
Germline Mutations in Are Associated with Familial Gastric Cancer
The underlying genetic mutations involved in 60% of inherited gastric cancer cases remain unknown. Here we present a large, extended pedigree with familial gastric cancer and an association in part of the family with a mutation in MAP3K6. The conservation, predicted pathogenicity of the variant, tissue distribution, and known function of MAP3K6 made this a strong candidate that warranted further investigation. Examination of an additional 115 unrelated probands identified additional mutations in MAP3K6, including a truncating mutation.
Published in the journal:
. PLoS Genet 10(10): e32767. doi:10.1371/journal.pgen.1004669
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004669
Summary
The underlying genetic mutations involved in 60% of inherited gastric cancer cases remain unknown. Here we present a large, extended pedigree with familial gastric cancer and an association in part of the family with a mutation in MAP3K6. The conservation, predicted pathogenicity of the variant, tissue distribution, and known function of MAP3K6 made this a strong candidate that warranted further investigation. Examination of an additional 115 unrelated probands identified additional mutations in MAP3K6, including a truncating mutation.
Introduction
Gastric cancer is the second leading cause of cancer-related death worldwide with 738,000 deaths per year [1]. Primary treatment consists of surgical resection of the tumor and may be followed by chemotherapy and/or radiotherapy. The 5-year survival rates after surgical resection are high if the disease is detected early (71% for stage 1A), however, they drop off quickly when the diagnosis is made at later stages (46% stage IIA, 20% stage IIIA, 4% stage IV) (National Cancer Institute's SEER database, October 2013). Unfortunately, because early symptoms of gastric cancer closely resemble other diseases, detection often does not occur until advanced stages have already been reached [2].
Classically, gastric cancer has been divided into two types: intestinal and diffuse [3]. The intestinal form occurs spontaneously and is most often found in elderly individuals, while the diffuse form often occurs in younger individuals and can be associated with a family history of gastric cancer. Populations with higher prevalence of chronic Helicobacter pylori infection tend to have higher gastric cancer burdens [4]. The majority of gastric cancers (90%) are sporadic, but approximately 10% show familial clustering [5]. Only 1% to 3% are caused by a hereditary syndrome, as opposed to environmental factors such as shared dietary practices [5]. The most well established familial form of gastric cancer is hereditary diffuse gastric cancer (HDGC [MIM 137215]), where approximately 40% of cases are attributed to germline mutations in the E-cadherin encoding gene, CDH1 [6]–[9].
We ascertained a large family from Maritime Canada with a history of Familial Gastric Cancer (FGC) displaying an apparent autosomal dominant pattern of inheritance, but bearing no variants in the coding region of the CDH1 gene. While the family displays many features typical of HDGC, there was diversity in the clinical presentation within the family as well as an advanced age of onset, therefore we have opted to simply refer to the condition as FGC over the more stringently defined HDGC. Genomic mapping of shared inherited regions among affected family members, followed by whole-exome sequencing, led to the identification of a germline single nucleotide variant (SNV) in mitogen-activated protein kinase kinase kinase 6 (MAP3K6, ASK2, MAPKKK6, MEKK6, ENSG00000142733), a gene encoding a member of the serine/threonine protein kinase family. Several in silico methods predicted the SNV in MAP3K6 to be damaging to the protein, and previous studies with MAP3K6 deficient mice [10], as well as the occurrence of mutations in this gene in both primary gastric cancer tumors and gastric cancer cell lines [11], were consistent with mutations in the MAP3K6 gene being the causative mutation. Sequencing of DNA isolated directly from a fixed tumor specimen of one individual demonstrated the presence of a de novo second-hit variant in MAP3K6. Screening of an additional 115 unrelated FGC samples, also negative for CDH1 mutations, revealed five individuals with four additional SNVs in MAP3K6 that were also predicted to be pathogenic, as well as an unrelated individual with the SNV identified in the family from Maritime Canada. The age of onset varied among MAP3K6 SNV carriers in the five families, and one had not developed cancer even at late stage of life, suggesting incomplete penetrance. This is the first report of a heritable cancer resulting from SNVs in MAP3K6.
Results
Clinical and Pathological Assessment
We ascertained a large Maritime Canadian family of European descent in the course of routine clinical assessment in the Hereditary Cancer Clinic as part of Maritime Medical Genetics Service at the IWK Health Centre in Halifax, Nova Scotia, Canada (Figure 1). Saliva, blood, or formalin-fixed paraffin-embedded (FFPE) samples were obtained from 6 family members with gastric cancer, as well as 27 unaffected relatives, and one married-in individual. No consanguinity was suspected in this pedigree.

The proband, affected individual 1884, was diagnosed with metastatic gastric carcinoma and underwent a total gastrectomy at age 51. Pathological examination revealed a poorly differentiated adenocarcinoma arising in the antrum of the stomach in a background of intestinal metaplasia and chronic gastritis. The tumor was composed of a sheet of signet ring cells (Figure 2C and D). The carcinoma penetrated through the entire thickness of the muscularis propria involving the serosal layer. No evidence of H. pylori was seen.

The proband's maternal aunt, individual 1826, was diagnosed with gastric carcinoma at age 80 and underwent a partial gastrectomy. Pathological examination revealed a moderately to poorly differentiated adenocarcinoma invading into muscularis propria (Figure 2A). The tumor was predominantly composed of cohesive nests of neoplastic cells with occasional glandular formation. Tumor cells with signet ring cell forms were seen in solid areas (Figure 2B). The gastric mucosa adjacent to the tumor showed focal intestinal metaplasia without evidence of H. pylori.
At age 76 a stomach biopsy of another maternal aunt to the proband, individual 1841, was reported to have a moderately differentiated adenocarcinoma with glandular formation. H. pylori was identified in the background gastric mucosa. A small biopsy section was available for re-examination. Although this sample was too small for a complete classification, it showed cohesive nests of neoplastic cells with small foci of glandular formation in keeping with a poorly differentiated adenocarcinoma. There were some tumor cells in the sample showing clear cytoplasm, but these could not be definitively classified as signet ring cells.
A stomach biopsy at age 82 of patient 1844, the proband's mother, showed a poorly differentiated adenocarcinoma with signet ring features. The background mucosa showed evidence of H. pylori and patchy intestinal metaplasia.
Patient 1845, a first-cousin-once-removed to the proband, was diagnosed at age 59 with an undifferentiated carcinoma without signet ring features. The tumor was associated with dense lymphoid infiltrate and was best classified as lymphoepithelial carcinoma. H. pylori was not seen in the adjacent normal mucosa. The tumor was positive for two intronic variants in CDH1 both of which are expected to be benign (NM_004360.3:c.688−83G>A and c.2439+52G>A).
Patient 2447, a third cousin, was diagnosed at age 44 with a poorly differentiated adenocarcinoma without signet ring cell features. The adjacent gastric mucosa showed extensive intestinal metaplasia. There was no evidence of H. pylori.
Following screening of a panel of 115 probands with non-CDH1 familial gastric cancer, an unrelated family from Portugal was added to our study (Figure 3A). Individual II-6 was diagnosed with gastric cancer at age 62, having poorly differentiated adenocarcinoma of the stomach and the presence of signet ring cells. Immunohistochemistry analysis showed positive membranous staining of E-cadherin in neoplastic cells (Figure 3C), including signet ring cells (Figure 3B). The related individuals I-4, II-1, and II-7 were diagnosed with gastric cancer (histology details unknown) at ages 53, 62 and 52 respectively. All four individuals in the Portuguese pedigree died from the disease within 5 years of diagnosis in this family. In the Maritime Canadian family, 1884, 1844 and 1841 died from the disease within one year of diagnosis.

