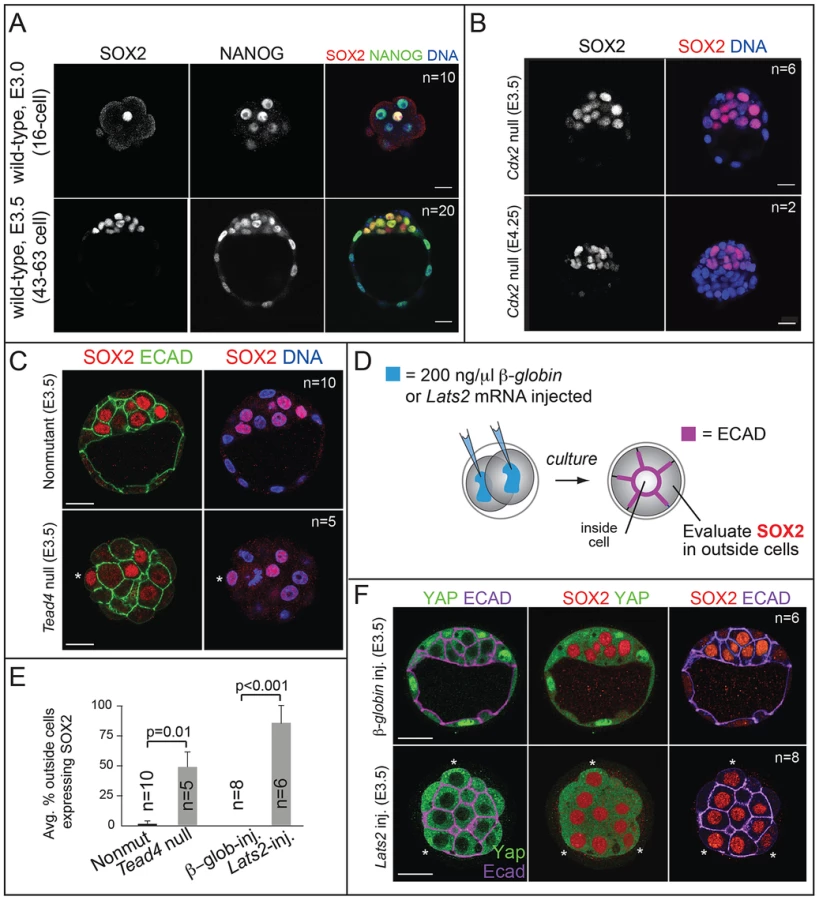
Next we examined the mechanism by which SOX2 expression is restricted to ICM. The TE-expressed transcription factor CDX2 restricts the expression of Oct4 and Nanog to the ICM by repressing Oct4 and Nanog expression in the TE after blastocyst formation [22]. We therefore asked whether CDX2 also restricts SOX2 to the ICM. Surprisingly, SOX2 remained restricted to the ICM in Cdx2 null embryos at early and late blastocyst stages (Fig. 1B), indicating that SOX2 expression is restricted to ICM progenitors through a Cdx2-independent mechanism. We therefore investigated whether the pathway that restricts CDX2 to the TE also restricts SOX2 to the ICM in parallel. We previously helped show that TEAD4 partners with YAP and WWTR1 to promote expression of CDX2 and GATA3 in TE cells [7], [24]–[26]. YAP and WWTR1 are localized to nuclei only in TE cells, where LATS kinase activity is lower [26]. We hypothesized that if TEAD4 regulates Sox2 in parallel to Cdx2, then we would detect ectopic SOX2 in the TE cells of Tead4 null embryos. To test this hypothesis, we examined SOX2 expression in Tead4 null embryos. Tead4 is essential for blastocyst formation, but not for polarization of TE cells [24], [25], enabling us to distinguish inside (apolar) and outside (polarized) cells in Tead4 null embryos. We observed that in Tead4 null embryos, ectopic SOX2 was detected in about half of outside cells, in contrast with control littermates, where SOX2 was not detected in outside cells (Fig. 1C, E). Thus, Tead4 is required to restrict SOX2 to ICM cells. This result is significant because although other pluripotency factors, such as OCT4 and NANOG are detected in outside cells of Tead4 null embryos when they die [24], [25], these factors are also detected in the outside cells of wild type embryos at this stage [22]. SOX2 is therefore the first pluripotency factor known to be restricted to ICM progenitors by TEAD4 and not by CDX2.
Finally, we misexpressed Lats2 mRNA in outside cells (Fig. 1D), which is sufficient to shift YAP and WWTR1 localization from nucleus to cytoplasm and downregulate CDX2 in TE cells [26]. As a control, we overexpressed β-Globin in a second group of embryos. As expected, overexpression of Lats2 disrupted nuclear YAP localization and led to ectopic SOX2 in outside cells, while ectopic of β-Globin did not (Fig. 1E, F). Therefore, LATS2 and TEAD4 regulate patterning of SOX2 and CDX2 in parallel, leading to the establishment of their complementary expression patterns in the blastocyst.
