
Gene Duplication Restores the Viability of Δ and Δ Mutants
Both replication polymerases and single-stranded DNA binding proteins (SSB, which associate with single-stranded DNA exposed transiently during replication) are ubiquitous and show high levels of functional and structural conservation across all species. Among the nine different polypeptides that compose the bacterial replicative polymerase, the HolC-HolD (χψ) complex interacts with SSB, and is crucial for normal growth in the model bacteria Escherichia coli. Interestingly, many bacterial species lack this complex, where its function is presumably carried out by other polymerase components. With the aim of better understanding HolC-HolD (χψ) complex function in E. coli, we isolated growth defect suppressor mutations of the holD mutant. We found that ssb gene duplication and the consequent doubling of SSB protein expression, renders the entire χψ complex dispensable for growth. We also show that growth-defect suppression requires the presence of the SSB C-terminal amino acids in both ssb gene copies. This C-terminal tail promotes interaction between SSB and its partner proteins. Thus, our results indicate that in vivo SSB concentration plays a key role in maintaining polymerase stability and replication efficiency, in a reaction that involves SSB interactions with protein partner(s) other than χψ.
Published in the journal:
. PLoS Genet 10(10): e32767. doi:10.1371/journal.pgen.1004719
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004719
Summary
Both replication polymerases and single-stranded DNA binding proteins (SSB, which associate with single-stranded DNA exposed transiently during replication) are ubiquitous and show high levels of functional and structural conservation across all species. Among the nine different polypeptides that compose the bacterial replicative polymerase, the HolC-HolD (χψ) complex interacts with SSB, and is crucial for normal growth in the model bacteria Escherichia coli. Interestingly, many bacterial species lack this complex, where its function is presumably carried out by other polymerase components. With the aim of better understanding HolC-HolD (χψ) complex function in E. coli, we isolated growth defect suppressor mutations of the holD mutant. We found that ssb gene duplication and the consequent doubling of SSB protein expression, renders the entire χψ complex dispensable for growth. We also show that growth-defect suppression requires the presence of the SSB C-terminal amino acids in both ssb gene copies. This C-terminal tail promotes interaction between SSB and its partner proteins. Thus, our results indicate that in vivo SSB concentration plays a key role in maintaining polymerase stability and replication efficiency, in a reaction that involves SSB interactions with protein partner(s) other than χψ.
Introduction
Chromosome replication is performed by the replisome, a molecular machine present in all living organisms with strong structural and functional similarities [1]–[4]. Replisomes combine the action of a primosome and a polymerase, for which the enzymes from Escherichia coli have proved an invaluable model for understanding their function. The E. coli primosome is itself composed of two interacting enzymes, the hexameric DnaB helicase that opens double-stranded DNA and the DnaG primase that synthesizes leading - and lagging-strand primers. DNA is synthesized by the DNA polymerase III holoenzyme (Pol III HE), composed of polypeptides encoded by 9 different genes. The holoenzyme is composed of three core polymerases [5], each made up of a polymerase α subunit (encoded by dnaE), a proofreading ε subunit (dnaQ), and a θ stability factor (holE). DNA binding by leading - and lagging - strand core polymerases is stabilized through interactions with the β-clamp (dnaN). Lagging-strand synthesis is discontinuous and Okazaki fragments (OF) are joined by the ligase. The role of the third core polymerase is still under investigation; current models suggest that it replaces the lagging-strand polymerase when needed [6]. β-clamps are loaded onto DNA for replication initiation and for the synthesis of each OF by a complex called the clamp loader. The minimal clamp loader core is a pentameric complex containing a δ (holA), a δ′ (holB) and three τ (dnaX) protein subunits. A χψ complex (holC, holD) connects this pentameric complex to DNA, as ψ (HolD) interacts with τ [7], [8] and χ (HolC) interacts with the single-stranded DNA binding proteins (SSB) that cover the lagging-strand template [9]–[12]. The three-dimensional structure of the χψ complex has been determined, identifying the sites of interaction between ψ and τ, and between χ and SSB [12], [13]. In vitro each clamp loader complex contains a single χψ complex, but four may be associated with the replisome in vivo [4], [5], [8]. How the three additional in vivo χψ complexes are organized is currently unknown.
The clamp loader complex ensures replisome cohesion through interactions between τ and Pol III, τ and the DnaB helicase, and χ and SSB [4]. Clamps and clamp-loaders are universally conserved in structure and function, for example PCNA and RFC, respectively, in eukaryotes [2]. In contrast, ψ and χ have only been found in proteobacteria [1], [14], [15] and no homologous proteins have been reported in eukaryotes. Using E. coli mutants to analyze the role of the χψ complex provides an alternative approach toward understanding chromosome replication and the molecular mechanisms that underlie clamp-loader function.
In vitro comparison of clamp loading and replication in the presence and absence of χψ have led to the identification of three main putative functions. Firstly, ψ-τ interactions stabilize the clamp loader complex, allowing it to form even at the low protein concentrations found in vivo [16]. The presence of ψ alone increases clamp-loader ATPase activity, and its affinity for DNA and the β-clamp [8], [17]. Secondly, in the presence of SSB, owing to χ-SSB interactions, χψ increases the affinity of the clamp-loader complex to primer-template DNA, stimulates clamp loading activity and increases Pol III processivity [9], [18]. Thirdly, χψ also promotes the in vitro displacement of primase from RNA primers, by switching from primase-SSB to χ-SSB interactions at the primer-template junction, and thus participating in primase recycling at replication forks [19].