The gastric cancer described for patients 1845 and 2447 had no signet rings observed and was diagnosed at an earlier average age (52 versus 72, although the proband was diagnosed at age 51). Based on differences in histology, particularly the lack of signet ring cells in 1845 and 2447 compared to the other affected individuals, it is possible that the disease in these two individuals represents a distinct condition. Alternatively it is possible that the family is displaying a more complex phenotypic pattern being driven by two (or more) genes.
Molecular Mapping and Exclusion of Known and Candidate Genes
Although 30–40% of HDGC cases are attributable to mutations in CDH1, no mutations in protein-coding exons of CDH1 were found in affected individuals from the Maritime Canadian family. To identify the pathogenic loci in this family, high density SNP-genotyping using Illumina arrays was performed on five affected individuals: the proband's mother (1844), two affected maternal aunts (1826 and 1841), and two distant cousins (1845 and 2447) as well as several related individuals with no reported incidence of cancer whose affection status was treated as unknown (1907, 1924, 1821, and 1822). For all individuals except 1845 genotyping data was available at 2.5 million markers. Individual 1845 had previously been genotyped at a density of 660K, and was not able to be re-genotyped at the higher density. No DNA suitable for SNP genotyping was obtained from the FFPE sample of the proband (1884). Using these data, we performed both non-parametric and parametric linkage analysis using Merlin [12]. Given the late age of onset in many affected family members, the penetrance in the Maritime pedigree is unknown. In order to be conservative in identifying genomic regions of interest, two dominant penetrance models (50% and 99% penetrance) using affected individuals 1826, 1841, 1844, and 2447 (and individuals 1907, 1924, 1821, and 1822 with unknown affection status) were used. Genomic regions identified under parametric linkage analysis were generally consistent with one another regardless of the penetrance parameter chosen (Table 1). The analysis was repeated with individual 2447 treated as unknown to analyze just the reduced pedigree where 2447 and 1845 were treated as potential phenocopies (Table 2). This resulted in lower overall LOD scores for all regions identified, as well as more and larger regions on average, encompassing a larger portion of the genome.
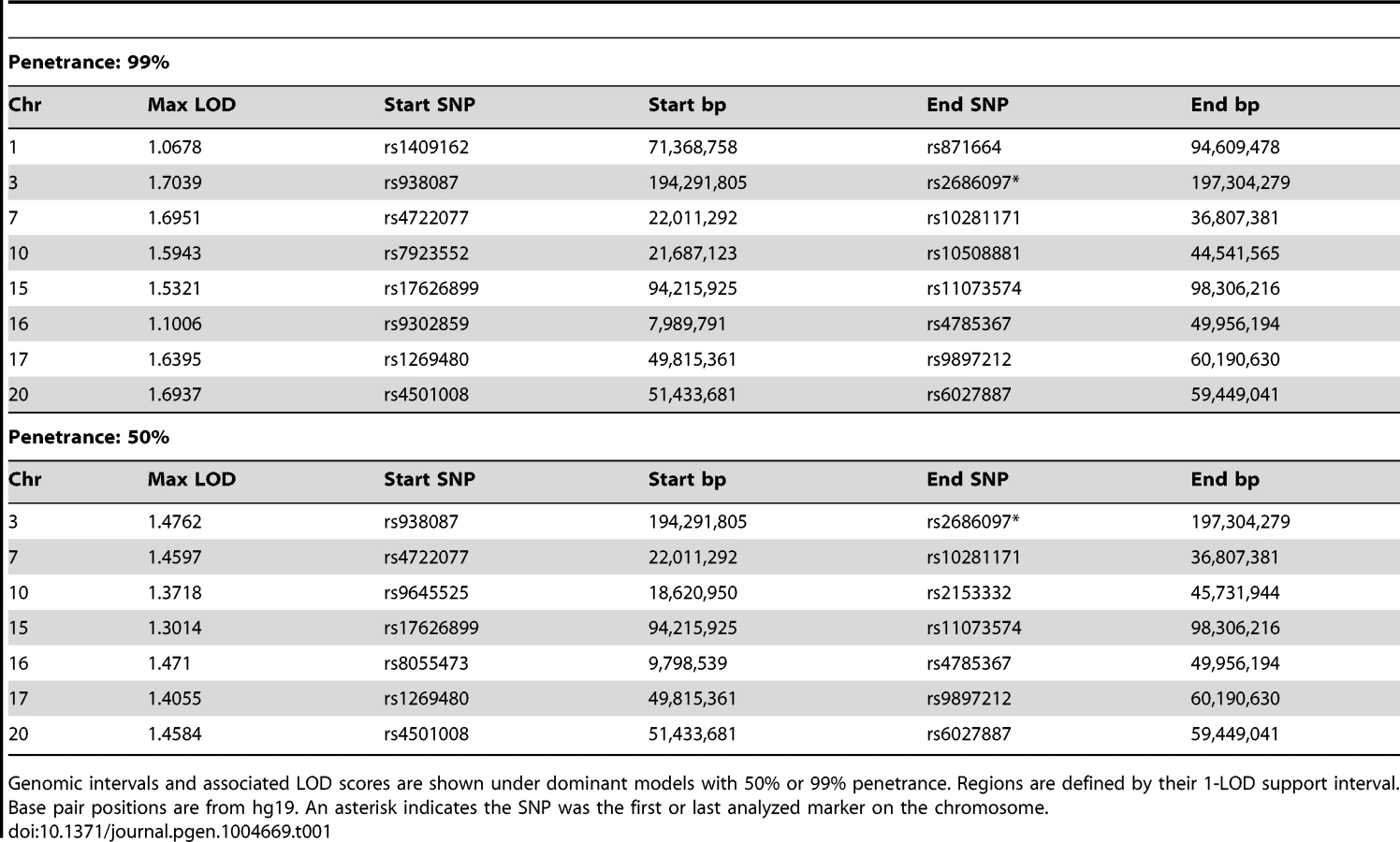

We also performed non-parametric linkage (NPL), a method with fewer underlying assumptions about the underlying inheritance model, using affected individuals 1826, 1841, 1844, and 2447 (pedigree-wide) or with the removal of 2447 as a potential phenocopy by specifying them to be of unknown status (sub-pedigree). Pedigree-wide genomic intervals were mostly consistent with those identified using the two parametric models (Table 3). Exclusion of 2447 resulted in a lower overall maximum score (1.204), which was found on several intervals throughout the genome (Table 3).

Genomic intervals identified in this manner were used for filtering the exome sequencing data to identify potential causative mutations. To be broad in identifying possible causative mutations, in both the pedigree-wide and sub-pedigree case the intervals from the respective parametric and non-parametric analyses were combined. For the pedigree-wide analyses this was the union of intervals described in Table 1 along with the appropriate intervals in Table 3 (including 2447) and for the sub-pedigree analysis the union of intervals found in Table 2 with appropriate intervals in Table 3 (excluding 2447).