Maternal Sox2 is not required for development
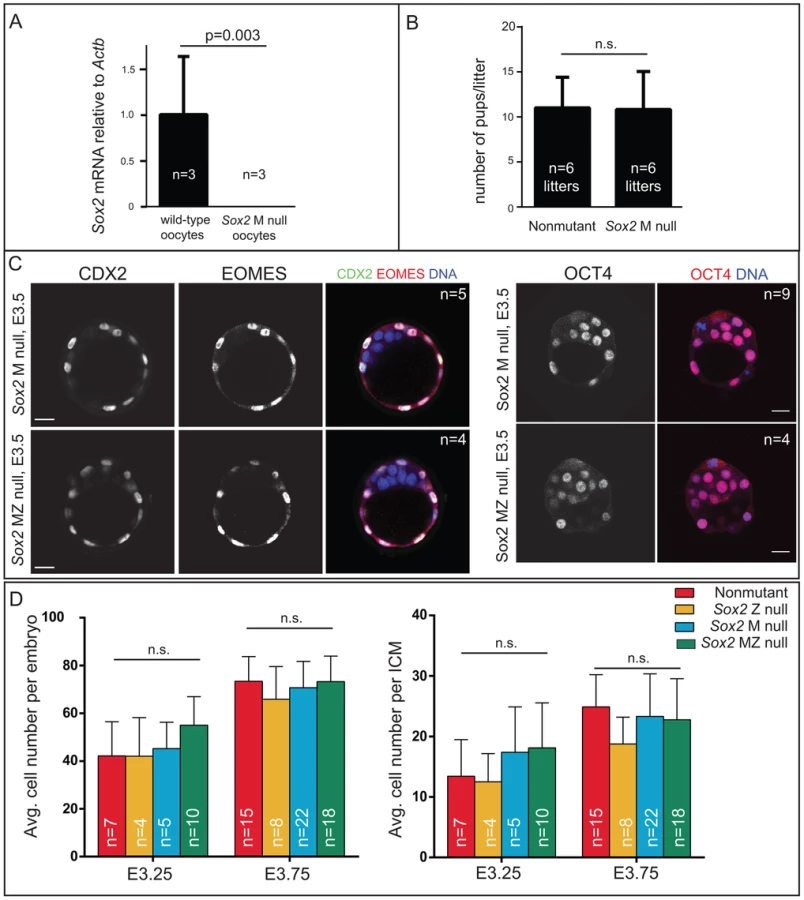
Our finding that SOX2 protein is restricted to ICM progenitors conflicted with prior reports suggesting that M SOX2 protein is present in TE cells and required for TE cell development [5], [6]. However, the requirement for MZ SOX2 in the TE has not been functionally evaluated using null alleles. We examined embryos lacking MZ Sox2 using a conditional Sox2 allele [27] and Zp3Cre, which is expressed in the female germ line [28]. We first confirmed that female germ line expression of Cre indeed deleted M Sox2 by quantitative RT-PCR (qPCR) analysis of oocytes from Zp3Cre; Sox2fl/fl or del females (Fig. 2A). Next, we mated these females to wild-type males to determine whether M Sox2 is required for development of their progeny. The number of offspring per litter did not differ significantly between Sox2 germ line-deleted females and control females (Fig. 2B), indicating that M Sox2 is not required for development, consistent with a recent report [29]. While variable levels of Sox2 mRNA have been detected in 1–2 cell embryos [14], we were unable to detect Sox2 mRNA or protein in wild type embryos at 1–2 cell stages, or in Sox2 Z null morulae (Fig. S2A), indicating that M SOX2 is neither present nor functional in the blastocyst.
We next examined whether loss of both M and Z Sox2 disrupts blastocyst formation by breeding Sox2 germ line-deleted females to males carrying a Sox2 null allele. Expression of TE (CDX2 and EOMES) and ICM (OCT4) markers was normal in the absence of MZ Sox2 (Fig. 2C), indicating that neither M nor Z Sox2 is required for TE specification or blastocyst formation, in contrast with the reported RNAi phenotype [6]. Moreover, quantification of the numbers of ICM and total cells in the blastocyst showed that there was no significant reduction in the average numbers of cells contributing to either lineage in the absence of MZ Sox2, compared to other genotypes (Fig. 2D). In ES cells, deletion of Sox2 leads to downregulation of Oct4 and upregulation of TE genes, including Eomes [16]. However, we did not detect ectopic expression of TE genes in ICM cells of Sox2 MZ null blastocysts at early (Fig. 2C) or late (Fig. S2B) time points. We therefore conclude that neither M nor Z Sox2 are required for segregation of ICM and TE lineages.
In the ICM, SOX2 is restricted to EPI cells by an FGF4/MEK-dependent mechanism
Our results indicated that Sox2 is not required for the first lineage decision, so we next asked if Sox2 is involved in the second lineage decision during mouse development: the subdivision of the ICM into EPI and PE cell fates. In the blastocyst at E3.75, EPI and PE cells are distributed in a salt-and-pepper fashion within the ICM. EPI cells express higher levels of NANOG, while PE cells express higher levels of SOX17, GATA6, PDGFRA, and GATA4 [4], [30]–[32]. In contrast, OCT4 is detected in both EPI and PE cells at this stage [4], [14], [23], [33]. It is not currently known whether SOX2 is restricted to EPI cells like NANOG, or if SOX2 is expressed throughout the ICM like OCT4. We therefore first examined the SOX2 pattern within the ICM in the E3.75 blastocyst.
Our analysis of early and late blastocysts indicated that the SOX2 expression pattern in the ICM resembles that of NANOG and not OCT4. At E3.75, SOX2 was detected in cells expressing NANOG and not in cells expressing SOX17 (Fig. 3A, B), although downregulation of NANOG in PE cells slightly preceded the downregulation of SOX2 in the PE cells (Fig. 3A, D). By E4.25, SOX2 was detected in EPI and not PE cells (Fig. 3C). These observations suggested that SOX2 and NANOG are restricted to EPI cells by a similar mechanism. NANOG has been shown to be restricted to EPI cells by FGF4/MEK [9], [34]–[37], so we evaluated whether FGF/MEK signaling is necessary and/or sufficient to repress SOX2 in the ICM. We cultured wild-type embryos in FGF4 and HEPARIN (FGF4/HEP) from E2.75 to E4.5, which leads to repression of NANOG, and upregulation of SOX17 in the ICM [3], [34]. Control embryos were cultured alongside in the absence of FGF4/HEP. We observed that FGF4/HEP was sufficient to repress SOX2 and upregulate SOX17 in wild-type embryos (Fig. 3E). Next, we cultured wild-type embryos in inhibitors of FGFR/MEK from E2.75 to E4.0, which leads to ectopic expression of NANOG and repression of SOX17 in the ICM of wild-type embryos [34], [36]. Treatment with FGFR/MEK inhibitors led to ectopic expression of SOX2 and repression of SOX17 throughout the ICM (Fig. 3F). We conclude that SOX2 expression is restricted to EPI cells through an FGFR/MEK-dependent mechanism.