The above properties indicate that χψ mainly acts on the lagging-strand template, which is SSB-coated and subject to clamp loading every 1–2 seconds. Accordingly, a holC deletion confers a hyper-recombination phenotype that can be explained by defective lagging-strand synthesis [20]. Moreover, a holD mutant was isolated in a screen for hyper-recombination mutants [21]. On the other hand, several lines of in vivo and in vitro evidence suggest that decreasing the cellular level of χ or ψ proteins affects both lagging - and leading-strand synthesis. A holD point mutation was shown to trigger replication fork reversal, which is caused by replication fork arrest and the subsequent annealing of leading - and lagging-strand ends to form a Holliday junction adjacent to a double-stranded DNA end [21], [22]. In addition, a mutation affecting holC suppressed the growth defects caused by replication over-initiation, possibly because it slows down replication fork progression [23]. Finally, in vitro studies using χ variants with impaired SSB interaction capacity revealed defects in leading-strand synthesis and resulted in the production of shorter OFs, in agreement with the idea that holC and/or holD impairment affects the synthesis of both strands during replication [12].
Deletion of the holD gene strongly affects E. coli growth at 30°C and is lethal at higher temperatures. These defects could be partially suppressed by blocking the SOS response [24]. The E. coli SOS response is triggered by the accumulation of RecA-coated single-stranded DNA (ssDNA) followed by proteolysis of the LexA repressor, inducing the expression of more than 40 LexA-repressed DNA repair genes [25], [26]. We have previously shown that the SOS-induced DinB and Pol II bypass polymerases are responsible for the deleterious effects of the SOS response on ΔholD mutant growth [24]. We proposed that a combination of replisome destabilization in the absence of ψ and displacement of the destabilized replisomes by these two SOS-induced polymerases was, at least in part, responsible for the poor growth of the ΔholD mutant [24].
In this work, we characterize a spontaneous suppressor mutation of the ΔholD growth defects and identify it as an ssb gene duplication. We find that this duplication also suppresses the growth defects of a ΔholC mutant but does not prevent ΔholD-induced expression of SOS response genes. We propose that ssb gene duplication directly compensates for the absence of the χψ complex by stabilizing the association of Pol III HE to DNA.
Results
Characterization of a ΔholD mutant growth defect suppressor
Because the ΔholD mutant is slow growing and accumulates suppressor mutations, we propagated it in the presence of a complementing plasmid that replicates from a conditional origin. The pAM-holD plasmid carries the wild-type holD gene and only replicates in the presence of the lac inducer isopropyl β-D thio-galactoside (IPTG) [24]. This plasmid was then cured prior to each experiment to restore the ΔholD mutant condition. Depending on the experiment, we either generated mixed cultures containing at least 99% cured cells by growing ΔholD [pAM-holD] cells in the absence of IPTG for about 15 generations, or isolated plasmid-less colonies by plating ΔholD [pAM-holD] cultures propagated in the absence of IPTG for 8 hours on IPTG-free medium. Cultures or colonies were confirmed to be free of both complementing plasmids and suppressor mutations. All strains used in this work carry a sfiA mutation (sfiA::MudAplacZ or sfiA11) to prevent SfiA-mediated cell division blockage upon SOS induction. When grown at 30°C on minimal medium supplemented with casamino acids (hereafter MM), ΔholD cells formed rare small colonies together with rapidly-growing colonies that we suspected to have acquired a suppressor mutation (Figure 1, Figure S1, Figure S3, [24]). Putative ΔholD suppressing clones were able to form normal sized colonies over two days when grown at 30°C on MM. We sequenced the genome of one such clone, designated JJC2394, and compared its sequence to that of the parental ΔholD mutant. Two mutations were found in JJC2394: a leuO point mutation (A233V) and a 10 kb duplication (Figure 2). Reversing the leuO mutation to Leu+ by P1 transduction did not affect JJC2394 viability. In contrast, removing the duplication abolished most of the suppression effect, indicating that it is important for the suppressor phenotype (Figure 1). The duplication is flanked by 6 base pairs (bp) microhomology DNA sequences and lies between positions 4 266 351 and 4 276 297 on the E. coli MG1655 chromosome used as the reference strain (Figure 2). The duplicated region contains 10 genes: five of unknown function, aphA encoding a periplasmic phosphatase, uvrA encoding a nucleotide excision repair protein, soxS and soxR encoding the superoxide response activator, and ssb.


Duplication of the ssb gene suppresses ΔholD mutant growth defects
Since ssb is the only one of the duplicated genes directly involved in DNA replication, we hypothesized that the presence of two copies of ssb might be responsible for the suppressor phenotype. To test this idea, we constructed a strain carrying an additional copy of ssb inserted into the argE locus, which is located approximately 120 kb from ssb. Strains harboring the argE::ssb insertion carry the same number of ssb genes as the ssb tandem duplication throughout the cell cycle, and the two ssb copies are stably maintained due to the distance separating them. Western analysis using anti-SSB antibodies showed that JJC2394 cells and cells containing the argE::ssb insertion expressed two to three times as much SSB protein as wild-type cells (Figure S2). Although over-production of SSB from a plasmid affects the growth of wild-type E. coli and induces the SOS response [27], ssb gene duplication appears to have no deleterious effect since argE::ssb did not affect the growth of wild-type cells and did not induce the SOS response (Figure 3, Table 1).


Next, we analyzed the growth properties of ΔholD argE::ssb cells. The presence of two ssb gene copies restored wild-type levels of colony formation to ΔholD mutants when grown on MM and LB, at 30°C, 37°C and 42°C (Figure 3, Figure S3). In fact, although the lexAind mutation allowed ΔholD cells to be propagated in liquid cultures [24], ΔholD lexAind colonies were slower growing and reduced in number compared to ΔholD argE::ssb (Figure 3, Figure S3).