Whole Exome Sequencing
We next performed whole-exome sequencing on two of the affected maternal aunts to the proband (1826 and 1841) and the affected third cousin (2447). We prioritized and filtered variants based on their frequency among European-descent populations (<2% and a stricter filter at <1%) from the 1000 Genomes and Exome Sequencing Project datasets as well as other exomes sequenced at the same sequencing provider, location within a genomic region of interest, and the functional consequence of the mutation (altering the protein coding sequence or splice site of at least one protein coding transcript). Variants of interest were then sequenced by Sanger sequencing in other affected individuals. Variant filtration based on genomic intervals was performed separately for each hypothesis (whole-pedigree and reduced-pedigree) (Tables S1 and S2). In addition to the identification and filtration of genetic variants, we assessed the sequencing depth of coverage of exons (defined by the Consensus CDS set) within genomic regions of interest and across individual exome sequencing results. Further, we searched for potentially shared variants that were “masked” by coverage issues. For all variants observed in one or more exomes, if no variant was observed in the remaining exome(s), we evaluated whether that was due to low coverage or coverage gaps within the exon. For variants where this was true we filtered using standard criteria (as above). Using these filtering criteria, several variants with low MAF and potentially having an effect at the protein-coding level were observed in the pedigree-wide genomic regions of interest that had been identified by parametric linkage analysis; however, none were present in all affected individuals. Further no “masked” candidate variants were identified by the same criteria.
We considered the possibility that individuals 1845 and 2447 have a distinct clinical condition, and examined the variants shared among the proband and immediate family. Using the same filtering criteria as above, but using only the exomes from individuals 1826 and 1841, a total of 127 variants were identified. Stricter filtering for rare variants (MAF <1%) reduced this to number to 85 (Table S2). A subset of these variants, based on a combination of factors (mutations in COSMIC [13], predicted effect of the mutation, conservation of the encoded amino acid, literature review, known expression patterns in normal tissues and tumors, disease phenotypes associated with the gene) were sequenced for follow-up in the proband and their mother. A variant in MAP3K6 (Chr1, NM_004672) was of particular interest. A mutation identified in MAP3K6 (c.[2837C>T];[ = ], p.P946L) was considered a strong candidate based on the known associations of other MAP kinases with cancer, and several publications elucidating a role for MAP3K6 in tumorigenesis [10], [11], [14], [15]. This variant has been reported previously (rs141787524) with a minor allele frequency of 0.7% in the 1000 Genomes Project (European descent group) and a frequency of 0.4% in the European-American population (Exome Variant Server (NHLBI GO Exome Sequencing Project (ESP): http://evs.gs.washington.edu/EVS [Accessed October, 2013]). It was seen as a heterozygous variant in 11 (of 1532) other exomes sequenced at the Genome Quebec Innovation Centre, corresponding to a MAF of 0.36%.
This SNV was present in four affected individuals in the Maritime family (1884, 1826, 1844, 1841), of which three clearly showed the presence of signet ring cells. Only a small punch biopsy was available for the maternal aunt, 1841, therefore we were not able to definitively confirm the presence or absence of signet ring cells. The MAP3K6 SNV was also present in five of the 27 currently unaffected relatives sampled, and it was not present in the married-in relative. One of the carriers was homozygous for the SNV and was over 80 years old with no reported cancer. Although no consanguinity was reported in the family, and no evidence of copy number variation was observed in the SNP genotype data, this individual was also homozygous for a 10 Mb region encompassing the locus. The remaining carriers ranged in age from 33 to 51, and as the age of onset of the cancer was generally later, their status was considered “unknown”. Both individuals, 1845 and 2447, with the phenotypically distinct gastric cancer were negative for the MAP3K6 SNV.
Somatic Variants within the Tumor
We next used the DNA isolated from a tumorous section of the FFPE sample of the affected MAP3K6 SNV carrier 1884 (the proband) to screen for additional somatic SNVs or loss of heterozygosity (LOH) within the tumor itself. In addition to the p.P946L variant, we identified a novel SNV in the MAP3K6 gene at position c.[1516C>T] leading to an amino acid change p.H506Y (Table 4), and were able to infer that the SNV was somatically acquired based on sequence data from the spouse and children.

Verification in Unrelated FGC Cases
We screened DNA samples from an additional 115 unrelated FGC individuals using a multiplexed targeted next generation sequencing assay. Samples were from unrelated families that met international gastric cancer linkage consortium (IGCLC) criteria for hereditary diffuse gastric cancer (106), but had previously tested negative for mutation of the CDH1 locus, or familial intestinal gastric cancer. Within this cohort, we identified five additional heterozygous SNVs in the MAP3K6 gene (Table 4): a truncating SNV (c.[2544delC], p.F849Sfs*142), three missense SNVs (c.[2872C>A], p.P958T; c.[598G>T], p.D200Y; and c.[620T>G], p.V207G), and another individual with the p.P946L (c.[2837C>T]) variant, previously found in the Maritime family (Figure 4). Mutations in MAP3K6 were only discovered in the individuals meeting the criteria for diffuse gastric cancer (106). The individual in this novel patient cohort carrying the p.P946L variant is thought to be unrelated to the Maritime family.

The truncating SNV was observed in a Portuguese individual with a family history of gastric cancer (Figure 3A). While this SNV has a dbSNP identifier (rs34008139), no population frequency has been associated with it from either the 1000 Genomes or Exome Variant Server projects. This SNV has also been reported in the COSMIC database [13] (somatic/germline status not specified) in a carcinoma sample of the large intestine. Histological examination of the proband's tumor showed a poorly differentiated gastric cancer with signet ring cells, retaining E-cadherin protein expression at the cell membrane (Figure 3B and 3C).
The p.D200Y variant was found in probands from two unrelated families, and has been observed within the 1000 genomes cohort (rs41291098), but is also rare with a minor allele frequency (MAF) of 0.4% in both the 1000 Genomes and Exome Sequencing Project European descent groups. The p.P958T variant (rs75893867) has no MAF reported in either the 1000 Genomes or Exome Sequencing Project European descent datasets, and was only identified in the 1000 Genomes among the Japanese cohort at a frequency of 2.2%. However, it has been identified in COSMIC (COSM99077) as a somatic mutation from a gastric carcinoma patient. The p.V207G variant has been identified in the Exome Sequencing Project European-American dataset with a MAF of 0.01%. It was not identified among European or European descent groups within the 1000 Genomes, but was observed within other population groups at a range of minor allele frequencies.
Along with MAP3K6, 50 additional genes previously suggested to be involved in risk for disease of the upper gastrointestinal tract were sequenced for this cohort using a custom panel-based assay (manuscript submitted). Genes for the custom MiSeq-based screen were selected based on literature review, as well as genes of interest in collaborative projects. In the cases where MAP3K6 missense variants were identified, no other candidate variants were found.