Sox2 is required for initial expression of PE genes
Our results showed that, like NANOG, SOX2 expression is restricted to EPI cells at E3.75 and later. We next asked whether Sox2 is required for the expression of NANOG or for the segregation of EPI and PE cell types at this stage. Prior analysis of Sox2 Z null embryos showed that formation of the ICM is independent of Z Sox2 [5], but the requirement for M Sox2 was not evaluated. To determine whether MZ Sox2 is required for NANOG expression, we evaluated whether the ICM contained normal numbers of NANOG-expressing cells in the absence of MZ Sox2. NANOG was detected at normal levels in the absence of MZ Sox2 at E3.75 (Fig. 4A), and in a normal number of ICM cells in Sox2 M, Z, and MZ null blastocysts at E3.75 (Fig. 4B). These observations indicate that Sox2 is dispensable for regulating the initial expression of NANOG.
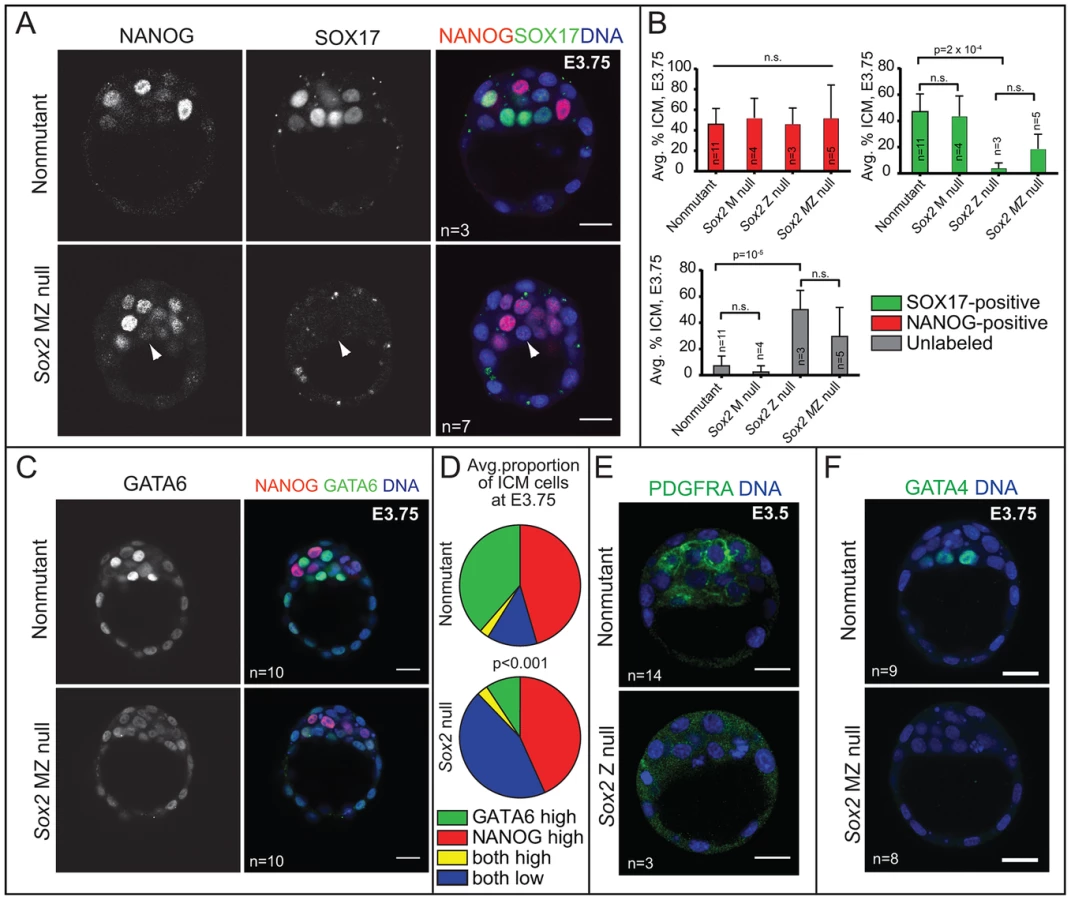
By contrast, the number of cells in which we detected the PE marker SOX17 was greatly reduced in Sox2 null blastocysts relative to control embryos at E3.75. In these embryos, the number of unlabeled cells, in which neither NANOG nor SOX17 was detected, was greatly increased in the absence of Sox2 (Fig. 4A, B). Notably, this phenotype was equivalent between Sox2 Z and MZ null embryos (Fig. 4B), consistent with the conclusion that there is no role for M Sox2. We also examined other PE markers and found that the proportion of ICM cells expressing a higher level of GATA6 was significantly reduced in the absence of Sox2 (Fig. 4C, D), and both PDGFRA and GATA4 were detected at much lower levels in the absence of Sox2 (Fig. 4E, F). These results indicate that Sox2 promotes PE gene expression at E3.5–E3.75.
Sox2 promotes PE gene expression via FGF4
To discover the mechanism by which Sox2 promotes PE development, we next examined the role of Sox2 in regulating Fgf4 expression, since Fgf4 is necessary and sufficient to induce PE gene expression in the ICM [34], [35], [37]. SOX2, together with OCT4, promotes expression of Fgf4 in pluripotent stem cell lines [38], [39], indicating that Sox2 may also promote expression of Fgf4 in the ICM. Consistent with this hypothesis, Fgf4 mRNA was reduced in Sox2 null blastocysts to about 30% of the wild-type level (Fig. 5A). Additionally, we found no requirement for M Sox2 in promoting expression of Fgf4 (Fig. S3A). Thus Z Sox2 is required for maximal expression of Fgf4 in the blastocyst, leading us to ask whether the observed defects in PE gene expression in Sox2 Z null blastocysts are due to reduced expression of Fgf4.

If the disruption in PE gene expression in Sox2 null embryos were due to reduced Fgf4 expression, then exogenous FGF4/HEP should restore PE gene expression. To test this prediction, we cultured Sox2 null embryos in FGF4/HEP from the 8-cell stage (E2.75) to E3.75 (Fig. 5B). As a positive control, we cultured non-mutant embryos in FGF4/HEP, and as a negative control we cultured Sox2 null embryos in the absence of exogenous FGF4/HEP. We first confirmed that embryos of all genotypes and treatment groups were equivalent to the E3.75 developmental stage in terms of cell number (Fig. 5B). Next, we evaluated expression of SOX17 and NANOG in each group. As predicted, FGF4/HEP treatment led to a significant increase in the proportion of SOX17-positive ICM cells in Sox2 null and non-mutant embryos, relative to untreated Sox2 null and non-mutant embryos (Fig. 5C, D). We also cultured Sox2 null and non-mutant embryos in FGF4/HEP for an extended period, after which 100% of ICM cells became SOX17-positive/NANOG-negative, irrespective of genotype (Fig. S3B, C), confirming that Sox2 null embryos respond to exogenous FGF4/HEP like non-mutant embryos. We conclude that Sox2 is not required for ICM cells to receive or respond to FGF4 signaling, but is required for maximal expression of Fgf4.