Duplication of the ssb gene does not prevent SOS response induction in the ΔholD mutant
As preventing SOS induction improves ΔholD mutant growth [24], we tested whether the ssb duplication acts by decreasing SOS-induction. SOS expression was measured using lacZ under the control of the SOS-induced promoter sfiA in ΔholD mutants and in suppressed strains. β-galactosidase expression was measured in cells propagated either at 30°C or after shifting to 42°C for 3 hours (Table 1). As reported previously, the ΔholD mutation induces the SOS response at both temperatures and this induction is prevented by recF inactivation [24]. In the JJC2394 spontaneous-suppression mutant, the SOS response was decreased compared to the ΔholD mutant at 30°C and 42°C, but was still significantly higher than in wild-type cells. The ΔholD argE::ssb mutant exhibited a similar SOS response to that of the ΔholD single mutant, showing that the suppression phenotype conferred by ssb duplication is not a consequence of SOS inactivation. SOS expression in ΔholD argE::ssb cells and in JJC2394 was largely RecF-dependent (Table 1), in the same way as in ΔholD cells. RecF, RecO and RecR proteins specifically promote RecA loading onto ssDNA gaps. Thus, RecF dependence suggests that the SOS response in ΔholD cells is induced by the accumulation of ssDNA gaps, possibly formed during lagging-strand synthesis. Inactivating the SOS response with a lexAind mutation did not further improve the capacity of ΔholD argE::ssb cells to form colonies at different temperatures (Figure 3, Figure S3). This is consistent with the fact that SOS induction is not required for ΔholD argE::ssb cell viability and that the viability of this double mutant is equivalent to wild-type bacteria.
For historical reasons, this work was realized in an AB1157 background. In the more commonly used MG1655 strain background, suppression of ΔholD growth defects by ssb gene duplication was observed at 30°C and 37°C but not at 42°C (Figure 3). In this case, introducing the lexAind mutation slightly improved ΔholD growth. Interestingly lexAind and argE::ssb suppressor mutations had additive effects on MG1655 ΔholD viability, with the MG1655 ΔholD lexAind argE::ssb mutant exhibiting similar plating efficiency to that of wild-type MG1655 at 30°C and 37°C. Nevertheless, in contrast to the AB1157 background, colonies were smaller than wild-type at 37°C and this strain was unable to propagate at 42°C (Figure 3).
The higher SOS response levels in ΔholD argE::ssb cells compared to the ΔholD sup cells of JJC2394 suggested the presence of an additional mutation in the latter strain (Table 1). However, the mutation(s) responsible for this difference could not be identified from the chromosome sequence. lexA and recombination genes were intact and, accordingly, survival to UV irradiation was unaffected in JJC2394 (Figure S4). To test the effect of the 10 kb tandem duplication, the argE::ssb allele was introduced into JJC2394 resulting in the spontaneous loss of the duplication, presumably by homologous recombination. The resulting strain carried the argE::ssb allele instead of the spontaneous 10 kb duplication but was otherwise identical to JJC2394. Significantly, the new strain showed the same SOS response levels as the ΔholD sup strain JJC2394 rather than the reconstructed ΔholD argE::ssb strain (Table 1, JJC6216). Therefore, the lower expression of the SOS response in JJC2394 compared to ΔholD argE::ssb is not caused by another gene within the 10 kb duplication and remained unexplained.
A ∼10-fold increase in DinB expression is not detrimental to ΔholD argE::ssb cell growth
We previously showed that inactivating either of the dinB and polB SOS-induced polymerase genes improves ΔholD mutant viability at 37°C, while inactivating both restored growth at 42°C [24]. These results suggested that DinB and PolB polymerases participate in the destabilization of HolD-less Pol III HE upon SOS response induction. One possibility is that the restoration of ΔholD mutant growth by ssb duplication is linked to the destabilizing effect of SOS-induced DinB on Pol III HE. DinB levels increase about 8 - to 10-fold upon SOS induction [25], [28], [29], and increase to similar levels when expressed from a pSC101-derived vector [29]. Thus, we used the pSC101-derived vector pGB2 to compare the effects of increased expression of the wild-type dinB gene (pGB-dinB). When tested in a lexAind background to prevent SOS induction, the transformation efficiency of [pGB-dinB] was similar for wild-type and ΔholD argE::ssb mutant bacteria in all conditions, confirming that 8-fold over-production of DinB is not deleterious to the ΔholD argE::ssb mutant (JJC6133 Figure 4, Table S2 and Figure S5). Moreover, pGB-dinB was not deleterious for growth in HolD+ LexA+ or LexAdef backgrounds, confirming previous results showing that DinB expressed from a pSC101 replicon is not deleterious for growth, even in the absence of the LexA repressor [29]. In these conditions, DinB is expressed at 8 - and 30-times the wild-type chromosomal level, respectively [29], and replication in wild-type cells is only sensitive to the higher levels of DinB over-expression [30], [31]. However, pGB-dinB could not be introduced into ΔholD argE::ssb or JJC2394 cells on MM (Figure 4, Table S2); on LB, ΔholD argE::ssb [pGB-dinB] clones were obtained at 37°C and 42°C but could not be propagated (Figure S5). These results suggest that ssb gene duplication does not stabilize ψ-less Pol III HE enough to compensate for the effect of pGB plasmid-mediated DinB overexpression combined with SOS response activation.