Second-Hit Analysis in the Tumor from the Portuguese Family
FFPE tumor tissue was available for the proband from the Portuguese family carrying the p.F849Sfs*142 germline truncating mutation. Somatic mutations were excluded in the complete coding sequence and intron-exon boundaries of MAP3K6 in the proband's tumor. Lack of LOH at the MAP3K6 gene could also be inferred in this tumor, as both wild-type and mutant alleles could be identified at the germline mutation site (Supplementary Figure S2). We therefore searched for putative alternative inactivating mechanisms. Hypermethylation of CpG islands within gene promoters and regulatory regions is a common phenomenon leading to decreased gene expression in cancer [16]. MAP3K6 regulation by promoter hypermethylation has been described for human bone marrow mesenchymal stem cell [17], although a correlation of hypermethylation and gene expression has not been established. We searched for MAP3K6 CpG islands [18] and found two CpG islands, one at the promoter region and another encompassing exon 10 and part of the downstream intron (Figure 5 and Supplementary Figure S1). The downstream CpG island (CpG island 2) is near a DNase hypersensitive site predicted to harbor promoter associated features (Ensembl regulatory feature ID ENSR00000533270, Figure 5). We bisulfite-treated DNA from: the proband's peripheral blood lymphocytes (PBLs); the proband's tumor; four different normal stomach control samples, and; seven gastric cancer cell lines. For the promoter CpG island (CpG island 1), no hypermethylation was detected using two different primer sets (Figure 5 and Suppl. Figure S1). Regarding CpG island 2, we observed complete methylation for the tumor DNA and no methylation for the PBLs DNA. Interestingly, the methylation analysis at CpG island 2 in normal stomach mucosa from controls displayed a partial methylation pattern. In line with the result obtained for tumor DNA, all seven gastric cancer cell lines displayed full methylation (Figure 5 and Suppl. Figure S1).

Pathogenicity of SNVs in MAP3K6
We applied a variety of in silico methods to predict the pathogenicity of the observed missense SNVs. Although there was no full consensus across programs (Table 5), all of the SNVs were considered deleterious by at least one program, and except p.V207G and p.P958T, the other four variants described in this report were predicted to be deleterious by at least 3 of the 7 methods. In addition, the EvoD [19] consensus prediction (based on a balanced combination of the EvoD, PolyPhen2 [20], and SIFT [21] scores) reported that three of the variants (p.D200Y, p.V207G, p.H506Y) were deleterious or likely to be deleterious. The two remaining variants, p.P946L and p.P958T, were predicted to likely be neutral changes although they were evolutionarily ultra-conserved and well-conserved (according to EvoD evolutionary rate classification) respectively; while only one of the other three variants (p.D200Y) was considered well-conserved and the other two were less-conserved. As (i) a MAP3K6 truncating variant was found, (ii) there was a second-hit variant identified in the individual for whom FFPE tissue was examined, and (iii) all of the programs tested are designed to predict pathogenicity based on loss of function, it is likely that the variants described lead to either a decrease in function or dominant negative phenotype.

Discussion
Here we present the first evidence that germline mutations in MAP3K6 are linked to inherited cancer. Four individuals with gastric cancer from a Maritime Canadian family were found to carry a heterozygous variant in the MAP3K6 gene, leading to a p.P946L amino acid change. This germline variant, located on chromosome 1, was identified in two of the three exome samples and was located within a region identified by parametric and non-parametric linkage analysis within the sub-pedigree (1845 and 2447 treated as “unknown” disease status). The significance of the MAP3K6 variant was supported by the identification of a somatic second-hit mutation in the MAP3K6 gene at p.H506Y present in DNA isolated directly from a tumorous section of a patient FFPE sample. Two individuals from the pedigree with gastric cancer, but with some phenotypic differences did not carry the mutation; however, no candidate variants were identified shared among all affected individuals within any region identified, by parametric linkage conducted pedigree-wide.
Screening of additional FGC families revealed five other MAP3K6 mutations, including a p.F849Sfs*142 germline mutation observed in the Portuguese proband, which is expected to lead to protein truncation. After excluding somatic mutations and LOH, a potential second-hit mechanism was found via hypermethylation at an intragenic CpG island near a predicted promoter-associated regulatory element (DNAse I hypersensitive site). The relevance of methylation at this MAP3K6 gene region could not be ascertained in terms of impact in gene expression, nevertheless the possibility of acting as a possible second-hit inactivation (partial or complete) mechanism is raised, due to the results obtained in normal stomach and cancer cell lines. If so, this may well represent another example of the increasingly recognized concept that DNA methylation in the gene body is not just a passive witness of gene transcription, but is actively involved in multiple gene regulation processes [22], warranting further investigation. Histopathology analysis of the individual from the Portuguese family carrying this truncating mutation featured signet-ring cells as part of the tumor phenotype, as did most individuals from the Maritime sub-pedigree (except 1841, where the signet ring status was inconclusive due to lack of sufficient material).
Although MAP3K6 mutations have not previously been identified in inherited cancer, there is a growing body of evidence that MAP3K6 has an important role in cancer pathogenesis. In mice, where MAP3K6 is normally expressed in gastric tissue and skin, the loss of MAP3K6 in homozygous knockout mice was found to increase susceptibility to induced skin cancer [10]. The mice did not develop cancer spontaneously; however, chemical induction performed in the presence of an inflammatory stimulus led to a greater number of skin tumors in the MAP3K6 deficient mice than in control animals. The number of tumors in heterozygous (MAP3K6+/−) mice, as well as their size, was intermediate between the wild-type and knock-out mice, suggesting a role for MAP3K6 dosage in its effects [10]. The susceptibility of these mice to gastric cancer was not assessed.
MAP3K6 is a member of the c-Jun N-terminal Kinase (JNK) and p38 signaling cascades [14] that is most strongly expressed in the skin, gastrointestinal tract, and lungs [10]. MAP3K6 forms a heteromeric complex with its paralog MAP3K5, through their coiled-coil domains, preventing constitutive MAP3K6 degradation and promoting its autophosphorylation and activation [14]. In the presence of oxidative stress and reactive oxygen species (ROS) such as occurs following chemical (eg. DMBA) or UVA induction, active phospho-MAP3K6 activates the JNK and p38 cascades and promotes apoptosis [10], [14]. These effects have been seen in chemically induced skin-cancer models in MAP3K6−/− mice as well as primary keratinocyte cell lines [10].
While MAP3K6 has a limited tissue and cell-type distribution, MAP3K5 is more broadly expressed. The interaction of these two proteins appears to be tightly regulated, with a balance necessary between the pro-apoptotic and tumor suppressing roles of MAP3K6 and the pro-inflammatory/anti-apoptotic roles of MAP3K5 [10], [14], [15], [23]. Expression of MAP3K6 is variable in many human tumors, with expression most significantly reduced in gastric cancer tumors compared to healthy gastric tissue [10], whereas MAP3K5 expression is increased [23]. Indeed MAP3K5−/− mice are more resistant to chemically induced gastric cancers than wild-type mice. It is clear that MAP3K5 and MAP3K6 are finely balanced to carry out both inflammatory and apoptotic roles, respectively in tissues where they are both expressed. Somatic mutations in MAP3K6 have been reported in screens of a panel of 532 kinase genes in apparent non-familial gastric cancer cases (p.S291L), as well as in two gastric cancer cell lines (N87 cells: p.R375Q, and IM95 cells: p.P958T) [11]. Somatic mutations in MAP3K6 have also been identified in other, non-gastric cancers, including ovarian (p.T968I [24]) and breast cancers (p.P869T[25], p.S648L, p.Q672* [24])). Furthermore, although expression of MAP3K6 is variable in cervical and ovarian cancer, there is a decrease in MAP3K6 expression, compared to normal tissue, in 75% out of 106 oral, esophageal, gastric and colorectal cancer cell lines tested [10] suggesting that MAP3K6 may have a tumor suppressive role in cancers of the gastrointestinal tract. Our results demonstrate that inherited mutations in MAP3K6 may predispose individuals to gastric cancer; however, regulation of this pathway could have a broader role in both sporadic gastric cancers and carcinogenesis in general that warrants further investigation.