Our observations that Sox2 promotes PE gene expression via FGF4 predicts that Sox2 promotes PE gene expression non cell-autonomously. We tested this hypothesis by examining expression of PE genes in chimeric embryos containing a Sox2 null PE and wild-type EPI. To generate these chimeras, we aggregated wild-type, YFP-expressing ES cells [40] with precompacted 8-cell Sox2 null or non-mutant host embryos, and then cultured these chimeras to E3.75 (Fig. 5E). Performing the aggregation at this stage allows the ES cells to completely colonize the EPI compartment, such that only PE and TE cells are host-derived [3], [41]. We observed that in Sox2 null host embryos, expression of SOX17 was rescued by wild type ES cells (Fig. 5F, G). These results indicate that Sox2 in EPI cells acts non cell-autonomously to promote expression of SOX17 in PE cells by E3.75.
Sox2 maintains EPI, but not PE, gene expression
We next examined whether Sox2 is required to maintain the expression of PE genes after E3.75. Surprisingly, we detected SOX17, GATA6, PDGFRA, and GATA4 in Sox2 null embryos at E4.25 (Fig. 6A). By examining the time course of SOX17 expression in Sox2 null embryos, we determined that SOX17 was detected in a progressively larger proportion of ICM cells in Sox2 null embryos starting from E3.25 until E4.25, when the proportion of SOX17-expressing cells was equivalent to wild type (Fig. 6B). Similarly, the proportion of cells expressing a high level of GATA6 was also normal in Sox2 null embryos at E4.25 (Fig. S4A). These results show that Sox2 is required for the initial, but not the later, expression of SOX17, GATA6, PDGFRA, and GATA4.

We hypothesized that PE gene expression is eventually induced in the cells that were originally unlabeled in Sox2 null embryos at E3.75 (Fig. 4B). Alternatively, rare, correctly specified PE cells may have proliferated to replace the unlabeled cells in Sox2 null embryos. This latter hypothesis predicts that unlabeled cells would undergo apoptosis in Sox2 null embryos to maintain ICM cell number from E3.75 to E4.25 (Fig. S4B). However, we did not observe a difference in the number of apoptotic cells in Sox2 null embryos during this window (Fig. 6C), suggesting that PE gene expression is eventually induced in cells that were originally unlabeled in Sox2 null embryos at E3.75. We hypothesized that the delayed expression of PE genes in Sox2 null embryos is due to the lower level of Fgf4 (Fig. 5A). Consistent with this hypothesis, the expression of SOX17 in Sox2 null embryos at E4.25 was indeed dependent on FGFR/MEK signaling (Fig. 6D), arguing that low levels of FGF4 can eventually induce expression of PE genes in Sox2 null embryos. To determine whether delayed PE gene expression also delayed PE maturation, we examined expression of SOX7, which is expressed only in mature PE cells [42]. At E4.25, SOX7 was detected in Sox2 null embryos (Fig. 6E), suggesting that PE cells had matured in an age-appropriate manner, in spite of the reduced Fgf4. We conclude that Sox2 is not required for maintaining PE gene expression in the blastocyst, consistent with the observation that PE-derived cells are detected in Sox2 null embryos postimplantation [5].
Curiously, we noted that in spite of the normal expression of PE genes in the absence of Sox2, PE cells were often mislocalized in Sox2 null embryos at E4.25. Rather than forming a single, contiguous hypoblast layer between EPI and blastocoel, PE cells were often observed between EPI and polar TE in Sox2 null embryos (Fig. 6A, E). These observations raised the possibility that Sox2 promotes expression of genes thought to regulate PE cohesion and sorting, such as LAMININ and DAB2 [43]–[45], which are normally detectable in the PE at E4.25 [31], [42], [46]. Consistent with this hypothesis, expression of both LAMA1 and DAB2 was reduced in PE cells at E4.25 in Sox2 null blastocysts (Fig. 6F). Notably, expression of DAB2 and PE sorting were rescued by wild type ES cells in Sox2 null E4.25 blastocysts (Fig. S4C), and expression of DAB2 was eventually restored in implantation-delayed blastocysts (Fig. S4D), consistent with SOX2 acting non cell-autonomously to promote the initiation, but not the maintenance, of PE gene expression.
Finally, we examined the status of the EPI in Sox2 null embryos around the time of implantation, because EPI cells are not detected in Sox2 null embryos postimplantation [5]. In Sox2 null embryos at E4.25, expression of the EPI marker PECAM1 [14], [47] was undetectable (4/5 Sox2 null embryos) or reduced (1/5 Sox2 null embryos), expression of OCT4 was undetectable (1/5 Sox2 null embryos) or reduced (3/5 Sox2 null embryos), and expression of NANOG was also undetectable (1/1 Sox2 null embryos) (Fig. 6G). These observations indicate that although Sox2 is dispensable for the initiation of EPI gene expression, Sox2 is required to maintain EPI gene expression. To evaluate the role of Sox2 in the EPI at later developmental stages, we prolonged preimplantation by inducing diapause. By two to four days of delayed implantation, the number of presumptive EPI cells was reduced in Sox2 null embryos relative to wild type, while the number of PE cells was largely maintained until the latest time point (Fig. S4D). We conclude that Sox2 is required for maintaining EPI cell fate at E4.25 and thereafter.