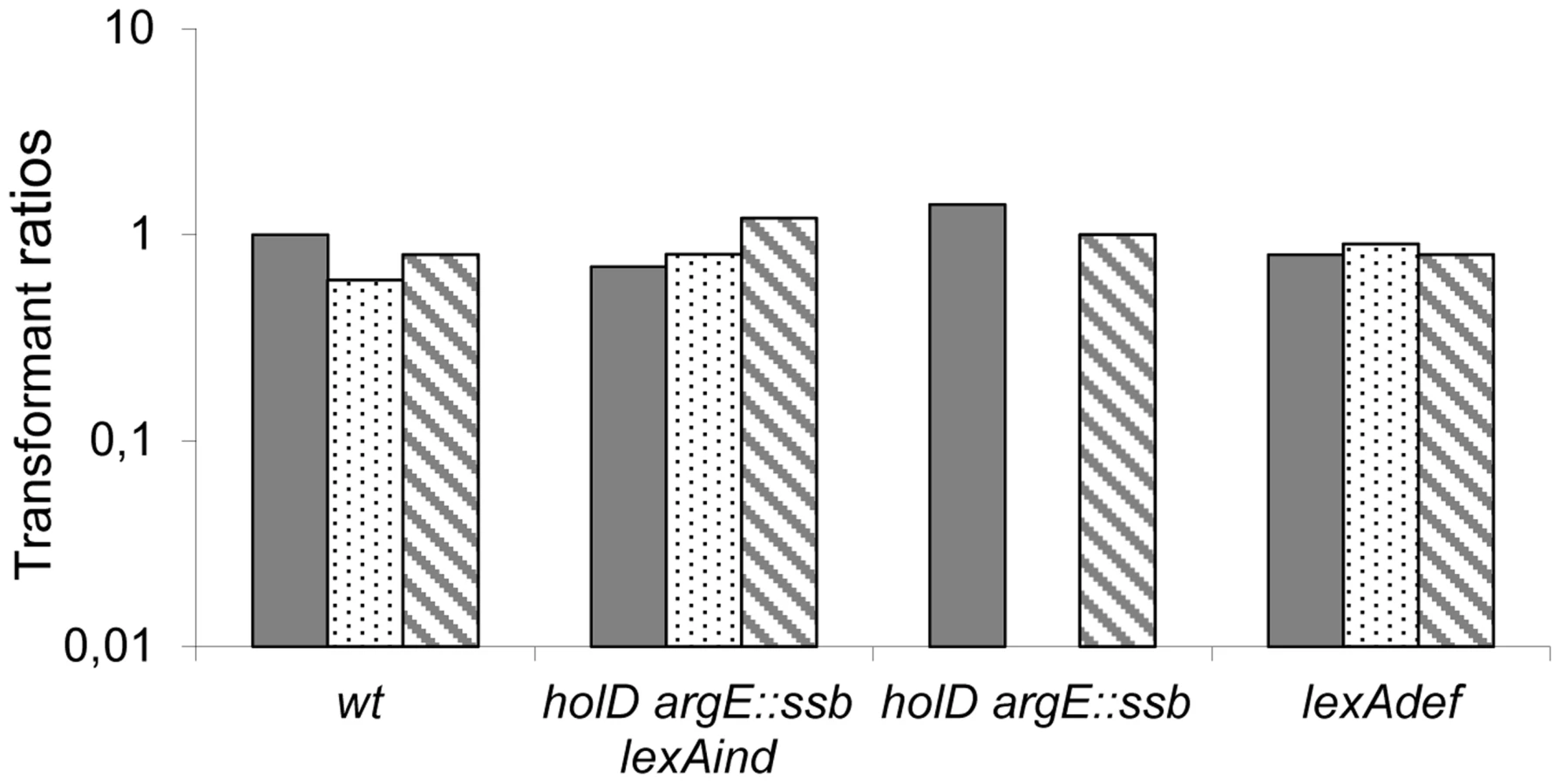
To explore the mechanism behind this effect, we performed the same assay using a dinB gene lacking 5 C-terminal amino acids required for interaction with the β-clamp (pGB-dinBΔC5 [32]). The absence of detrimental effects following expression of this deletion mutant (pGB-dinBΔC5, Figure 4, Table S2 and Figure S5) demonstrates that a functional interaction between DinB and DnaN is required for the effects of 30-fold DinB overproduction. Moreover, it shows that this interaction induces the substitution of β-clamp DnaN-bound Pol III by DinB.
Altogether, these results indicate that doubling SSB concentration in vivo protects the ΔholD mutant against the deleterious effects of a 8 - to 10-fold increase in DinB, regardless of whether this increase is caused by SOS induction, or increased dinB gene expression from a ∼10 copy-number plasmid in the absence of SOS induction. These results suggest that doubling the amount of SSB stabilizes Pol III HE DNA binding in the absence of HolD and consequently improves resistance to physiological increases in DinB levels, such as those produced by SOS induction. However, the ΔholD argE::ssb mutant remains sensitive to ∼30-fold increases in DinB production, showing that even in the presence of twice the normal amount of SSB, the HolD-less Pol III HE complex is more sensitive than the wild-type holoenzyme to non-physiological DinB amounts.
A five amino acid ssb C-terminal deletion prevents ssb-mediated suppression of ΔholD growth defects
SSB interacts with a large number of DNA replication, recombination and repair proteins via its C-terminus in both E. coli and Bacillus subtilis (reviewed in [33], [34]). In order to test whether these interactions play a role in the suppression of ΔholD defects by ssb duplication, we constructed a strain in which the additional copy of ssb inserted into argE contains a five amino acid C-terminal deletion (ssb-ΔC5; see Materials and Methods). The argE::ssb-ΔC5 allele did not affect wild-type growth and did not induce the SOS response (Figure 5, Table 1), showing that expression of this SSB truncated protein does not affect the function of the wild-type protein. Growth of the ΔholD argE::ssb-ΔC5 mutant was tested on MM and on LB at different temperatures. Compared to the ΔholD single mutant, ΔholD argE::ssb-ΔC5 was only slightly more viable on MM at 30°C and rapidly acquired suppressor mutations (Figure 5, Figure S3). We conclude from these experiments that ΔholD mutant growth defects can only be suppressed by an additional copy of ssb carrying an intact C-terminus. This result suggests that interaction(s) with SSB partner(s) are crucial for the rescue of ΔholD mutant by increased ssb gene dosage.

ssb duplication suppresses ΔholC and ΔholC ΔholD mutant growth defects
The χψ complex (HolC-HolD) bridges the minimal clamp loader complex to SSB. We hypothesized that χ (HolC) might be the SSB interacting protein required for ΔholD growth defect suppression. If doubling the amount of SSB allows χ to act without ψ, introduction of ΔholC should abolish the suppression. Alternatively, if ssb gene duplication bypasses the need for the entire χψ complex, it should also suppress the growth defects of ΔholC and ΔholC ΔholD mutants. We tested these ideas using the ΔholC102::CmR deletion mutant [20].