The penetrance of the disease in individuals carrying the p.P946L mutation is incomplete, with the mutation identified in 5 out of the 27 as yet unaffected individuals tested. Surprisingly, this included one individual who was homozygous for the mutation and was over the age of 80 with no reported cancer. However, there is significant variability in the age of onset (51–82 at diagnosis) in this family, unlike many other hereditary cancer syndromes, which feature an early age of onset. In particular, the average age of onset in this family is significantly higher than that typically diagnostic of HDGC. The Maritime Canadian p.P946L mutation may be hypomorphic, resulting in reduced function but not a complete loss of activity, contributing to the late age of onset and incomplete penetrance within the family, and offering a possible explanation for the individual who is homozygous for the mutation, but has not yet demonstrated signs of gastric cancer at the age of 80.
The complexity between genotype and phenotype is illustrated in Familial Adenomatous Polyposis 2 (FAP2, MUTYH-associated Polyposis), where even in recessive cancer-predisposing syndromes the penetrance and age of onset can vary significantly between pathogenic mutations within the same gene. FAP2 is a recessive disorder caused by compound heterozygous or homozygous mutations in the mismatch repair gene mutY homolog (MUTYH). FAP2 is characterized by an extremely elevated risk of colorectal cancer (CRC). Average age of onset of CRC in FAP2 is 48–56. Penetrance for CRC among biallelic carriers has been estimated at approximately 80% by age 80 [26] due to the presence of homozygous carriers of of the common pathogenic p.T165C (p.T179C, rs34612342) mutation in the control group of at least two case-control studies [26], [27]. This mutation is known to be pathogenic both in the homozygous state as well as a compound heterozygous mutation with other pathogenic variants. A homozygous individual for the other common FAP2 mutation, p.G396D (rs36053993), is also found in the Exome Sequencing Project (European-American) dataset. Interestingly, among CRC studies of MUTYH mutations while the p.T165C homozygous mutation is associated with a lower odds ratio it is generally also associated with a lower mean age of onset (48.9 versus 56.7) and more severe phenotype [28].
Incomplete penetrance of other recessive Mendelian disease is known in other cases including Leber Congenital Amaurosis [29], Schimke immuno-osseous dysplasia [30], and Bardet-Biedl syndrome [31], [32]. In those cases it is expected that the homozygous carrier of a hypomorphic allele retains more protein function than more severe combinations of compound heterozygous or the hypomorphic allele in combination with complete loss of the other allele or its expression. In addition to hypomorphism, the homozygous combination of p.P946L alleles may also be displaying over-dominance as during stress signaling MAP3K6 molecules likely form homo-dimers as well as heteromeric complexes with MAP3K5 [14], [33]. If the p.P946L variant impacts this self-interaction the homozygous state may confer some protection over the heterozygous state.
Alternatively, the mutation may confer risk, but as with other cancers, additional environmental and genetic factors likely play a role in the progression to gastric cancer. This is consistent with experiments in MAP3K6 knock-out mice where cancer was only observed after the administration of both a carcinogen and an inflammatory agent, suggesting that the presence of additional stimuli are required [10]. Other variants segregating within the family may modulate both overall and individual risk of gastric cancer.
We do not know the importance of H. pylori in driving gastric cancer in the setting of this gene, as H. pylori infection was not a consistent finding among affected individuals from the Maritime Canadian family, and the rate was not different from what would be expected in the general population in this region [34], [35]. It is possible that a second, as yet unidentified genetic abnormality not detected in the exome sequencing data is also contributing to disease in that pedigree. Several regions of the genome, including in the pedigree-wide analysis, displayed significant LOD or NPL scores. The region on chromosome 1, where MAP3K6 is located, was only identified in the reduced pedigree, where 2447 and 1845 were treated as unknown. Several regions identified pedigree-wide with high LOD scores (Table 1) are still of potential interest, and may indicate a second allele also segregating within the family contributing to the disease, with MAP3K6 mutations modifying the phenotype in the Maritime pedigree. Regions on chromosomes 3, 7, 17, and 20 displayed consistently high LOD scores in the pedigree-wide analysis, regardless of the specified penetrance (99% or 50%) while one on chromosome 16 was identified under both models but whose LOD score was more strongly affected by the penetrance parameter.
The truncating SNV identified in the Portuguese pedigree is much more likely to result in complete loss of function of MAP3K6, as the frameshift and truncation occur in the region of the protein involved in activation and interaction with MAP3K5. While penetrance of this mutation and segregation within the pedigree is unknown, penetrance is expected to be higher than that for the p.P946L mutation. This individual was diagnosed with Hereditary Diffuse Gastric Cancer at the age of 62, intermediate between early-onset HDGC and the late-onset observed in the Maritime Canadian pedigree. The impact of the other germline variants discovered in the additional probands is not clear.
The identification of new germline mutations that appear to predispose individuals to familial gastric cancer can aid in identifying at risk individuals in affected families that are negative for CDH1 mutations. It may also help to shed light on the underlying mechanisms leading to cancer development. Further, somatic mutations and altered expression of MAP3K6 in sporadic cancers and gastric cancer cell lines may suggest MAP3K6 as a potential therapeutic target for exploration. Its binding partner in the heteromeric complex, MAP3K5, is already being investigated for therapeutic potential [36]–[39].
The presence of MAP3K6 mutations in the probands of six unrelated families, somatic mutations in sporadic cancers (particularly those of the gastrointestinal tract), evidence from MAP3K6 knockout mice, second-hit mutations or hypermethylation of the wild-type allele in the tumors tested, and its molecular role in inflammation and apoptosis, all suggest that MAP3K6 is an interesting candidate for mutations associated with Familial Gastric Cancer, warranting further study in additional cohorts of CDH1 mutation-negative familial gastric cancer cases.
Methods
Ethics Statement
Approval for the research study was obtained from the IWK Health Centre research ethics board (project approval number 1005367). Informed consent was obtained from individuals or their guardians for all samples used in this study. DNA was obtained from blood, saliva or FFPE samples using standard methods
Data Availability
Due to the small number of samples and nature of the study, genetic information can reveal potentially identifiable and unrelated health data of individuals from the family, including individuals who were not enrolled in this study. For this reason, the research ethics approval of this study and informed consent signed by participants does not allow for data to be deposited in public databases. Data used in this study are available upon request from the corresponding author pending approval from the Maritime Medical Genetics Service at the IWK Health Science Centre at: Maritime Medical Genetics Service, PO Box 9700, Halifax, Nova Scotia, Canada, B3K 6R8. Phone +1-902-470-8754.
SNP Genotyping and Genomic Mapping
Whole genome high-density single nucleotide polymorphism (SNP) genotype scanning was performed at the McGill University and Genome Quebec Centre for Innovation, using the Illumina Human610-Quadv1_B chip (Illumina, Inc., San Diego, CA) panel with 620,901 markers and the Illumina HumanOmni 2.5M panel. DNA for SNP genotyping was isolated from either saliva or blood collected from patients. Genotype arrays were scanned using the Bead Array Reader (Illumina, Inc.), plate Crane Ex, and Illumina BeadLab software (Illumina, Inc.). Initial quality control and export of data was done using Illumina's GenomeStudio software.