Discussion
Here we have examined the roles and regulation of SOX2 in the preimplantation embryo, with the goal of deepening our understanding of the origins of pluripotency during development. We showed that SOX2 is a unique, early marker of ICM progenitors, consistent with the reported early enrichment of Sox2 mRNA in ICM progenitors [14]. However, the significance of this early expression is not obvious, since the cell-autonomous role for Sox2 in regulating cell fate does not become apparent until late blastocyst stage. It is possible that Sox2 is initially genetically redundant with other pluripotency factors, such as Oct4 or Nanog. Although phenotypes resulting from disruption of multiple pluripotency genes have not yet been reported in mice, there is evidence of genetic redundancy among zebrafish Oct/Sox/Nanog orthologues [48]. Genetic redundancy between these factors is consistent with our observations that Fgf4 expression is reduced, but not eliminated, in the absence of either Sox2 or Oct4 [3]. Thus, in the embryo, OCT4 and SOX2 may promote expression of Fgf4, and possibly other targets, synergistically, as has been demonstrated in pluripotent stem cell lines [38], [39].
Our evidence suggests that SOX2 and CDX2 are patterned by HIPPO pathway components in parallel (Fig. 7A), but it is not yet clear whether SOX2 and CDX2 are regulated by HIPPO pathway components in the same way. In the TE, expression of CDX2 is activated by a YAP/WWTR1/TEAD complex [24]–[26]. Here we showed that TEAD4 represses expression of SOX2, but we do not yet know if TEAD4 regulates expression of Sox2 directly or indirectly. In ES cells, the YAP/TEAD complex been shown to bind upstream of Sox2 to promote its expression [49], arguing that TEAD4 could, in principle, work together with a transcriptional repressor to repress expression of Sox2 in TE cells. It will be exciting to address this hypothesis in future studies in addition to examining whether position, polarity, and/or contact regulate Sox2 expression, as has been shown for Cdx2 [26], [50]–[54]. Interestingly, the HIPPO pathway can be activated in a position-independent manner in blastomeres [53], [55], raising the possibility that multiple upstream inputs could regulate expression of genes such as Sox2 and Cdx2 in the embryo. Finally, our observations are also consistent with LATS regulating the activity of an as-yet unidentified transcription factor that promotes expression of SOX2 in ICM progenitors. This hypothesis is also supported by qPCR evidence that Lats1/2 maintains expression of Sox2 in the blastocyst [56]. Identification of LATS targets in the preimplantation embryo will therefore provide exciting new inroads into understanding the origins of pluripotency.

Our study provides insight into the regulation of extraembryonic cell types during preimplantation development. In terms of the TE lineage, we showed that SOX2 is not detected in TE cells during preimplantation, nor is it required for their specification. These observations suggest that expression of Sox2 is activated de novo in the trophoblast lineage postimplantation, where it promotes trophoblast development [5], and raise the possibility that HIPPO pathway components participate in regulation of Sox2 expression postimplantation as well. Investigation of the mechanisms by which Sox2 expression becomes activated in the extraembryonic ectoderm is an exciting opportunity to learn about the origins of trophoblast stem (TS) cells, which are derived from extraembryonic ectoderm [57], and dependent on Sox2 and Tead4 [15], [26].
While SOX2 does not activate TE gene expression, SOX2 also does not appear to repress TE gene expression in the ICM. This is in contrast to OCT4, which is known to repress expression of TE genes in the ICM and in ES cell lines [3], [7], [10], [58], [59]. Moreover, we have shown that SOX2 is neither expressed nor functional in PE cells at the time when OCT4 represses TE gene expression in PE cells [3]. These observations support the idea that SOX2 and OCT4 have important non-overlapping roles in the embryo and stem cell lines [15], [60], in addition to their widely appreciated overlapping functions. Thus in the ICM, OCT4 may act alone or with partners other than SOX2 to repress transcription of TE genes in EPI cells.
Our analysis led us to explore the genetic regulation of PE specification as well. We have shown that in the ICM, SOX2 becomes expressed in a salt-and-pepper fashion, similar to NANOG. We have also shown that the salt-and-pepper distribution of SOX2 in the ICM depends on FGFR/MEK signaling, but it is not yet clear whether FGFR/MEK signaling regulates SOX2 expression directly, or whether FGFR/MEK signaling maintains cell fate, which in turn regulates SOX2 expression. Alternatively, NANOG or GATA6 could help regulate SOX2 expression in the ICM. While the expression pattern of SOX2 in Nanog null embryos has not yet been reported, in Gata6 null embryos, SOX2 is expressed in all ICM cells [61], suggesting that GATA6 helps mediate FGFR/MEK signaling to repress SOX2 in the E3.75 ICM (Fig. 7B). Further studies of SOX2 in Gata6 and Nanog null embryos, with and without manipulations to the FGFR/MEK signaling pathway will help clarify the direct and indirect mechanisms regulating Sox2 expression in the ICM.
We also showed that SOX2 helps to maintain the appropriate level of FGF4 that is essential for timely creation of the hypoblast layer. Curiously, Sox2 null embryos do not completely phenocopy Fgf4 null embryos, since NANOG was not upregulated in Sox2 null embryos as it is in Fgf4 null embryos [35]. We hypothesize that the intermediate level of Fgf4 in Sox2 null embryos are sufficient to repress NANOG, as we have shown for Oct4 null embryos [3]. In addition, we did not observe reduced total cell number in Sox2 null embryos as has been observed in Fgf4 null embryos [35], arguing that a moderate level of FGF4 can maintain cell proliferation during preimplantation. Finally, the moderate level of FGF4 produced by Sox2 null embryos is eventually able to restore expression of PE genes (Fig 7C), which does not occur in embryos completely lacking Fgf4 [35], [37], or downstream signaling components [4]. Interestingly, the timing of PE gene expression has also been shown to be sensitive to dose of Gata6 [61], suggesting Fgf4 may be regulated by GATA6 as well. By contrast, PE gene expression is not eventually restored in Oct4 null or in Nanog null embryos [3], [9]. In light of evidence that Oct4 is not required for later expression of PE genes [59], our observations predict that Fgf4 levels, or levels of an as-yet unidentified, later-acting signal essential for maintaining PE gene expression, may be more rapidly and/or dramatically lost in Oct4 and possibly Nanog null embryos, than in Sox2 null embryos.