Growth of the ΔholC mutant was strongly affected at 42°C and only slightly affected at 30°C and 37°C, both on MM and LB (R. Maurer personal communication, Figure 6 and Figure S6). It is worth noting that ΔholC is less deleterious for growth at 30°C and 37°C than ΔholD, suggesting a role for ψ-τ (HolD-DnaX) interaction in Pol III HE stability. We constructed pAM-holC and pAM-holCD plasmids that carry wild-type copies of holC or both holC and holD genes respectively, which were cured at the onset of each experiment (see Materials and Methods). We analyzed ΔholC single, ΔholC argE::ssb double and ΔholC ΔholD argE::ssb triple mutants (Figure 6 and Figure S6) and obtained similar results regardless of whether the strains were originally constructed in the presence of pAM-holC or pAM-holCD. The ssb gene duplication conferred viability to both ΔholC single and ΔholC ΔholD double mutants at all temperatures, although at 42°C ΔholC argE::ssb and ΔholC ΔholD argE::ssb colonies were slightly smaller than wild-type. Thus, doubling the amount of SSB suppresses growth defects caused by the absence of the entire χψ complex, regardless of whether χψ function is affected by the inactivation of holC, holD or both genes. We conclude that ssb duplication suppresses the growth defects caused by a HolCD-less Pol III holoenzyme via SSB interactions with a replisome protein other than χ.

Discussion
In this work, we show that ssb gene duplication restores the viability of ΔholD cells at all temperatures. Since the SOS response remains induced in ΔholD argE::ssb cells, ssb gene duplication renders the HolD-less Pol III holoenzyme insensitive to SOS-induced levels of DinB and Pol II proteins. This observation suggests that doubling the amount of SSB stabilizes HolD-less Pol III HE DNA binding at replication forks, which increases its resistance to competing SOS-induced polymerases. Suppression was only observed when both ssb gene copies were intact and not if the second ssb copy carried a five C-terminal amino acid deletion. These results suggest that suppression bypasses the entire χψ complex and requires SSB interaction with a replisome partner other than χ.
ΔholD mutant growth defects are mainly caused by the intrinsic instability of HolD-less Pol III holoenzyme DNA binding
In vivo, the ΔholD mutant accumulates gaps, as deduced from RecF-dependent constitutive SOS expression, and suffers from replication arrest and polymerase loss, as deduced from the occurrence of replication fork reversal and from its sensitivity to SOS-induced polymerases [21], [24]. Accordingly, purified χ proteins containing a mutation that specifically affects SSB interaction were clearly impaired for both leading - and lagging-strand synthesis and for replisome stability [12]. The well-documented importance of χψ for lagging-strand synthesis in vitro [16], [17], [19] suggests that the gaps that induce the SOS-response in vivo are formed on the lagging strand. We observe that ssb gene duplication restores the viability of ΔholD mutant cells but does not prevent RecF-dependent SOS induction, thus does not suppress gap formation. Therefore, excessive gap formation is not directly responsible for the poor viability of the ΔholD mutant. Furthermore, ssb duplication restores normal ΔholD growth in the presence of an 8 to 10-fold excess of DinB, expressed either from the SOS-induced chromosomal copy or from a low copy plasmid in the absence of SOS induction. Consequently, we propose that an intrinsic lack of stability of HolD-less Pol III HE bound to DNA is responsible for the growth defects of the ΔholD mutant, and that ssb gene duplication acts by stabilizing the HolD-less Pol III holoenzyme. It is worth noting that competition by SOS-induced polymerases is not the only reason for HolD-less Pol III HE instability, as lexAind mutation does not suppress ΔholD growth defects as efficiently as ssb gene duplication.
In the MG1655 background, suppression of ΔholD growth defects by either lexAind or ssb duplication is partial, showing that the effects of these two suppressor mutations are additive. We propose that a combination of decreased expression of SOS-induced polymerases (lexAind) and increased Pol III HE DNA stability (ssb duplication) is necessary to restore viability in this background. In AB1157, where ssb duplication suppresses ΔholD growth defects quite efficiently, the additive effects of lexAind and ssb duplication are not directly detectable. The thermosensitivity of the ΔholD argE::ssb lexAind MG1655 mutant, also observed for the ΔholC AB1157 single mutant, is interesting since no protein is intrinsically sensitive to high temperature in these mutants. It cannot be accounted for solely by a higher number of replication forks per chromosome at 42°C compared to 30°C, since the number of replication forks per chromosome is also increased in rich medium and these mutants show no rich medium sensitivity. High temperature affects protein-protein and protein-DNA interactions and the sensitivity of these mutants to high temperature supports the idea that the primary defect of the HolCD-less Pol III holoenzyme is its intrinsic instability on DNA. In agreement with a direct role for HolD in clamp loader complex stability in vitro [8], [17], growth is clearly more affected at both 30°C and 37°C in ΔholD mutant cells than in cells lacking holC.
ssb duplication stabilizes the HolCD-less Pol III holoenzyme
In vitro SSB binds ssDNA in multiple binding modes, among which the two major forms are (SSB)35 and (SSB)65, where 35 and 65 nucleotides, respectively, are wrapped around a SSB tetramer [35]. SSB proteins are also mobile on ssDNA, undergoing random diffusion along ssDNA mainly in the (SSB)65 binding mode (Zhou et al. 2011). The (SSB)35 binding mode is less mobile, more stable, and highly cooperative, forming protein clusters or filaments on DNA [35]–[37]. The binding mode is determined by salt concentration and by the SSB protein to ssDNA ratio. Increasing SSB concentration in vitro shifts the binding mode toward the (SSB)35 form [36]. It has been proposed that the binding mode could also be influenced by protein interactors, and actually interaction between PriC and the C-terminal tail of SSB can also shift the ssDNA binding mode from (SSB)65 to (SSB)35 [38]. The primary effect of ssb gene duplication and the resulting increase in SSB protein concentration could involve a shift from the (SSB)65 to the (SSB)35 binding mode on the lagging-strand template at in vivo salt concentration. This phenomenon could compensate for the absence of the χψ complex if χ-SSB interaction is normally responsible for the shift, as has been hypothesized [36]. Nevertheless, it should be noted that to date the existence of different SSB binding modes, and their dependence on SSB concentration and on SSB-protein interactions have only been demonstrated in vitro and remain to be tested in vivo.