Before linkage analysis was performed, the set of SNPs was pruned to obtain a subset that was appropriate for linkage analysis. Out of the 2,391,739 markers passing QC, only markers on the autosomes were retained. Markers with alleles ambiguous for strand information (A/T and G/C variants) were removed, in order to facilitate strand matching with HapMap data. Markers that were monomorphic in all genotyped samples were removed. At this point, 1,008,604 markers remained. This set of SNPs was merged with HapMap3 CEU data; only markers present in both sets of data were kept, matching on marker name. This resulted in 476,551 markers. From this set, only markers with MAF>0.4 were kept. In order to avoid inconsistencies between the observed data and the genetic map, markers with unique positions on the genetic map were selected. Arbitrarily, for a group of markers with the same genetic position, the marker with the lowest physical position was retained, and the remaining markers in the group were discarded. Finally, since linkage disequilibrium (LD) between the SNPs can arbitrarily inflate multipoint LOD scores [40], the markers were pruned to remove strong pairwise LD (r2<0.1 on a chromosome). This resulted in a set of 8,472 SNPs across the genome that were roughly independent, and should have high informativity for linkage analysis. For this set, the average intermarker distance was 0.57 cM, or 451 kb. Filtering was performed using a combination of PLINK v1.07 [41] and in-house scripts.
Merlin 1.1.2 [12] was used to perform multipoint non-parametric linkage on the family, using the set of 8,472 SNPs selected above. Only affected individuals will contribute to this analysis; the individuals with unknown affection status will only help infer phase. The Sall statistic was used, which looks for allele sharing in all affected individuals, and tends to perform well for dominant traits [42]. The exponential model of Kong and Cox was used, which is preferred when a small number of families are analyzed [43]. CEU genotypes from HapMap3 release 28 [44] were used to estimate allele frequencies. Since it is possible that sample 2447, a distant cousin to the other affected individuals, had a slightly different phenotype, the analysis was repeated, coding this individual as having an unknown affection status, effectively removing him from the analysis. For the pedigree-wide analysis regions with an NPL score >2 were selected, with boundaries defined by markers flanking a set of SNPs with p<0.05. For the reduced pedigree with sample 2447 excluded regions with an NPL score >1 were selected.
Parametric linkage analysis was also performed, under two dominant models, one with 50% and the other with 99% penetrance. Both models used a disease allele frequency of 0.001 and 2% phenocopy rate. The analyses were performed using Merlin. Regions with LOD>1 and with SNP boundaries defined by 1 – LOD support interval were used for further analysis. Similarly to the NPL analysis above, two sets of analyses were performed with 2447 coded as either affected or unknown.
Whole Exome Sequencing
A total of 3 µg of DNA was used for exome capture with the Agilent SureSelect All Exon 38 Mb kit. Sequencing was performed with 100-bp paired-end reads using the Illumina HiSeq 2000 at the McGill University and Genome Quebec Centre for Innovation as previously described [45]. Reads were assembled against the human genomic reference sequence (hg19) using the Burrows-Wheeler Aligner (BWA) [46]. Genomic variants were called using the Genome Analysis Toolkit (GATK) pipeline [47], and annotated with SnpEff [48] and GEMINI [49]. All variants were compared against dbSNP [50], 1000 Genomes Project [51], the Exome Sequencing Project (Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP): http://evs.gs.washington.edu/EVS [Accessed October, 2013]), and a pool of exomes sequenced at the Genome Quebec Innovation Centre (1532 samples). Potentially damaging variants included non-synonymous mutations (missense and nonsense), splice-site variants, and frameshift changes due to insertions and/or deletions (indels). Exome variants were further filtered by their location in regions of the genome not excluded by linkage analysis. Variants were further selected based on their predicted impact by snpEff and GEMINI (Medium and High Impact) as well as their minor allele frequency in public databases and additional exomes sequenced at Genome Quebec (MAF< = 2%). Selected mutations in candidate genes were verified and screened in additional family members by Sanger sequencing.
PCR and Direct Sanger Sequencing
Selected regions were amplified from genomic DNA by PCR. Amplified fragments were purified from an agarose gel, and sequenced using Sanger fluorescent sequencing and capillary electrophoresis. Sequence traces were analyzed using MutationSurveyor V.3.97 (Soft Genetics, Inc.). Because of a lack of a fresh DNA sample, initially the three children of 1884 and his spouse, 1885 were used for genotyping. The presence of the heterozygous mutation (c.[2837C>T];[ = ]) at the genomic level could be inferred for the affected individual 1884 by the absence of the mutation in his spouse, 1885, but the presence of the mutation in their child, 1903 and not in their children 1902 and 1879. The mutation was then confirmed in DNA derived from a tumorous section of an FFPE sample from this individual. An additional second-hit mutation was observed in this FFPE-derived DNA, which could be inferred to be somatic based on its absence in all three children despite both paternal copies being represented among them. In the Portuguese family, FFPE-derived DNA was extracted from the family proband's tumor (macrodissected to guarantee a minimum of 75% of tumor cells) and was used for somatic mutation analysis of the full MAP3K6 coding sequence and intron-exon boundaries. LOH was inferred from the Sanger sequencing data, using the germline mutation site as an intragenic marker. LOH would have been considered if only the mutant allele had been found in the proband's tumor.
Validation in an Unrelated Population
DNA previously isolated from the blood of 115 FGC patients that had been demonstrated to be negative for CDH1 mutations were sequenced for the presence of mutations in MAP3K6 using a multiplexed TruSeq Custom Amplicon (TSCA) sequencing assay (Illumina, San Diego). Samples were first quantified using Qubit dsDNA broad ranged assay kit (Life Technologies) and custom oligos were pooled and hybridized to individual samples. Sample indices were then added to each template library by PCR using the TSCA reagents and protocol (Illumina, San Diego). A post-PCR bead-based normalization technique was used according to the TSCA protocol to avoid the necessity of laborious and time-consuming quantification methods. Samples were then pooled prior to loading onto the MiSeq platform for simultaneous cluster generation, bi-directional sequencing and data analysis. Amplicons were designed only for coding regions (exons) of the targeted areas of interest plus up to 25 base pairs of padding around each individual amplicon. Coverage across each amplicon varied depending on location and quality of sample. In the sample where the germline MAP3K6 frameshift variant was found, there was 80× coverage of the amplicon of interest.
Detection of MAP3K6 Hypermethylation
We have submitted the MAP3K6 genomic sequence to a CpG island searcher (http://cpgislands.usc.edu/, [18]) and identified two CpG islands that were also annotated in ENSEMBL (release 75; www.ensembl.org). CpG island 1 was located at the promoter region and CpG island 2 encompassed exon 10 and part of the downstream intron. MAP3K6 methylation analysis was performed in the Portuguese family proband, for which FFPE tumor and peripheral blood lymphocytes (PBLs) DNA was available. Additionally, we analyzed DNA from normal stomach mucosa from controls (n = 4), and 7 gastric cancer cell lines DNA (MKN28, MKN45, NCI-N87, Kato III, AGS, GP202 and IPA220). The EpiTect Bisulfite Kit (Qiagen, Valencia, Calif) was used to treat 300 ng of DNA per sample. Unmethylated cytosines were converted to uracil, whereas methylated ones remained unmodified. A fraction of CpG sites contained within CpG islands 1 and 2 were PCR amplified using flanking primers, specifically designed for bisulfite treated DNA sequences without CpG sites, and sequenced for methylation status determination (primer sequences available upon request). Independent PCR reactions were performed at least twice for each sample.