Our observation that SOX2 is one of the earliest known unique markers of ICM progenitors is supported by the observation that SOX2 is also one of the first pluripotency genes to localize to ICM progenitors in the morula in other mammals [62]. While Sox2 is not initially required for expression of pluripotency genes in the mouse, Sox2 eventually does promote expression of pluripotency genes in the ICM, as in ES cells. Thus, in the ICM, the role of Sox2 appears to be to maintain, and not to initiate, pluripotency (Fig. 7C). This idea is consistent with observations that pluripotency genes are initially normal in Oct4 and Nanog null embryos [3], [12], [33]. The idea of a later role for Sox2 in maintaining expression of pluripotency genes in the ICM is also consistent with evidence that both the genetic regulation and the transcriptional profile of ES cells are more similar to late than to early EPI cells [7], [63]. Recent studies have shown that HIPPO and IL6/JAK/LIF/STAT3 pathways also maintain expression of pluripotency genes in the ICM around implantation stage [50]–[52], [64]. Discovering the mechanisms of crosstalk between pluripotency pathway members at the implantation stage and shortly thereafter will therefore provide exciting new insight into the origins of pluripotent stem cells.
Materials and Methods
Mouse strains and genotyping
All animal research was conducted in accordance with the guidelines of the University of California Santa Cruz Institutional Animal Care and Use Committee or by RIKEN CDB and Kumamoto University. The following alleles or transgenes were used in this study: Sox2tm1.1Lan [27], Tg(Zp3-cre)93Knw [28], Tead4tm1Hssk [24], and Cdx2tm1.1Aral [21]. Mice carrying the Sox2 null allele (Sox2del+) were generated by crossing mice carrying Sox2tm1.1Lan with 129-Alpltm1(cre)Nagy [65].
Embryo collection and manipulation
As described previously [3], [21], mice were maintained on a 12-hour light/dark cycle. Embryos were collected from timed natural matings by flushing dissected oviducts or uteri with M2 medium. Cultured embryos were cultured in KSOM (Millipore) alone, or KSOM with a final concentration of 1 µg/ml recombinant human FGF4 (R&D Systems) and 1 U/mL Heparin (Sigma), or 100 nM PD173074 and 500 nM PD0325901 (Stemgent) at 37°C and 6% CO2. Microinjection of mRNA was performed as described [26], [50]. To delay implantation, diapause was induced as previously described [32], [66], [67].
Immunofluorescence and confocal microscopy
Embryos were fixed, stained, imaged, and recovered for genotyping as previously described [68]. Primary and secondary antibody sources were described previously [3], [50], and also included rabbit anti-EOMES (Abcam), rabbit anti-DAB2 (Santa Cruz Biotech), rabbit anti-LAMA1 (Sigma), and rat anti-PECAM1 (BD Biosciences).
Chimeras
Chimeras were performed using Sox2 MZ null embryos as previously described [3]. Chimeras were subsequently genotyped by PCR using primers that distinguished wild-type, floxed, and deleted Sox2 alleles [27].
RNA isolation and cDNA preparation
RNA isolation and single blastocyst qPCR was performed as previously described [21]. qPCR primers included Sox2 (GCGGAGTGGAAACTTTTGTCC and CGGGAAGCGTGTACTTATCCTT), Fgf4 (AGCAGGGGCAAGCTCTTC and GGGTACGCGTAGGATTCG), Oct4 (AGCTGCTGAAGCAGAAGAGG and AGATGGTGGTCTGGCTGAAC), and Actb (CTGAACCCTAAGGCCAACC and CCAGAGGCATACAGGGACAG).
Supporting Information
Zdroje
1. XenopoulosP, KangM, HadjantonakisAK (2012) Cell lineage allocation within the inner cell mass of the mouse blastocyst. Results Probl Cell Differ 55 : 185–202.
2. YamanakaY, RalstonA (2010) Early embryonic cell fate decisions in the mouse. Adv Exp Med Biol 695 : 1–13.
3. FrumT, HalbisenMA, WangC, AmiriH, RobsonP, et al. (2013) Oct4 cell-autonomously promotes primitive endoderm development in the mouse blastocyst. Dev Cell 25 : 610–622.
4. ChazaudC, YamanakaY, PawsonT, RossantJ (2006) Early lineage segregation between epiblast and primitive endoderm in mouse blastocysts through the Grb2-MAPK pathway. Dev Cell 10 : 615–624.
5. AvilionAA, NicolisSK, PevnyLH, PerezL, VivianN, et al. (2003) Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev 17 : 126–140.
6. KeramariM, RazaviJ, IngmanKA, PatschC, EdenhoferF, et al. (2010) Sox2 is essential for formation of trophectoderm in the preimplantation embryo. PLoS One 5: e13952.
7. RalstonA, CoxB, NishiokaN, SasakiH, CheaE, et al. (2010) Gata3 regulates trophoblast development downstream of Tead4 and in parallel to Cdx2. Development 137 : 395–403.
8. MesserschmidtDM, KemlerR (2010) Nanog is required for primitive endoderm formation through a non-cell autonomous mechanism. Dev Biol 344 : 129–137.
9. FrankenbergS, GerbeF, BessonnardS, BelvilleC, PouchinP, et al. (2011) Primitive Endoderm Differentiates via a Three-Step Mechanism Involving Nanog and RTK Signaling. Dev Cell 21 : 1005–1013.
10. NicholsJ, ZevnikB, AnastassiadisK, NiwaH, Klewe-NebeniusD, et al. (1998) Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell 95 : 379–391.
11. MitsuiK, TokuzawaY, ItohH, SegawaK, MurakamiM, et al. (2003) The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell 113 : 631–642.
12. WuG, HanD, GongY, SebastianoV, GentileL, et al. (2013) Establishment of totipotency does not depend on Oct4A. Nat Cell Biol 15 : 1089–1097.
13. AksoyI, JauchR, ChenJ, DylaM, DivakarU, et al. (2013) Oct4 switches partnering from Sox2 to Sox17 to reinterpret the enhancer code and specify endoderm. EMBO J 32 : 938–953.
14. GuoG, HussM, TongG, WangC, Li SunL, et al. (2010) Resolution of cell fate decisions revealed by single-cell gene expression analysis from zygote to blastocyst. Dev Cell 18 : 675–685.
15. AdachiK, NikaidoI, OhtaH, OhtsukaS, UraH, et al. (2013) Context-dependent wiring of Sox2 regulatory networks for self-renewal of embryonic and trophoblast stem cells. Mol Cell 52 : 380–392.