A C-terminal SSB truncation promotes ssDNA binding and shifts the equilibrium toward the (SSB)35 mode in vitro [10], [36], [39], [40]. Therefore, it is unlikely that this deletion prevents a putative shift from (SSB)65 to (SSB)35 in cells expressing both wild-type and truncated SSB. Thus, the requirement for two intact ssb genes to suppress ΔholD mutant growth defects may instead reflect a need for SSB interaction(s) with one or more protein partner(s) [33]. Three replisome proteins have been reported to interact with SSB: χ, primase and the α polymerase (DnaE) [33], [41]. χ is not required for suppression since ΔholC and ΔholC ΔholD mutants are also fully suppressed by the ssb gene duplication. The SSB and the primase appear to interact via SSB C-terminal amino acids and a specific region of the primase [42]. Primase could be a key SSB interacting protein for stabilization of the HolD-less Pol III holoenzyme, although its requirement for the OF synthesis and the high level of SOS induction in ΔholD argE::ssb cells suggests that gap formation during OF synthesis is not suppressed by ssb gene duplication. The SSB-DnaE interaction was detected in a Tap-Tag high-throughput analysis of E. coli proteins using DnaE as bait and SSB as prey [41]. Even though the protein regions involved in SSB and DnaE interaction have not yet been identified, this interaction could also be crucial for the growth of HolD-less Pol III containing cells. Interaction between the SSB C-terminus and a Pol III holoenzyme component other than χ has been shown to stimulate initiation complex formation [43]. In cells lacking χψ, DnaE-SSB interaction could be needed for OF initiation, and throughout lagging-strand synthesis for a stabilizing effect of the (SSB)35 binding mode on the Pol III holoenzyme. In vitro experiments would be required to test these various hypotheses.
It is remarkable that simply doubling the amount of SSB has such a large effect on viability, even though the ssb gene is not known to be regulated and strong SSB over-production is deleterious [27]. The striking effects of increased SSB expression on viability suggest that the in vivo SSB-DNA complex equilibrium is finely balanced between binding modes and can be switched by different factors, including SSB concentration and SSB interactors. It is noteworthy that ψ is present only in a few bacterial species [14], and although χ is more widely distributed, it is not universal [1], [14], [15]. Since stabilization of Pol III can apparently be achieved by SSB interaction with a Pol III holoenzyme component other than HolC provided that the amount of SSB is doubled, bacteria which lack χψ may tune the stability of their Pol III holoenzyme via one of the minimal clamp-loader subunits, for example through SSB interactions that do not exist in E. coli, by a stronger interaction with DnaE, or by naturally expressing higher amounts of SSB than in E. coli, together with lower levels of competing SOS-induced polymerases.
Materials and Methods
Strains and constructions
Strains, plasmids and oligonucleotides used in this work are described in Table S1. New mutations were constructed by recombineering as described in [44] and using DY330 [45]. All other strains were constructed by P1 transduction. pAM-holD plasmid was cured prior to each experiment by growing cells in the absence of IPTG and plasmid-less colonies were isolated on minimal medium glucose casaminoacids (MM) plates. We checked that less than 5% of cells in the culture contained pAM-holD and less than 1% had acquired a suppressor mutation. All mutations introduced by P1 transduction were checked by PCR and all new mutations constructed by recombineering were checked by PCR and sequencing. lexAind and recF mutations were tested by measuring UV sensitivity. For argE::ssb construction, DY330 was transformed by electroporation with a ssb-KanR PCR fragment flanked by 50 bp of homology with argE. For argE::ssbΔC5, DY330 - argE::ssb (JJC5953) was transformed by electroporation with a PCR CmR fragment flanked by 50 bp homology with SSB C-ter sequence lacking the five last residues and 50 bp of homology with argE. In this construction the 5 last SSB residues are replaced by two stop codons and the KanR gene in argE::ssb is replaced by CmR. For pAM-holC construction, holC was PCR amplified from the chromosome and cloned into the pAM34 vector after digestion with PstI and HindIII; the resulting plasmid was verified by sequencing and complementation of the holC mutant. For pAM-holCD construction, holC was cloned from pAHM101 [12] in pAM-holD using Ssp1/BsaB1 and Xba1; the resulting plasmid was verified by PCR and complementation of the holC mutant.
Viability measurement
For spot assays, colonies formed in three days on MM at 30°C were suspended in MM salt medium. Serial 10-fold dilutions were then performed and 7 µl of dilutions 10−2 to 10−6 were spotted on three MM and three LB plates that were placed at 30°C, 37°C and 42°C. For pGB-dinB transformants, 10−2 to 10−5 dilutions of colonies obtained overnight on LB were used. In all cases, plates were scanned after 16–24 hours of incubation at 37°C and 42°C, and after 2 days of incubation at 30°C. Spot assays were performed at least twice for each strain. In addition, for viable strains colony forming units (cfu) were determined by plating appropriate dilutions of overnight MM cultures on MM and LB plates. The number of colony was counted after 16–24 hours of incubation for plates at 37°C and 42°C and after 48 hours of incubation for plates at 30°C. Each strain was tested at least three times and results confirmed the full viability observed in spot assays. Non-viable mutants could not be grown overnight and therefore the lack of viability was also checked by streaking several isolated plasmid-less colonies on LB and MM at 30°C, 37°C and 42°C.
Genome sequencing
Chromosomal DNA was extracted by SDS-proteinase K cell lysis, followed with phenol-chloroform, chloroform-isoamyl alcohol chromosome purification and isopropanol precipitation. 5 µg of DNA were used to generate a genomic library according to Illumina's protocol. The sequencing was performed at the High-throughput Sequencing Department of IMAGIF platform (https://www.imagif.cnrs.fr/plateforme-36-High-throughput_Sequencing_Platform.html, CNRS, Gif-Sur-Yvette, France). The library was sequenced paired-ends, with a read length of 36b, on a GAIIx to an expected depth of 50×. Sequence of the isogenic wild-type strain JJC40 was determined in parallel and reads from mutant and wild-type genomes were aligned using Illumina's package CASAVA-1.7.0. The point mutation was detected by Illumina's package CASAVA-1.7.0, and the 10 kb duplication using Illumina's software GenomeStudio.