Predicting Pathogenicity of Detected Mutations
For all mutations detected in this work, either germline or somatic, the potential pathogenicity was predicted using a variety of published bioinformatic methods. Because individual tools are known to have differing performance profiles (both in terms of false positives and false negatives), we employed a variety of different tools that use differing algorithms for our assessment. In this work we evaluated all relevant mutations using PMut [52], PolyPhen2 [20], [53], SIFT [21], Provean [54], MutationTaster [55], EvoD [19], and FATHMM [56]. In particular FATHMM has been updated [57] with an algorithm specific to somatic mutations in cancer, which has also been used here. Unless otherwise noted default options were used with all prediction programs for their default web interface. Both PROVEAN and SIFT scores were provided through the PROVEAN interface.
Supporting Information
Zdroje
1. FerlayJ, ShinH-R, BrayF, FormanD, MathersC, et al. (2010) Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. 127 : 2893–2917 doi:10.1002/ijc.25516
2. WadhwaR, SongS, LeeJ-S, YaoY, WeiQ, et al. (2013) Gastric cancer-molecular and clinical dimensions. Nat Rev Clin Oncol 10 : 643–655 doi:10.1038/nrclinonc.2013.170
3. LaurenP (1965) The two histological main types of gastric carcinoma, an attempt at a histoclinical classification. Acta Pathol Microbiol Scand 64 : 31–49.
4. ParkinDM (2006) The global health burden of infection-associated cancers in the year 2002. Int J Cancer 118 : 3030–3044 doi:10.1002/ijc.21731
5. OliveiraC, SerucaR, CarneiroF (2009) Hereditary gastric cancer. 23 : 147–157 doi:10.1016/j.bpg.2009.02.003
6. GuilfordP, HopkinsJ, HarrawayJ, McLeodM, McLeodN, et al. (1998) E-cadherin germline mutations in familial gastric cancer. Nature 392 : 402–405 doi:10.1038/32918
7. FitzgeraldRC, HardwickR, HuntsmanD, CarneiroF, GuilfordP, et al. (2010) Hereditary diffuse gastric cancer: updated consensus guidelines for clinical management and directions for future research. J Med Genet 47 : 436–444 doi:10.1136/jmg.2009.074237
8. ParkJ-G, YangH-K, KimWH, CaldasC, YokotaJ, et al. (2000) Report on the first meeting of the International Collaborative Group on Hereditary Gastric Cancer. J Natl Cancer Inst 92 : 1781–1782 doi:10.1093/jnci/92.21.1781
9. OliveiraC, SenzJ, KaurahP, PinheiroH, SangesR, et al. (2009) Germline CDH1 deletions in hereditary diffuse gastric cancer families. Hum Mol Genet 18 : 1545–1555 doi:10.1093/hmg/ddp046
10. IriyamaT, TakedaK, NakamuraH, MorimotoY, KuroiwaT, et al. (2009) ASK1 and ASK2 differentially regulate the counteracting roles of apoptosis and inflammation in tumorigenesis. The EMBO Journal 28 : 843–853 doi:10.1038/emboj.2009.32
11. ZangZJ, OngCK, CutcutacheI, YuW, ZhangSL, et al. (2011) Genetic and structural variation in the gastric cancer kinome revealed through targeted deep sequencing. Cancer Res 71 : 29–39 doi:10.1158/0008-5472.CAN-10-1749
12. AbecasisGR, ChernySS, CooksonWO, CardonLR (2002) Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nature Genetics 30 : 97–101 doi:10.1038/ng786
13. ForbesSA, BindalN, BamfordS, ColeC, KokCY, et al. (2011) COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 39: D945–D950 doi:10.1093/nar/gkq929
14. TakedaK, ShimozonoR, NoguchiT, UmedaT, MorimotoY, et al. (2007) Apoptosis signal-regulating kinase (ASK) 2 functions as a mitogen-activated protein kinase kinase kinase in a heteromeric complex with ASK1. Journal of Biological Chemistry 282 : 7522–7531 doi:10.1074/jbc.M607177200
15. EtoN, MiyagishiM, InagiR, FujitaT, NangakuM (2009) Mitogen-activated protein 3 kinase 6 mediates angiogenic and tumorigenic effects via vascular endothelial growth factor expression. Am J Pathol 174 : 1553–1563 doi:10.2353/ajpath.2009.080190
16. BaylinSB, EstellerM, RountreeMR, BachmanKE, SchuebelK, et al. (2001) Aberrant patterns of DNA methylation, chromatin formation and gene expression in cancer. Hum Mol Genet 10 : 687–692 doi:10.1093/hmg/10.7.687
17. ChoiMR, InY-H, ParkJ, ParkT, JungKH, et al. (2012) Genome-scale DNA methylation pattern profiling of human bone marrow mesenchymal stem cells in long-term culture. Exp Mol Med 44 : 503 doi:10.3858/emm.2012.44.8.057
18. TakaiD, JonesPA (2003) The CpG island searcher: a new WWW resource. In silico biology 3 : 235–240.
19. KumarS, SanderfordM, GrayVE, YeJ, LiuL (2012) Evolutionary diagnosis method for variants in personal exomes. Nat Methods 9 : 855–856 doi:10.1038/nmeth.2147
20. AdzhubeiIA, SchmidtS, PeshkinL, RamenskyVE, GerasimovaA, et al. (2010) A method and server for predicting damaging missense mutations. Nat Methods 7 : 248–249 doi:10.1038/nmeth0410-248
21. NgPC, HenikoffS (2003) SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31 : 3812–3814 doi:10.1093/nar/gkg509
22. KulisM, QueirósAC, BeekmanR, Martín-SuberoJI (2013) Intragenic DNA methylation in transcriptional regulation, normal differentiation and cancer. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms 1829 : 1161–1174 doi:10.1016/j.bbagrm.2013.08.001
23. HayakawaY, HirataY, NakagawaH (2011) Apoptosis signal-regulating kinase 1 and cyclin D1 compose a positive feedback loop contributing to tumor growth in gastric cancer Vol. 108. 780–785 doi:10.1073/pnas.1011418108/-/DCSupplemental
24. GreenmanC, StephensP, SmithR, DalglieshGL, HunterC, et al. (2007) Patterns of somatic mutation in human cancer genomes. Nature 446 : 153–158 doi:10.1038/nature05610
25. SjöblomT, JonesS, WoodLD, ParsonsDW, LinJ, et al. (2006) The consensus coding sequences of human breast and colorectal cancers. Science 314 : 268–274 doi:10.1126/science.1133427
26. LubbeSJ, Di BernardoMC, ChandlerIP, HoulstonRS (2009) Clinical implications of the colorectal cancer risk associated with MUTYH mutation. Journal of Clinical Oncology 27 : 3975–3980 doi:10.1200/JCO.2008.21.6853
27. ClearySP, CotterchioM, JenkinsMA, KimH, BristowR, et al. (2009) Germline MutY Human Homologue Mutations and Colorectal Cancer: A Multisite Case-Control Study. Gastroenterology 136 : 1251–1260 doi:10.1053/j.gastro.2008.12.050