16. MasuiS, NakatakeY, ToyookaY, ShimosatoD, YagiR, et al. (2007) Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nat Cell Biol 9 : 625–635.
17. LiJ, PanG, CuiK, LiuY, XuS, et al. (2007) A dominant-negative form of mouse SOX2 induces trophectoderm differentiation and progressive polyploidy in mouse embryonic stem cells. J Biol Chem 282 : 19481–19492.
18. Okumura-NakanishiS, SaitoM, NiwaH, IshikawaF (2005) Oct-3/4 and Sox2 regulate Oct-3/4 gene in embryonic stem cells. J Biol Chem 280 : 5307–5317.
19. KurodaT, TadaM, KubotaH, KimuraH, HatanoS, et al. (2005) Octamer and Sox elements are required for transcriptional cis regulation of Nanog gene expression. Mol Cell Biol 25 : 2475–2485.
20. ChewJL, LohYH, ZhangW, ChenX, TamWL, et al. (2005) Reciprocal transcriptional regulation of Pou5f1 and Sox2 via the Oct4/Sox2 complex in embryonic stem cells. Mol Cell Biol 25 : 6031–6046.
21. BlijS, FrumT, AkyolA, FearonE, RalstonA (2012) Maternal Cdx2 is dispensable for mouse development. Development 139 : 3969–3972.
22. StrumpfD, MaoCA, YamanakaY, RalstonA, ChawengsaksophakK, et al. (2005) Cdx2 is required for correct cell fate specification and differentiation of trophectoderm in the mouse blastocyst. Development 132 : 2093–2102.
23. PalmieriS, PeterW, HessH, SchölerH (1994) Oct-4 transcription factor is differentially expressed in the mouse embryo during establishment of the first two extraembryonic cell lineages involved in implantation. Dev Biol 166 : 259–267.
24. NishiokaN, YamamotoS, KiyonariH, SatoH, SawadaA, et al. (2008) Tead4 is required for specification of trophectoderm in pre-implantation mouse embryos. Mech Dev 125 : 270–283.
25. YagiR, KohnM, KaravanovaI, KanekoK, VullhorstD, et al. (2007) Transcription factor TEAD4 specifies the trophectoderm lineage at the beginning of mammalian development. Development 134 : 3827–3836.
26. NishiokaN, InoueK, AdachiK, KiyonariH, OtaM, et al. (2009) The Hippo signaling pathway components Lats and Yap pattern Tead4 activity to distinguish mouse trophectoderm from inner cell mass. Dev Cell 16 : 398–410.
27. SmithAN, MillerLA, RadiceG, Ashery-PadanR, LangRA (2009) Stage-dependent modes of Pax6-Sox2 epistasis regulate lens development and eye morphogenesis. Development 136 : 2977–2985.
28. de VriesWN, BinnsLT, FancherKS, DeanJ, MooreR, et al. (2000) Expression of Cre recombinase in mouse oocytes: a means to study maternal effect genes. Genesis 26 : 110–112.
29. CampoloF, GoriM, FavaroR, NicolisS, PellegriniM, et al. (2013) Essential role of Sox2 for the establishment and maintenance of the germ cell line. Stem Cells 31 : 1408–1421.
30. PlusaB, PiliszekA, FrankenbergS, ArtusJ, HadjantonakisA (2008) Distinct sequential cell behaviours direct primitive endoderm formation in the mouse blastocyst. Development 135 : 3081–3091.
31. NiakanKK, JiH, MaehrR, VokesSA, RodolfaKT, et al. (2010) Sox17 promotes differentiation in mouse embryonic stem cells by directly regulating extraembryonic gene expression and indirectly antagonizing self-renewal. Genes Dev 24 : 312–326.
32. ArtusJ, PanthierJJ, HadjantonakisAK (2010) A role for PDGF signaling in expansion of the extra-embryonic endoderm lineage of the mouse blastocyst. Development 137 : 3361–3372.
33. SilvaJ, NicholsJ, TheunissenT, GuoG, van OostenA, et al. (2009) Nanog is the gateway to the pluripotent ground state. Cell 138 : 722–737.
34. YamanakaY, LannerF, RossantJ (2010) FGF signal-dependent segregation of primitive endoderm and epiblast in the mouse blastocyst. Development 137 : 715–724.
35. KangM, PiliszekA, ArtusJ, HadjantonakisAK (2013) FGF4 is required for lineage restriction and salt-and-pepper distribution of primitive endoderm factors but not their initial expression in the mouse. Development 140 : 267–279.
36. NicholsJ, SilvaJ, RoodeM, SmithA (2009) Suppression of Erk signalling promotes ground state pluripotency in the mouse embryo. Development 136 : 3215–3222.
37. KrawchukD, Honma-YamanakaN, AnaniS, YamanakaY (2013) FGF4 is a limiting factor controlling the proportions of primitive endoderm and epiblast in the ICM of the mouse blastocyst. Dev Biol 384 : 65–71.
38. YuanH, CorbiN, BasilicoC, DaileyL (1995) Developmental-specific activity of the FGF-4 enhancer requires the synergistic action of Sox2 and Oct-3. Genes Dev 9 : 2635–2645.
39. AmbrosettiDC, BasilicoC, DaileyL (1997) Synergistic activation of the fibroblast growth factor 4 enhancer by Sox2 and Oct-3 depends on protein-protein interactions facilitated by a specific spatial arrangement of factor binding sites. Mol Cell Biol 17 : 6321–6329.
40. GeorgeSH, GertsensteinM, VinterstenK, Korets-SmithE, MurphyJ, et al. (2007) Developmental and adult phenotyping directly from mutant embryonic stem cells. Proc Natl Acad Sci U S A 104 : 4455–4460.
41. PoueymirouWT, AuerbachW, FrendeweyD, HickeyJF, EscaravageJM, et al. (2007) F0 generation mice fully derived from gene-targeted embryonic stem cells allowing immediate phenotypic analyses. Nat Biotechnol 25 : 91–99.