β-galactosidase assays
β-galactosidase assays for measures of SOS induction were performed as described [46]. Since isolated JJC2067 colonies could not be propagated owing to the growth advantage of suppressor mutations, pAM-holD containing clones were grown overnight in MM lacking IPTG and diluted 50 fold in MM for the experiment. Cultures were checked for the loss of pAM-holD and for containing at most 1% suppressor mutations. The same procedure was used for JJC2068 as a control.
Immunodetection assays
SSB and FtsZ proteins were detected in cell extracts using polyclonal chicken antibodies against SSB (gift from MM Cox, University of Wisconsin-Madison) and polyclonal rabbit antibodies against FtsZ (gift from J Camberg, National Institutes of Health, Bethesda, Maryland). Cell extracts were prepared from a fixed amount of exponentially growing cells. The cells were resuspended in 100 µl of Laemmli Buffer (Bio-Rad #161-0737) and incubated for 10 min at 100°C. Total cellular proteins were fractionated by SDS-PAGE on 12.5% gels and transferred to a Hybond Nitrocellulose membrane (Amersham) by electroblotting using a semidry transfer system. Immunodetection was carried out as described in the ECL+ kit (Amersham). Western blots were revealed using LAS-3000 FujiFilm and quantified with ImageQuant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. RobinsonA, CauserRJ, DixonNE (2012) Architecture and conservation of the bacterial DNA replication machinery, an underexploited drug target. Curr Drug Targets 13 : 352–372.
2. IndianiC, O'DonnellM (2006) The replication clamp-loading machine at work in the three domains of life. Nat Rev Mol Cell Biol 7 : 751–761.
3. LangstonLD, IndianiC, O'DonnellM (2009) Whither the replisome: emerging perspectives on the dynamic nature of the DNA replication machinery. Cell Cycle 8 : 2686–2691.
4. McHenryCS (2011) DNA replicases from a bacterial perspective. Annu Rev Biochem 80 : 403–436.
5. Reyes-LamotheR, SherrattDJ, LeakeMC (2010) Stoichiometry and architecture of active DNA replication machinery in Escherichia coli. Science 328 : 498–501.
6. GeorgescuRE, KurthI, O'DonnellME (2012) Single-molecule studies reveal the function of a third polymerase in the replisome. Nat Struct Mol Biol 19 : 113–116.
7. XiaoH, DongZ, O'DonnellM (1993) DNA polymerase III accessory proteins. IV. Characterization of chi and psi. J Biol Chem 268 : 11779–11784.
8. SimonettaKR, KazmirskiSL, GoedkenER, CantorAJ, KelchBA, et al. (2009) The mechanism of ATP-dependent primer-template recognition by a clamp loader complex. Cell 137 : 659–671.
9. GloverBP, McHenryCS (1998) The chi psi subunits of DNA polymerase III holoenzyme bind to single-stranded DNA-binding protein (SSB) and facilitate replication of an SSB-coated template. Journal of Biological Chemistry 273 : 23476–23484.
10. KozlovAG, CoxMM, LohmanTM (2010) Regulation of single-stranded DNA binding by the C termini of Escherichia coli single-stranded DNA-binding (SSB) protein. J Biol Chem 285 : 17246–17252.
11. NaueN, FedorovR, PichA, MansteinDJ, CurthU (2010) Site-directed mutagenesis of the chi subunit of DNA polymerase III and single-stranded DNA-binding protein of E. coli reveals key residues for their interaction. Nucleic Acids Res 39 : 1398–1407.
12. MarceauAH, BahngS, MassoniSC, GeorgeNP, SandlerSJ, et al. (2011) Structure of the SSB-DNA polymerase III interface and its role in DNA replication. Embo J 30 : 4236–4247.
13. GulbisJM, KazmirskiSL, FinkelsteinJ, KelmanZ, O'DonnellM, et al. (2004) Crystal structure of the chi:psi sub-assembly of the Escherichia coli DNA polymerase clamp-loader complex. Eur J Biochem 271 : 439–449.
14. BrezellecP, HoebekeM, HietMS, PasekS, FeratJL (2006) DomainSieve: a protein domain-based screen that led to the identification of dam-associated genes with potential link to DNA maintenance. Bioinformatics 22 : 1935–1941.
15. JarvisTC, BeaudryAA, BullardJM, OchsnerU, DallmannHG, et al. (2005) Discovery and characterization of the cryptic psi subunit of the pseudomonad DNA replicase. J Biol Chem 280 : 40465–40473.
16. OlsonMW, DallmannHG, McHenryCS (1995) DnaX complex of Escherichia coli DNA polymerase III holoenzyme. The chi psi complex functions by increasing the affinity of tau and gamma for delta.delta' to a physiologically relevant range. J Biol Chem 270 : 29570–29577.
17. AndersonSG, WilliamsCR, O'DonnellM, BloomLB (2007) A function for the psi subunit in loading the Escherichia coli DNA polymerase sliding clamp. J Biol Chem 282 : 7035–7045.
18. KelmanZ, YuzhakovA, AndjelkovicJ, ODonnellM (1998) Devoted to the lagging strand - the chi subunit of DNA polymerase III holoenzyme contacts SSB to promote processive elongation and sliding clamp assembly. EMBO Journal 17 : 2436–2449.
19. YuzhakovA, KelmanZ, ODonnellM (1999) Trading places on DNA-a three-point switch underlies primer handoff from primase to the replicative DNA polymerase. Cell 96 : 153–163.