28. TheodoratouE, CampbellH, TenesaA, HoulstonR, WebbE, et al. (2010) A large-scale meta-analysis to refine colorectal cancer risk estimates associated with MUTYH variants. Br J Cancer 103 : 1875–1884 doi:10.1038/sj.bjc.6605966
29. SiemiatkowskaAM, Schuurs-HoeijmakersJHM, BoschDGM, BoonstraFN, RiemslagFCC, et al. (2014) Nonpenetrance of the Most Frequent Autosomal Recessive Leber Congenital Amaurosis Mutation in NMNAT1. JAMA Ophthalmol 132 : 1002–4 doi:10.1001/jamaophthalmol.2014.983
30. Baradaran-HeraviA, ChoKS, TolhuisB, SanyalM, MorozovaO, et al. (2012) Penetrance of biallelic SMARCAL1 mutations is associated with environmental and genetic disturbances of gene expression. Hum Mol Genet 21 : 2572–2587 doi:10.1093/hmg/dds083
31. Estrada-CuzcanoA, KoenekoopRK, SenechalA, De BaereEBW, de RavelT, et al. (2012) BBS1 Mutations in a Wide Spectrum of Phenotypes Ranging From Nonsyndromic Retinitis Pigmentosa to Bardet-Biedl Syndrome. Arch Ophthalmol 130 : 1425–1432 doi:10.1001/archophthalmol.2012.2434
32. BealesPL, BadanoJL, RossAJ, AnsleySJ (2003) Genetic Interaction of BBS1 Mutations with Alleles at Other BBS Loci Can Result in Non-Mendelian Bardet-Biedl Syndrome. Am J Hum Gen doi:10.1086/375178
33. OrtnerE, MoellingK (2007) Heteromeric complex formation of ASK2 and ASK1 regulates stress-induced signaling. Biochemical and Biophysical Research Communications 362 : 454–459 doi:10.1016/j.bbrc.2007.08.006
34. van ZantenSJOV, PollakPT, BestLM, BezansonGS, MarrieT (1994) Increasing Prevalence of Helicobacter pylori Infection with Age: Continuous Risk of Infection in Adults Rather than Cohort Effect. J Infect Dis 169 : 434–437 doi:10.1093/infdis/169.2.434
35. GoodmanKJ, JacobsonK, Veldhuyzen van ZantenS (2008) Helicobacter pylori infection in Canadian and related Arctic Aboriginal populations. Can J Gastroenterol 22 : 289–295.
36. SinghO, ShillingsA, CraggsP, WallI, RowlandP, et al. (2013) Crystal structures of ASK1-inhibtor complexes provide a platform for structure-based drug design. Protein Science 22 : 1071–1077 doi:10.1002/pro.2298
37. JinL, LiD, LeeJS, ElfS, AlesiGN, et al. (2013) p90 RSK2 Mediates Antianoikis Signals by both Transcription-Dependent and-Independent Mechanisms. … and cellular biology. doi:10.1128/MCB.01677-12
38. CaiD-T, JinH, XiongQ-X, LiuW-G, GaoZ-G, et al. (2013) ER stress and ASK1-JNK activation contribute to oridonin-induced apoptosis and growth inhibition in cultured human hepatoblastoma HuH-6 cells. Mol Cell Biochem 379 : 161–169 doi:10.1007/s11010-013-1638-2
39. HayakawaR, HayakawaT, TakedaK (2012) Therapeutic targets in the ASK1-dependent stress signaling pathways Vol. 88. 434–453 doi:10.2183/pjab.88.434
40. HuangQ, SheteS, AmosCI (2004) Ignoring linkage disequilibrium among tightly linked markers induces false-positive evidence of linkage for affected sib pair analysis. Am J Hum Gen 75 : 1106–1112 doi:10.1086/426000
41. PurcellS, NealeB, Todd-BrownK, ThomasL, FerreiraMAR, et al. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Gen 81 : 559–575 doi:10.1086/519795
42. WhittemoreAS, HalpernJ (1994) A class of tests for linkage using affected pedigree members. Biometrics 50 : 118–127.
43. KongA, CoxNJ (1997) Allele-sharing models: LOD scores and accurate linkage tests. Am J Hum Gen 61 : 1179–1188 doi:10.1086/301592
44. International HapMap 3 Consortium (2010) AltshulerDM, GibbsRA, PeltonenL, DermitzakisE, et al. (2010) Integrating common and rare genetic variation in diverse human populations. Nature 467 : 52–58 doi:10.1038/nature09298
45. AlfaresA, NunezLD, Al-ThihliK, MitchellJ, MelançonS, et al. (2011) Combined malonic and methylmalonic aciduria: exome sequencing reveals mutations in the ACSF3 gene in patients with a non-classic phenotype. 48 : 602–605 doi:10.1136/jmedgenet-2011-100230
46. LiH, DurbinR (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 : 1754–1760 doi:10.1093/bioinformatics/btp324
47. McKennaA, HannaM, BanksE, SivachenkoA, CibulskisK, et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research 20 : 1297–1303 doi:10.1101/gr.107524.110
48. CingolaniP, PlattsA, WangLL, CoonM, NguyenT, et al. (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6 : 80–92 doi:10.4161/fly.19695
49. PailaU, KirchnerR, ChapmanBA, QuinlanAR (2013) GEMINI: integrative exploration of genetic variation and genome annotations. PLoS Comput Biol 9: e1003153 doi:10.1371/journal.pcbi.1003153
50. SherryST, WardMH, KholodovM, BakerJ, PhanL, et al. (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29 : 308–311 doi:10.1093/nar/29.1.308
51. The 1000 Genomes Project Consortium (2010) A map of human genome variation from population-scale sequencing. Nature 467 : 1061–1073 doi:10.1038/nature09534
52. Ferrer-CostaC, GelpíJL, ZamakolaL, ParragaI, la Cruz deX, et al. (2005) PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics 21 : 3176–3178 doi:10.1093/bioinformatics/bti486
53. AdzhubeiI, JordanDM, SunyaevSR (2013) Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet Chapter 7: Unit7.20–7.20.41 doi:10.1002/0471142905.hg0720s76
54. ChoiY, SimsGE, MurphyS, MillerJR, ChanAP (2012) Predicting the functional effect of amino acid substitutions and indels. 7: e46688 doi:10.1371/journal.pone.0046688
55. SchwarzJM, RödelspergerC, SchuelkeM, SeelowD (2010) MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 7 : 575–576 doi:10.1038/nmeth0810-575
56. ShihabHA, GoughJ, CooperDN, StensonPD, BarkerGLA, et al. (2013) Predicting the Functional, Molecular, and Phenotypic Consequences of Amino Acid Substitutions using Hidden Markov Models. Hum Mutat 34 : 57–65 doi:10.1002/humu.22225
57. ShihabHA, GoughJ, CooperDN, DayINM, GauntTR (2013) Predicting the functional consequences of cancer-associated amino acid substitutions. Bioinformatics 29 : 1504–1510 doi:10.1093/bioinformatics/btt182
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 10
Nejčtenější v tomto čísle
- The Master Activator of IncA/C Conjugative Plasmids Stimulates Genomic Islands and Multidrug Resistance Dissemination
- A Splice Mutation in the Gene Causes High Glycogen Content and Low Meat Quality in Pig Skeletal Muscle
- Keratin 76 Is Required for Tight Junction Function and Maintenance of the Skin Barrier
- A Role for Taiman in Insect Metamorphosis