42. ArtusJ, PiliszekA, HadjantonakisAK (2011) The primitive endoderm lineage of the mouse blastocyst: sequential transcription factor activation and regulation of differentiation by Sox17. Dev Biol 350 : 393–404.
43. MorrisS, TallquistM, RockC, CooperJ (2002) Dual roles for the Dab2 adaptor protein in embryonic development and kidney transport. EMBO J 21 : 1555–1564.
44. YangDH, SmithER, RolandIH, ShengZ, HeJ, et al. (2002) Disabled-2 is essential for endodermal cell positioning and structure formation during mouse embryogenesis. Dev Biol 251 : 27–44.
45. SmythN, VatanseverHS, MurrayP, MeyerM, FrieC, et al. (1999) Absence of basement membranes after targeting the LAMC1 gene results in embryonic lethality due to failure of endoderm differentiation. J Cell Biol 144 : 151–160.
46. KlaffkyE, WilliamsR, YaoCC, ZioberB, KramerR, et al. (2001) Trophoblast-specific expression and function of the integrin alpha 7 subunit in the peri-implantation mouse embryo. Dev Biol 239 : 161–175.
47. RobsonP, SteinP, ZhouB, SchultzRM, BaldwinHS (2001) Inner cell mass-specific expression of a cell adhesion molecule (PECAM-1/CD31) in the mouse blastocyst. Dev Biol 234 : 317–329.
48. LeeMT, BonneauAR, TakacsCM, BazziniAA, DiVitoKR, et al. (2013) Nanog, Pou5f1 and SoxB1 activate zygotic gene expression during the maternal-to-zygotic transition. Nature 503 : 360–364.
49. LianI, KimJ, OkazawaH, ZhaoJ, ZhaoB, et al. (2010) The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev 24 : 1106–1118.
50. HirateY, HiraharaS, InoueK, SuzukiA, AlarconVB, et al. (2013) Polarity-dependent distribution of angiomotin localizes Hippo signaling in preimplantation embryos. Curr Biol 23 : 1181–1194.
51. LeungCY, Zernicka-GoetzM (2013) Angiomotin prevents pluripotent lineage differentiation in mouse embryos via Hippo pathway-dependent and -independent mechanisms. Nat Commun 4 : 2251.
52. CockburnK, BiecheleS, GarnerJ, RossantJ (2013) The Hippo pathway member Nf2 is required for inner cell mass specification. Curr Biol 23 : 1195–1201.
53. LorthongpanichC, DorisTP, LimviphuvadhV, KnowlesBB, SolterD (2012) Developmental fate and lineage commitment of singled mouse blastomeres. Development 139 : 3722–3731.
54. StephensonRO, YamanakaY, RossantJ (2010) Disorganized epithelial polarity and excess trophectoderm cell fate in preimplantation embryos lacking E-cadherin. Development 137 : 3383–3391.
55. AnaniS, BhatS, Honma-YamanakaN, KrawchukD, YamanakaY (2014) Initiation of Hippo signaling is linked to polarity rather than to cell position in the pre-implantation mouse embryo. Development 141 : 2813–2824.
56. LorthongpanichC, MesserschmidtDM, ChanSW, HongW, KnowlesBB, et al. (2013) Temporal reduction of LATS kinases in the early preimplantation embryo prevents ICM lineage differentiation. Genes Dev 27 : 1441–1446.
57. TanakaS, KunathT, HadjantonakisAK, NagyA, RossantJ (1998) Promotion of trophoblast stem cell proliferation by FGF4. Science 282 : 2072–2075.
58. NiwaH, ToyookaY, ShimosatoD, StrumpfD, TakahashiK, et al. (2005) Interaction between Oct3/4 and Cdx2 determines trophectoderm differentiation. Cell 123 : 917–929.
59. Le BinGC, Muñoz-DescalzoS, KurowskiA, LeitchH, LouX, et al. (2014) Oct4 is required for lineage priming in the developing inner cell mass of the mouse blastocyst. Development 141 : 1001–1010.
60. DeVealeB, BrokhmanI, MohseniP, BabakT, YoonC, et al. (2013) Oct4 is required ∼E7.5 for proliferation in the primitive streak. PLoS Genet 9: e1003957.
61. SchrodeN, SaizN, Di TaliaS, HadjantonakisAK (2014) GATA6 Levels Modulate Primitive Endoderm Cell Fate Choice and Timing in the Mouse Blastocyst. Dev Cell 29 : 454–467.
62. GoissisMD, CibelliJB (2014) Functional characterization of SOX2 in bovine preimplantation embryos. Biol Reprod 90 : 30.
63. BoroviakT, LoosR, BertoneP, SmithA, NicholsJ (2014) The ability of inner-cell-mass cells to self-renew as embryonic stem cells is acquired following epiblast specification. Nat Cell Biol 16 : 516–528.
64. DoDV, UedaJ, MesserschmidtDM, LorthongpanichC, ZhouY, et al. (2013) A genetic and developmental pathway from STAT3 to the OCT4-NANOG circuit is essential for maintenance of ICM lineages in vivo. Genes Dev 27 : 1378–1390.
65. LomelíH, Ramos-MejíaV, GertsensteinM, LobeCG, NagyA (2000) Targeted insertion of Cre recombinase into the TNAP gene: excision in primordial germ cells. Genesis 26 : 116–117.
66. Batlle-MoreraL, SmithA, NicholsJ (2008) Parameters influencing derivation of embryonic stem cells from murine embryos. Genesis 46 : 758–767.
67. NicholsJ, ChambersI, TagaT, SmithA (2001) Physiological rationale for responsiveness of mouse embryonic stem cells to gp130 cytokines. Development 128 : 2333–2339.
68. RalstonA, RossantJ (2008) Cdx2 acts downstream of cell polarization to cell-autonomously promote trophectoderm fate in the early mouse embryo. Dev Biol 313 : 614–629.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 10
Nejčtenější v tomto čísle
- The Master Activator of IncA/C Conjugative Plasmids Stimulates Genomic Islands and Multidrug Resistance Dissemination
- A Splice Mutation in the Gene Causes High Glycogen Content and Low Meat Quality in Pig Skeletal Muscle
- Keratin 76 Is Required for Tight Junction Function and Maintenance of the Skin Barrier
- A Role for Taiman in Insect Metamorphosis