20. SavesonCJ, LovettST (1997) Enhanced deletion formation by aberrant DNA replication in Escherichia coli. Genetics 146 : 457–470.
21. FloresMJ, BierneH, EhrlichSD, MichelB (2001) Impairment of lagging strand synthesis triggers the formation of a RuvABC substrate at replication forks. Embo J 20 : 619–629.
22. BaharogluZ, PetranovicM, FloresMJ, MichelB (2006) RuvAB is essential for replication forks reversal in certain replication mutants. Embo J 25 : 596–604.
23. NordmanJ, SkovgaardO, WrightA (2007) A novel class of mutations that affect DNA replication in E. coli. Mol Microbiol 64 : 125–138.
24. VigueraE, PetranovicM, ZahradkaD, GermainK, EhrlichDS, et al. (2003) Lethality of bypass polymerases in Escherichia coli cells with a defective clamp loader complex of DNA polymerase III. Mol Microbiol 50 : 193–204.
25. CourcelleJ, KhodurskyA, PeterB, BrownPO, HanawaltPC (2001) Comparative gene expression profiles following UV exposure in wild - type and SOS-deficient Escherichia coli. Genetics 158 : 41–64.
26. MichelB (2005) After 30 years of study, the bacterial SOS response still surprises us. PLoS Biol 3: e255.
27. MoreauPL (1988) Overproduction of single-stranded-DNA-binding protein specifically inhibits recombination of UV-irradiated bacteriophage DNA in Escherichia coli. J Bacteriol 170 : 2493–2500.
28. KimSR, MatsuiK, YamadaM, GruzP, NohmiT (2001) Roles of chromosomal and episomal dinB genes encoding DNA pol IV in targeted and untargeted mutagenesis in Escherichia coli. Mol Genet Genomics 266 : 207–215.
29. HeltzelJM, MaulRW, WolffDW, SuttonMD (2012) Escherichia coli DNA polymerase IV (Pol IV), but not Pol II, dynamically switches with a stalled Pol III* replicase. J Bacteriol 194 : 3589–3600.
30. IndianiC, LangstonLD, YurievaO, GoodmanMF, O'DonnellM (2009) Translesion DNA polymerases remodel the replisome and alter the speed of the replicative helicase. Proc Natl Acad Sci U S A 106 : 6031–6038.
31. IkedaM, ShinozakiY, UchidaK, OhshikaY, FurukohriA, et al. (2012) Quick replication fork stop by overproduction of Escherichia coli DinB produces non-proliferative cells with an aberrant chromosome. Genes Genet Syst 87 : 221–231.
32. Lenne-SamuelN, WagnerJ, EtienneH, FuchsRP (2002) The processivity factor beta controls DNA polymerase IV traffic during spontaneous mutagenesis and translesion synthesis in vivo. EMBO Rep 3 : 45–49.
33. SheredaRD, KozlovAG, LohmanTM, CoxMM, KeckJL (2008) SSB as an organizer/mobilizer of genome maintenance complexes. Crit Rev Biochem Mol Biol 43 : 289–318.
34. CostesA, LecointeF, McGovernS, Quevillon-CheruelS, PolardP (2010) The C-terminal domain of the bacterial SSB protein acts as a DNA maintenance hub at active chromosome replication forks. PLoS Genet 6: e1001238.
35. LohmanTM, FerrariME (1994) Escherichia coli single-stranded DNA-binding protein: Multiple DNA-binding modes and cooperativities. Annual Review of Biochemistry 63 : 527–570.
36. RoyR, KozlovAG, LohmanTM, HaT (2007) Dynamic structural rearrangements between DNA binding modes of E. coli SSB protein. J Mol Biol 369 : 1244–1257.
37. ZhouR, KozlovAG, RoyR, ZhangJ, KorolevS, et al. (2011) SSB Functions as a Sliding Platform that Migrates on DNA via Reptation. Cell 146 : 222–232.
38. WesselSR, MarceauAH, MassoniSC, ZhouR, HaT, et al. (2013) PriC-mediated DNA replication restart requires PriC complex formation with the single-stranded DNA-binding protein. J Biol Chem 288 : 17569–17578.
39. AntonyE, WeilandE, YuanQ, ManhartCM, NguyenB, et al. (2013) Multiple C-terminal tails within a single E. coli SSB homotetramer coordinate DNA replication and repair. J Mol Biol 425 : 4802–4819.
40. SuXC, WangY, YagiH, ShishmarevD, MasonCE, et al. (2014) Bound or free: interaction of the C-terminal domain of Escherichia coli single-stranded DNA-binding protein (SSB) with the tetrameric core of SSB. Biochemistry 53 : 1925–1934.
41. ButlandG, Peregrin-AlvarezJM, LiJ, YangW, YangX, et al. (2005) Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature 433 : 531–537.
42. NaueN, BeerbaumM, BogutzkiA, SchmiederP, CurthU (2013) The helicase-binding domain of Escherichia coli DnaG primase interacts with the highly conserved C-terminal region of single-stranded DNA-binding protein. Nucleic Acids Res 41 : 4507–4517.
43. DowneyCD, McHenryCS (2010) Chaperoning of a replicative polymerase onto a newly assembled DNA-bound sliding clamp by the clamp loader. Mol Cell 37 : 481–491.
44. DatsenkoKA, WannerBL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97 : 6640–6645.
45. YuD, EllisHM, LeeEC, JenkinsNA, CopelandNG, et al. (2000) An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci U S A 97 : 5978–5983.
46. Miller JH (1992) A short course in bacterial genetic: Cold Spring Harbor, New York Cold Spring Harbor Press.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 10
Nejčtenější v tomto čísle
- The Master Activator of IncA/C Conjugative Plasmids Stimulates Genomic Islands and Multidrug Resistance Dissemination
- A Splice Mutation in the Gene Causes High Glycogen Content and Low Meat Quality in Pig Skeletal Muscle
- Keratin 76 Is Required for Tight Junction Function and Maintenance of the Skin Barrier
- A Role for Taiman in Insect Metamorphosis